Nanoparticles Targeting Receptors on Breast Cancer for Efficient Delivery of Chemotherapeutics



Abstract
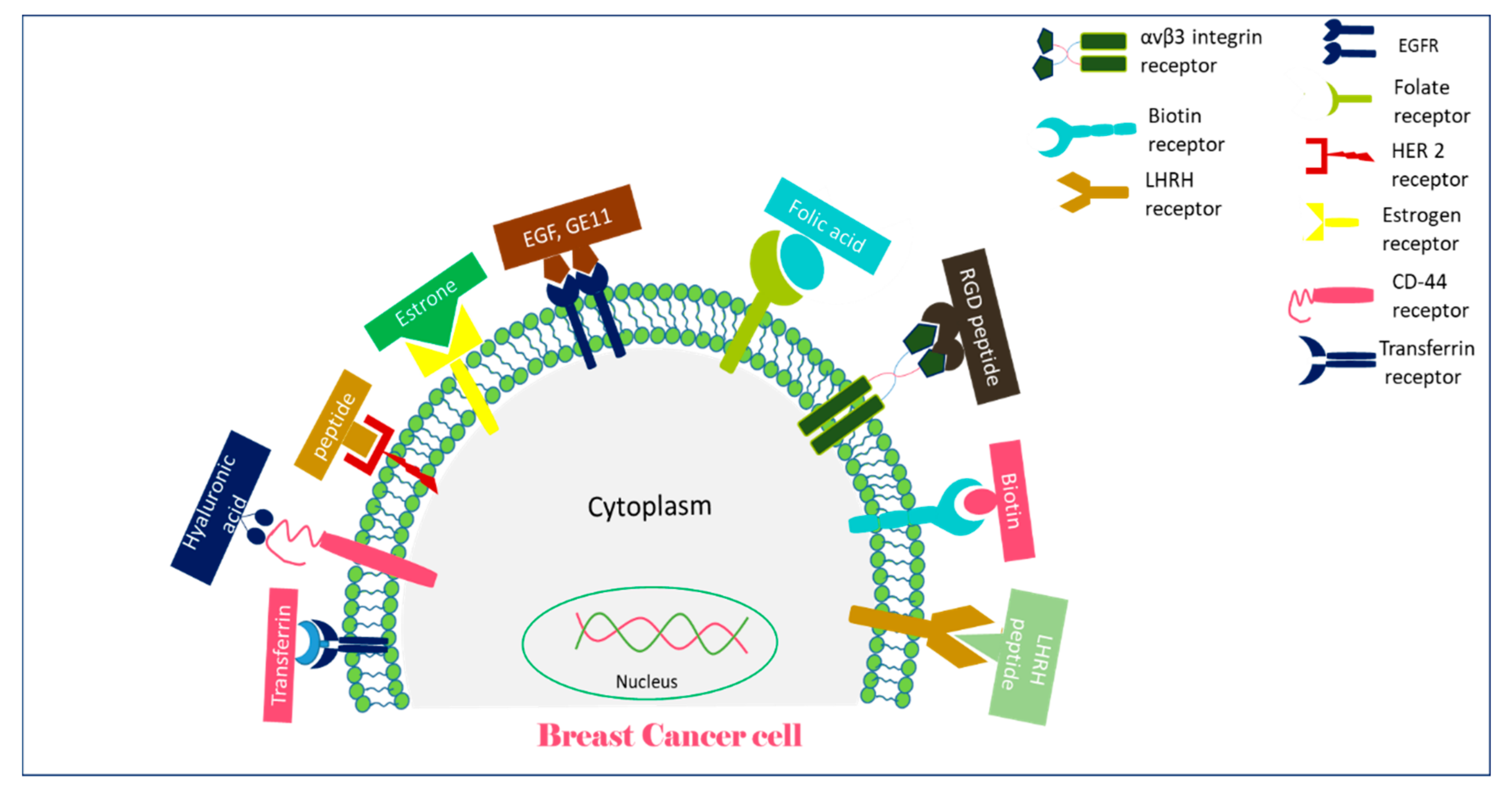
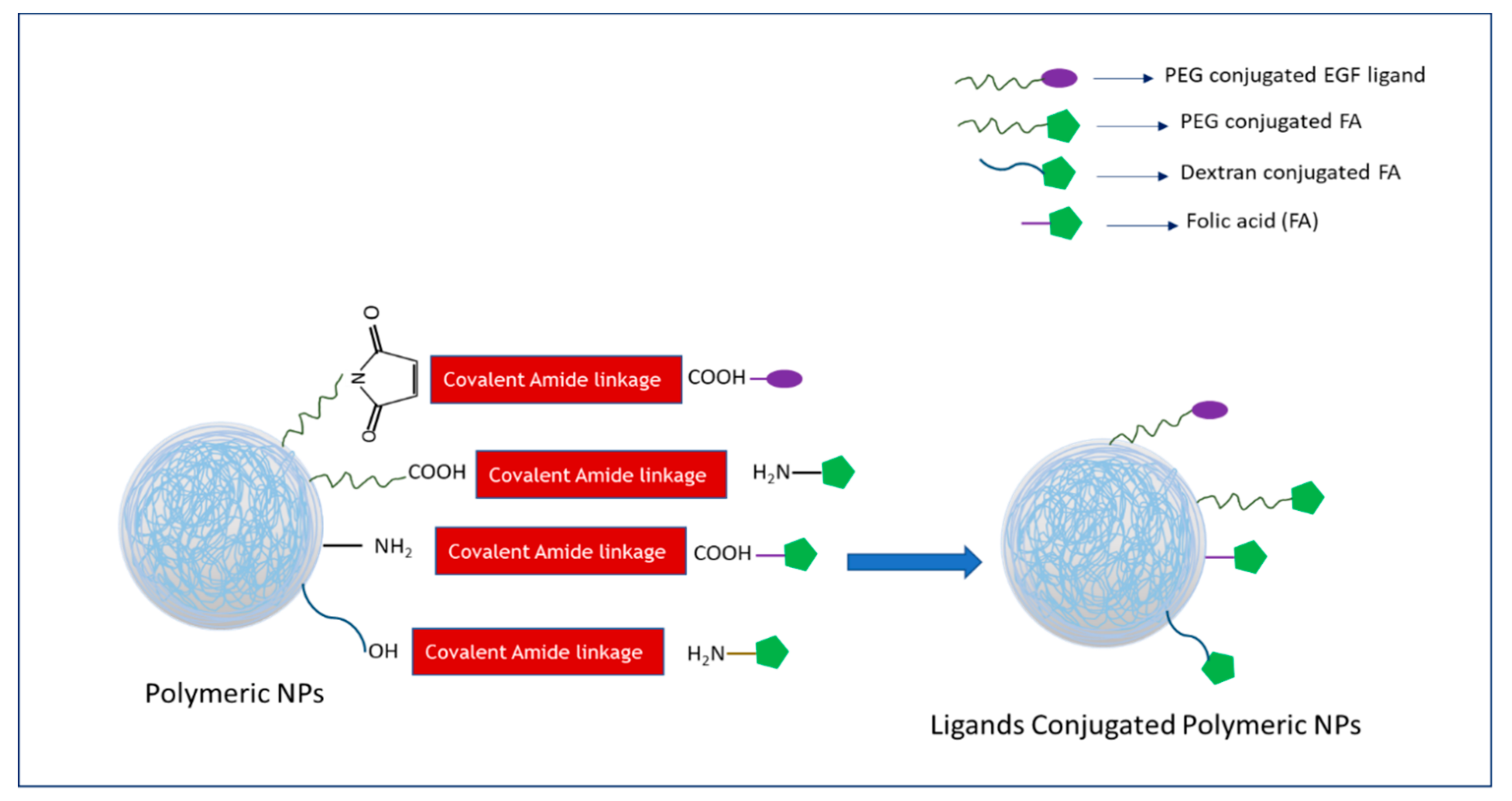
:1. Introduction
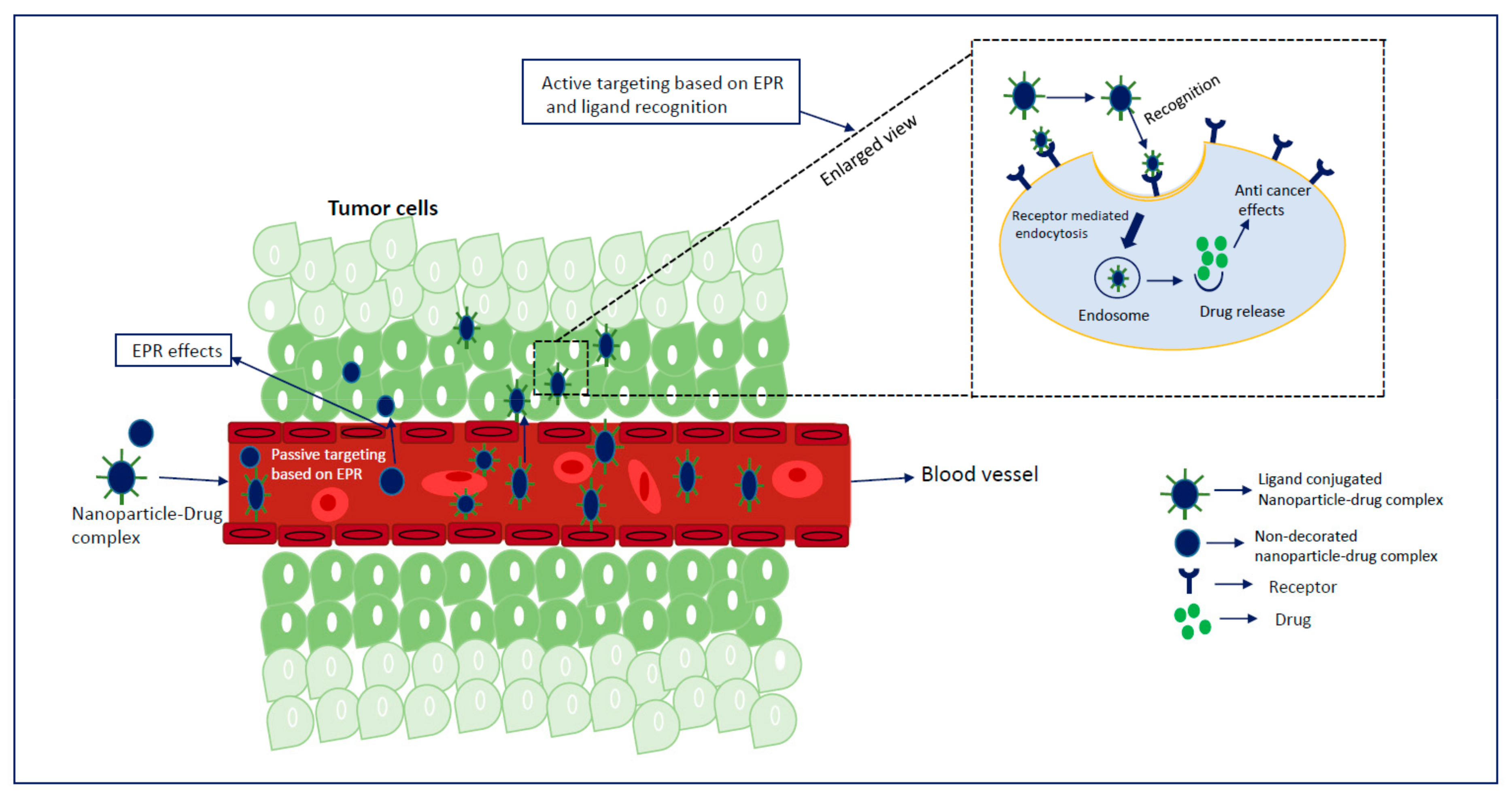
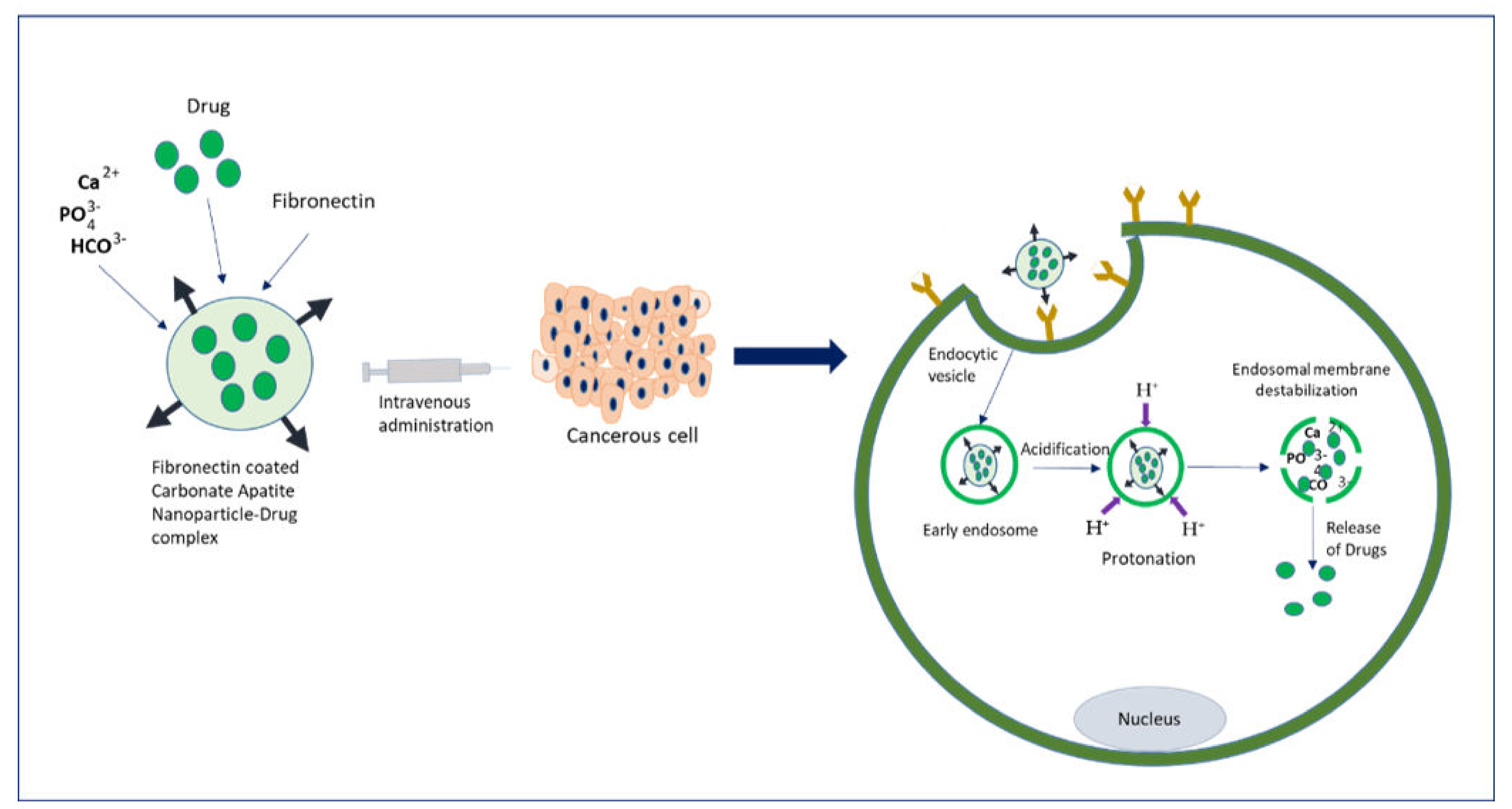
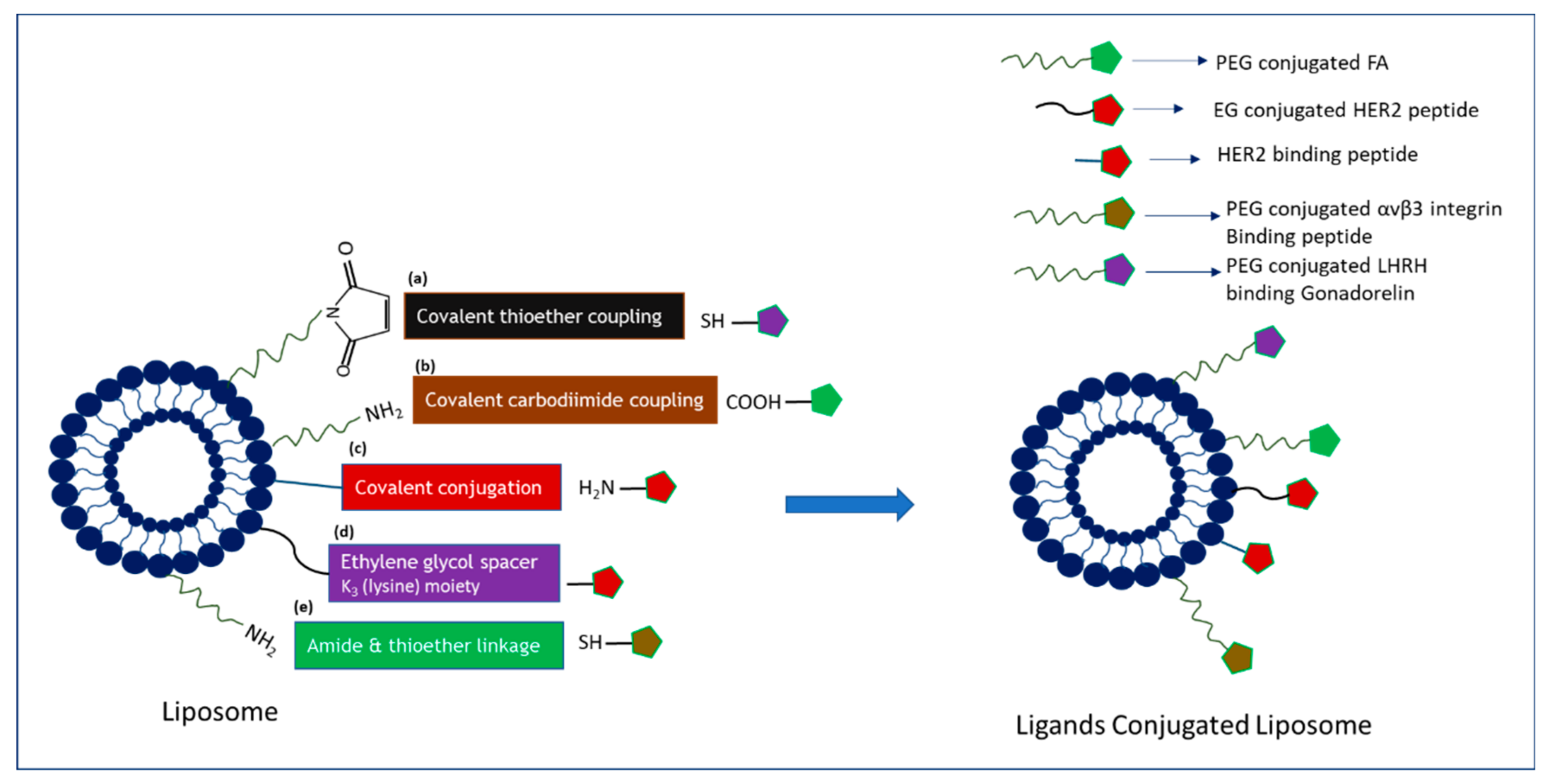
2. Insights of NP-Based Targeted Drug Delivery
3. Receptor-Targeted Delivery of Chemotherapeutic Agents in Mouse Breast Cancer Models
3.1. EGFR-Targeted Drug-Loaded NPs
3.2. Folate Receptor-Targeted Drug-Loaded NPs
3.3. HER-2 Targeted Drug-Loaded NPs
3.4. Estrogen Receptors-Targeted Drug-Loaded NPs
3.5. CD-44 Targeted Drug-Loaded NPs
3.6. Transferrin-Receptor Targeted Drug-Loaded NPs
3.7. αvβ3 Integrin-Targeted Drug-Loaded NPs
3.8. Biotin Receptor-Targeted Drug-Loaded NPs
3.9. LHRH Receptor-Targeted Drug-Loaded NPs
4. Therapeutic Evaluation and Translational Gap of Receptor-Targeted Drug Delivery
5. Conclusions and Future Prospective
Author Contributions
Funding
Conflicts of Interest
References
- Pashayan, N.; Antoniou, A.C.; Ivanus, U.; Esserman, L.J.; Easton, D.F.; French, D.; Sroczynsk, G.; Hall, P.; Cuzick, J.; Evans, D.G.; et al. Personalized early detection and prevention of breast cancer: ENVISION consensus statement. Nat. Rev. Clin. Oncol. 2020, 17, 687–705. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer (Primer). Nat. Rev. Dis. Primers 2019. [Google Scholar] [CrossRef] [PubMed]
- Muley, H.; Fadó, R.; Rodríguez-Rodríguez, R.; Casals, N. Drug uptake-based chemoresistance in breast cancer treatment. Biochem. Pharmacol. 2020, 177, 113959. [Google Scholar] [CrossRef] [PubMed]
- Amjad, E.; Asnaashari, S.; Sokouti, B.; Dastmalchi, S. Systems biology comprehensive analysis on breast cancer for identification of key gene modules and genes associated with TNM-based clinical stages. Sci. Rep. 2020, 10, 10816. [Google Scholar] [CrossRef]
- Tran, P.; Lee, S.-E.; Kim, D.-H.; Pyo, Y.-C.; Park, J.-S. Recent advances of nanotechnology for the delivery of anticancer drugs for breast cancer treatment. J. Pharm. Investig. 2020, 50, 261–270. [Google Scholar] [CrossRef]
- Mittal, P.; Singh, S.; Singh, A.; Singh, I.K. Current advances in drug delivery systems for treatment of Triple negative breast cancer (TNBC). Chem. Biol. Lett. 2020, 7, 1–12. [Google Scholar]
- Koh, J.; Kim, M.J. Introduction of a new staging system of breast cancer for radiologists: An emphasis on the prognostic stage. Korean J. Radiol. 2019, 20, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Cserni, G.; Chmielik, E.; Cserni, B.; Tot, T. The new TNM-based staging of breast cancer. Virchows Arch. 2018, 472, 697–703. [Google Scholar] [CrossRef]
- Grobmyer, S.R.; Zhou, G.; Gutwein, L.G.; Iwakuma, N.; Sharma, P.; Hochwald, S.N. Nanoparticle delivery for metastatic breast cancer. Nanomed. Nanotechnol. Biol. Med. 2012, 8, S21–S30. [Google Scholar] [CrossRef]
- Mutebi, M.; Anderson, B.O.; Duggan, C.; Adebamowo, C.; Agarwal, G.; Ali, Z.; Bird, P.; Bourque, J.M.; DeBoer, R.; Gebrim, L.H.; et al. Breast cancer treatment: A phased approach to implementation. Cancer 2020, 126, 2365–2378. [Google Scholar] [CrossRef]
- Sridharan, S.; Howard, C.M.; Tilley, A.M.; Subramaniyan, B.; Tiwari, A.K.; Ruch, R.J.; Raman, D. Novel and Alternative Targets Against Breast Cancer Stemness to Combat Chemoresistance. Front. Oncol. 2019, 9, 1003. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Chen, Z.; Shin, D.M. Nanoparticle therapeutics: An emerging treatment modality for cancer. Nanosci. Technol. A Collect. Rev. Nat. J. World Sci. 2010, 239–250. [Google Scholar] [CrossRef]
- Alqaraghuli, H.G.J.; Kashanian, S.; Rafipour, R. A Review on Targeting Nanoparticles for Breast Cancer. Curr. Pharm. Biotechnol. 2019, 20, 1087–1107. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update. Bioeng. Transl. Med. 2019, 4, e10143. [Google Scholar] [CrossRef] [Green Version]
- Van der Meel, R.; Sulheim, E.; Shi, Y.; Kiessling, F.; Mulder, W.J.; Lammers, T. Smart cancer nanomedicine. Nat. Nanotechnol. 2019, 14, 1007–1017. [Google Scholar] [CrossRef]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef]
- Kumari, P.; Ghosh, B.; Biswas, S. Nanocarriers for cancer-targeted drug delivery. J. Drug Target. 2016, 24, 179–191. [Google Scholar] [CrossRef]
- Piddock, L.J. Multidrug-resistance efflux pumps? Not just for resistance. Nat. Rev. Microbiol. 2006, 4, 629–636. [Google Scholar] [CrossRef]
- Prasad, R.; Jain, N.K.; Yadav, A.S.; Chauhan, D.S.; Devrukhkar, J.; Kumawat, M.K.; Shinde, S.; Gorain, M.; Thakor, A.S.; Kundu, G.C.; et al. Liposomal nanotheranostics for multimode targeted in vivo bioimaging and near-infrared light mediated cancer therapy. Commun. Biol. 2020, 3, 1–14. [Google Scholar] [CrossRef]
- Nag, O.K.; Delehanty, J.B. Active cellular and subcellular targeting of nanoparticles for drug delivery. Pharmaceutics 2019, 11, 543. [Google Scholar] [CrossRef] [Green Version]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, M.; Azadi, A.; Rafiei, P.; Ashrafi, H. A pharmacokinetic overview of nanotechnology-based drug delivery systems: An ADME-oriented approach. Crit. Rev. Ther. Drug Carr. Syst. 2013, 30, 435–467. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Wei, T.; Goldberg, H.; Wang, W.; Cullion, K.; Kohane, D.S. Getting drugs across biological barriers. Adv. Mater. 2017, 29, 1606596. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, A.C.; Mitragotri, S. An overview of clinical and commercial impact of drug delivery systems. J. Control. Release 2014, 190, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Bender, A.; Scheiber, J.; Glick, M.; Davies, J.W.; Azzaoui, K.; Hamon, J.; Urban, L.; Whitebread, S.; Jenkins, J.L. Analysis of pharmacology data and the prediction of adverse drug reactions and off-target effects from chemical structure. ChemMedChem 2007, 2, 861–873. [Google Scholar] [CrossRef]
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef]
- Mo, R.; Jiang, T.; Gu, Z. Recent progress in multidrug delivery to cancer cells by liposomes. Nanomedicine 2014, 9, 1117–1120. [Google Scholar] [CrossRef]
- Shih, H.; Lin, C.-C. Photoclick hydrogels prepared from functionalized cyclodextrin and poly (ethylene glycol) for drug delivery and in situ cell encapsulation. Biomacromolecules 2015, 16, 1915–1923. [Google Scholar] [CrossRef] [Green Version]
- Palanikumar, L.; Al-Hosani, S.; Kalmouni, M.; Nguyen, V.P.; Ali, L.; Pasricha, R.; Barrera, F.N.; Magzoub, M. pH-responsive high stability polymeric nanoparticles for targeted delivery of anticancer therapeutics. Commun. Biol. 2020, 3, 1–17. [Google Scholar] [CrossRef]
- Karim, M.; Shetty, J.; Islam, R.A.; Kaiser, A.; Bakhtiar, A.; Chowdhury, E.H. Strontium Sulfite: A New pH-Responsive Inorganic Nanocarrier to Deliver Therapeutic siRNAs to Cancer Cells. Pharmaceutics 2019, 11, 89. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Pi, J.; Yang, F.; Jiang, J.; Wang, X.; Bai, H.; Shao, M.; Huang, L.; Zhu, H.; Yang, P.; et al. Folate-chitosan nanoparticles loaded with ursolic acid confer anti-breast cancer activities in vitro and in vivo. Sci. Rep. 2016, 6, 30782. [Google Scholar] [CrossRef] [PubMed]
- Khalid, M.; El-Sawy, H.S. Polymeric nanoparticles: Promising platform for drug delivery. Int. J. Pharm. 2017, 528, 675–691. [Google Scholar]
- Brannon-Peppas, L.; Blanchette, J. Nanoparticle and targeted systems for cancer therapy. Adv. Drug Deliv. 2012, 56, 1649–1659. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, C.; Praça, C.; Ferreira, R.; Santos, T.; Ferreira, L.; Bernardino, L. Nanoparticle-mediated brain drug delivery: Overcoming blood–brain barrier to treat neurodegenerative diseases. J. Control. Release 2016, 235, 34–47. [Google Scholar] [CrossRef] [Green Version]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Koo, O.M.; Rubinstein, I.; Onyuksel, H. Role of nanotechnology in targeted drug delivery and imaging: A concise review. Nanomed. Nanotechnol. Biol. Med. 2005, 1, 193–212. [Google Scholar] [CrossRef]
- Anselmo, A.; Mitragotri, S. Nanoparticles in the clinic. Bioeng Transl Med 2016, 1, 10–29. [Google Scholar] [CrossRef]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20. [Google Scholar] [CrossRef]
- Haque, S.T.; Karim, M.; Abidin, S.A.Z.; Othman, I.; Holl, M.M.B.; Chowdhury, E.H. Fe/Mg-Modified Carbonate Apatite with Uniform Particle Size and Unique Transport Protein-Related Protein Corona Efficiently Delivers Doxorubicin into Breast Cancer Cells. Nanomaterials 2020, 10, 834. [Google Scholar] [CrossRef]
- Karim, E.; Rosli, R.; HChowdhury, E. Systemic delivery of nanoformulations of anti-cancer drugs with therapeutic potency in animal models of cancer. Curr. Cancer Ther. Rev. 2016, 12, 204–220. [Google Scholar] [CrossRef]
- Karim, M.; Tha, K.K.; Othman, I.; Borhan Uddin, M.; Chowdhury, E.H. Therapeutic potency of nanoformulations of siRNAs and shRNAs in animal models of cancers. Pharmaceutics 2018, 10, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mura, S.; Nicolas, J.; Couvreur, P. Stimuli-responsive nanocarriers for drug delivery. Nat. Mater. 2013, 12, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Foy, S.P.; Manthe, R.L.; Foy, S.T.; Dimitrijevic, S.; Krishnamurthy, N.; Labhasetwar, V. Optical imaging and magnetic field targeting of magnetic nanoparticles in tumors. ACS Nano 2010, 4, 5217–5224. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.J.; David, A.E.; Wang, J.; Galbán, C.J.; Hill, H.L.; Yang, V.C. Polyethylene glycol modified, cross-linked starch-coated iron oxide nanoparticles for enhanced magnetic tumor targeting. Biomaterials 2011, 32, 2183–2193. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Fan, T.M.; Borst, L.B.; Cheng, J. Synthesis and biological response of size-specific, monodisperse drug–silica nanoconjugates. ACS Nano 2012, 6, 3954–3966. [Google Scholar] [CrossRef] [Green Version]
- Toy, R.; Peiris, P.M.; Ghaghada, K.B.; Karathanasis, E. Shaping cancer nanomedicine: The effect of particle shape on the in vivo journey of nanoparticles. Nanomedicine 2014, 9, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Anselmo, A.C.; Zhang, M.; Kumar, S.; Vogus, D.R.; Menegatti, S.; Helgeson, M.E.; Mitragotri, S. Elasticity of nanoparticles influences their blood circulation, phagocytosis, endocytosis, and targeting. ACS Nano 2015, 9, 3169–3177. [Google Scholar] [CrossRef]
- Rosenblum, D.; Peer, D. Omics-based nanomedicine: The future of personalized oncology. Cancer Lett. 2014, 352, 126–136. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46 Pt 1, 6387–6392. [Google Scholar]
- Soussan, E.; Cassel, S.; Blanzat, M.; Rico-Lattes, I. Drug delivery by soft matter: Matrix and vesicular carriers. Angew. Chem. Int. Ed. 2009, 48, 274–288. [Google Scholar] [CrossRef]
- Hobbs, S.K.; Monsky, W.L.; Yuan, F.; Roberts, W.G.; Griffith, L.; Torchilin, V.P.; Jain, R.K. Regulation of transport pathways in tumor vessels: Role of tumor type and microenvironment. Proc. Natl. Acad. Sci. USA 1998, 95, 4607–4612. [Google Scholar] [CrossRef] [Green Version]
- Li, S.-D.; Huang, L. Pharmacokinetics and biodistribution of nanoparticles. Mol. Pharm. 2008, 5, 496–504. [Google Scholar] [CrossRef]
- Yu, B.; Tai, H.C.; Xue, W.; Lee, L.J.; Lee, R.J. Receptor-targeted nanocarriers for therapeutic delivery to cancer. Mol. Membr. Biol. 2010, 27, 286–298. [Google Scholar] [CrossRef] [Green Version]
- Wisse, E.; Braet, F.; Luo, D.; De Zanger, R.; Jans, D.; Crabbe, E.; Vermoesen, A. Structure and function of sinusoidal lining cells in the liver. Toxicol. Pathol. 1996, 24, 100–111. [Google Scholar] [CrossRef]
- Sadauskas, E.; Wallin, H.; Stoltenberg, M.; Vogel, U.; Doering, P.; Larsen, A.; Danscher, G. Kupffer cells are central in the removal of nanoparticles from the organism. Part. Fibre Toxicol. 2007, 4, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 1–12. [Google Scholar] [CrossRef]
- Byrne, J.D.; Betancourt, T.; Brannon-Peppas, L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Advanced Drug Deliv. Rev. 2008, 60, 1615–1626. [Google Scholar] [CrossRef]
- Kurmi, B.D.; Patel, P.; Paliwal, R.; Paliwal, S.R. Molecular approaches for targeted drug delivery towards cancer: A concise review with respect to nanotechnology. J. Drug Deliv. Sci. Technol. 2020, 57, 101682. [Google Scholar] [CrossRef]
- Das, M.; Mohanty, C.; Sahoo, S.K. Ligand-based targeted therapy for cancer tissue. Expert Opin. Drug Deliv. 2009, 6, 285–304. [Google Scholar] [CrossRef]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.; Park, C.; Yi, G.; Lee, D.; Koo, H. Active targeting strategies using biological ligands for nanoparticle drug delivery systems. Cancers 2019, 11, 640. [Google Scholar] [CrossRef] [Green Version]
- Vhora, I.; Patil, S.; Bhatt, P.; Gandhi, R.; Baradia, D.; Misra, A. Receptor-targeted drug delivery: Current perspective and challenges. Ther. Deliv. 2014, 5, 1007–1024. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, R.I.; Gee, J.M.W.; Harper, M.E. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37, 9–15. [Google Scholar] [CrossRef]
- Verbeek, B.S.; Adriaansen-Slot, S.S.; Vroom, T.M.; Beckers, T.; Rijksen, G. Overexpression of EGFR and c-erbB2 causes enhanced cell migration in human breast cancer cells and NIH3T3 fibroblasts. FEBS Lett. 1998, 425, 145–150. [Google Scholar] [CrossRef]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; Feo, G.D.; Caponigro, F.; Salomon, D.S.; et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Yarden, R.I.; Wilson, M.A.; Chrysogelos, S.A. Estrogen suppression of EGFR expression in breast cancer cells: A possible mechanism to modulate growth. J. Cell. Biochem. 2001, 81 (Suppl. 36), 232–246. [Google Scholar] [CrossRef]
- Cappuzzo, F.; Hirsch, F.R.; Rossi, E.; Bartolini, S.; Ceresoli, G.L.; Bemis, L.; Haney, J.; Witta, S.; Danenberg, K.; Domenichini, I.; et al. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non–small-cell lung cancer. J. Natl. Cancer Inst. 2005, 97, 643–655. [Google Scholar] [CrossRef]
- Spano, J.-P.; Lagorce, C.; Atlan, D.; Milano, G.; Domont, J.; Benamouzig, R.; Attar, A.; Benichou, J.; Martin, A.; Morere, J.-F.; et al. Impact of EGFR expression on colorectal cancer patient prognosis and survival. Ann. Oncol. 2005, 16, 102–108. [Google Scholar] [CrossRef]
- Erjala, K.; Sundvall, M.; Junttila, T.T.; Zhang, N.; Savisalo, M.; Mali, P.; Kulmala, J.; Pulkkinen, J.; Grenman, R.; Elenius, K.; et al. Signaling via ErbB2 and ErbB3 associates with resistance and epidermal growth factor receptor (EGFR) amplification with sensitivity to EGFR inhibitor gefitinib in head and neck squamous cell carcinoma cells. Clin. Cancer Res. 2006, 12, 4103–4111. [Google Scholar] [CrossRef] [Green Version]
- Brinkman, A.M.; Chen, G.; Wang, Y.; Hedman, C.J.; Sherer, N.M.; Havighurst, T.C.; Gong, S.; Xu, W. Aminoflavone-loaded EGFR-targeted unimolecular micelle nanoparticles exhibit anti-cancer effects in triple negative breast cancer. Biomaterials 2016, 101, 20–31. [Google Scholar] [CrossRef] [Green Version]
- Gumuskaya, B.; Alper, M.; Hucumenoglu, S.; Altundag, K.; Uner, A.; Guler, G. EGFR expression and gene copy number in triple-negative breast carcinoma. Cancer Genet. Cytogenet. 2010, 203, 222–229. [Google Scholar] [CrossRef]
- Reis-Filho, J.S.; Pinheiro, C.; Lambros, M.; Milanezi, F.; Carvalho, S.; Savage, K.; Simpson, P.T.; Jones, C.; Swift, S.; Mackay, A.; et al. EGFR amplification and lack of activating mutations in metaplastic breast carcinomas. J. Pathol. A J. Pathol. Soc. Great Br. Irel. 2006, 209, 445–453. [Google Scholar] [CrossRef]
- Shimada, T.; Ueda, M.; Jinno, H.; Chiba, N.; Wada, M.; Watanabe, J. Development of targeted therapy with paclitaxel incorporated into EGF-conjugated nanoparticles. Anticancer Res. 2009, 29, 1009–1014. [Google Scholar]
- Crown, J.; O’Leary, M. The taxanes: An update. Lancet 2000, 355, 1176–1178. [Google Scholar] [CrossRef]
- Gelderblom, H.; Verweij, J.; Nooter, K.; Sparreboom, A. Cremophor EL: The drawbacks and advantages of vehicle selection for drug formulation. Eur. J. Cancer 2001, 37, 1590–1598. [Google Scholar] [CrossRef]
- Rowinsky, E.K.; Donehower, R.C. Paclitaxel (taxol). N. Engl. J. Med. 1995, 332, 1004–1014. [Google Scholar] [CrossRef]
- D’souza, A.A.; Shegokar, R. Polyethylene glycol (PEG): A versatile polymer for pharmaceutical applications. Expert Opin. Drug Deliv. 2016, 13, 1257–1275. [Google Scholar] [CrossRef] [PubMed]
- Mozar, F.S.; Chowdhury, E.H. Pegylation of carbonate apatite nanoparticles prevents opsonin binding and enhances tumor accumulation of gemcitabine. J. Pharm. Sci. 2018, 107, 2497–2508. [Google Scholar] [CrossRef]
- Milane, L.; Duan, Z.-F.; Amiji, M. Pharmacokinetics and biodistribution of lonidamine/paclitaxel loaded, EGFR-targeted nanoparticles in an orthotopic animal model of multi-drug resistant breast cancer. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 435–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torchilin, V.P. Micellar nanocarriers: Pharmaceutical perspectives. Pharm. Res. 2007, 24, 1. [Google Scholar] [CrossRef]
- Nishiyama, N.; Kataoka, K. Current state, achievements, and future prospects of polymeric micelles as nanocarriers for drug and gene delivery. Pharmacol. Ther. 2006, 112, 630–648. [Google Scholar] [CrossRef] [PubMed]
- Jaskula-Sztul, R.; Xu, W.; Chen, G.; Harrison, A.; Dammalapati, A.; Nair, R.; Cheng, Y.; Gong, S.; Chen, H. Thailandepsin A-loaded and octreotide-functionalized unimolecular micelles for targeted neuroendocrine cancer therapy. Biomaterials 2016, 91, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Hong, H.; Javadi, A.; Engle, J.W.; Xu, W.; Yang, Y. Multifunctional unimolecular micelles for cancer-targeted drug delivery and positron emission tomography imaging. Biomaterials 2012, 33, 3071–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, L.; Soto, U.; Agama, K.; Francis, J.; Jimenez, R.; Pommier, Y.; Sowers, L.; Brantley, E. Aminoflavone induces oxidative DNA damage and reactive oxidative species-mediated apoptosis in breast cancer cells. Int. J. Cancer 2008, 122, 1665–1674. [Google Scholar] [CrossRef] [Green Version]
- Brinkman, A.M.; Wu, J.; Ersland, K.; Xu, W. Estrogen receptor α and aryl hydrocarbon receptor independent growth inhibitory effects of aminoflavone in breast cancer cells. BMC Cancer 2014, 14, 344. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Pi, J.; Zhao, Y.; Jiang, J.; Li, T.; Zeng, X.; Yang, P.; Evans, C.E.; Cai, J. EGFR-targeting PLGA-PEG nanoparticles as a curcumin delivery system for breast cancer therapy. Nanoscale 2017, 9, 16365–16374. [Google Scholar] [CrossRef]
- Tiwari, S.K.; Agarwal, S.; Seth, B.; Yadav, A.; Nair, S.; Bhatnagar, P.; Karmakar, M.; Kumari, M.; Chauhan, L.K.S.; Patel, D.K.; et al. Curcumin-loaded nanoparticles potently induce adult neurogenesis and reverse cognitive deficits in Alzheimer’s disease model via canonical Wnt/β-catenin pathway. ACS Nano 2014, 8, 76–103. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Chen, H.-W.; Kuo, Y.-C.; Chang, Y.-F.; Lee, Y.-J.; Hwang, J.-J. Therapeutic efficacy evaluation of curcumin on human oral squamous cell carcinoma xenograft using multimodalities of molecular imaging. Am. J. Chin. Med. 2010, 38, 343–358. [Google Scholar] [CrossRef]
- Sharma, R.A.; Euden, S.A.; Platton, S.L.; Cooke, D.N.; Shafayat, A.; Hewitt, H.R. Phase I clinical trial of oral curcumin: Biomarkers of systemic activity and compliance. Clin. Cancer Res. 2004, 10, 6847–6854. [Google Scholar] [CrossRef] [Green Version]
- Sabharanjak, S.; Mayor, S. Folate receptor endocytosis and trafficking. Advanced Drug Deliv. Rev. 2004, 56, 1099–1109. [Google Scholar] [CrossRef]
- Assaraf, Y.G.; Leamon, C.P.; Reddy, J.A. The folate receptor as a rational therapeutic target for personalized cancer treatment. Drug Resist. Updates 2014, 17, 89–95. [Google Scholar] [CrossRef]
- Boogerd, L.S.; Boonstra, M.C.; Beck, A.-J.; Charehbili, A.; Hoogstins, C.E.; Prevoo, H.A.; Singhal, S.; Low, P.S.; Van de Velde, C.J.H.; Vahrmeijer, A.L.; et al. Concordance of folate receptor-α expression between biopsy, primary tumor and metastasis in breast cancer and lung cancer patients. Oncotarget 2016, 7, 17442. [Google Scholar] [CrossRef] [Green Version]
- Elnakat, H.; Ratnam, M. Distribution, functionality and gene regulation of folate receptor isoforms: Implications in targeted therapy. Adv. Drug Deliv. Rev. 2004, 56, 1067–1084. [Google Scholar] [CrossRef]
- Dixit, V.; Van den Bossche, J.; Sherman, D.M.; Thompson, D.H.; Andres, R.P. Synthesis and grafting of thioctic acid− PEG− folate conjugates onto Au nanoparticles for selective targeting of folate receptor-positive tumor cells. Bioconjugate Chem. 2006, 17, 603–609. [Google Scholar] [CrossRef]
- Yang, C.; Chen, H.; Zhao, J.; Pang, X.; Xi, Y.; Zhai, G. Development of a folate-modified curcumin loaded micelle delivery system for cancer targeting. Colloids Surf. B Biointerfaces 2014, 121, 206–213. [Google Scholar] [CrossRef]
- Muralidharan, R.; Babu, A.; Amreddy, N.; Basalingappa, K.; Mehta, M.; Chen, A.; Zhao, Y.D.; Kompella, U.B.; Munshi, A.; Ramesh, R.; et al. Folate receptor-targeted nanoparticle delivery of HuR-RNAi suppresses lung cancer cell proliferation and migration. J. Nanobiotechnol. 2016, 14, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Soe, Z.C.; Thapa, R.K.; Ou, W.; Gautam, M.; Nguyen, H.T.; Jin, S.G. Folate receptor-mediated celastrol and irinotecan combination delivery using liposomes for effective chemotherapy. Colloids Surf. B Biointerfaces 2018, 170, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Madni, A.; Sarfraz, M.; Rehman, M.; Ahmad, M.; Akhtar, N.; Ahmad, S.; Tahir, N.; Ijaz, S.; Al-Kassas, R.; Löbenberg, R.; et al. Liposomal drug delivery: A versatile platform for challenging clinical applications. J. Pharm. Pharm. Sci. 2014, 17, 401–426. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Zhang, L.; Song, C. Luteinizing hormone-releasing hormone receptor-mediated delivery of mitoxantrone using LHRH analogs modified with PEGylated liposomes. Int. J. Nanomed. 2010, 5, 697. [Google Scholar]
- Karanth, H.; Murthy, R. pH-Sensitive liposomes-principle and application in cancer therapy. J. Pharm. Pharmacol. 2007, 59, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhu, D.; Dong, X.; Sun, H.; Song, C.; Wang, C.; Kong, D. Folate-modified lipid–polymer hybrid nanoparticles for targeted paclitaxel delivery. Int. J. Nanomed. 2015, 10, 2101. [Google Scholar] [CrossRef]
- Min, H.-K.; Kim, C.-S.; Han, J.; Park, J.-O.; Choi, E. Folate receptor-targeted liposomal nanocomplex for effective synergistic photothermal-chemotherapy of breast cancer in vivo. Colloids Surf. B Biointerfaces 2019, 173, 539–548. [Google Scholar]
- Iwamoto, T. Clinical application of drug delivery systems in cancer chemotherapy: Review of the efficacy and side effects of approved drugs. Biol. Pharm. Bull. 2013, 36, 715–718. [Google Scholar] [CrossRef] [Green Version]
- Tavassolian, F.; Kamalinia, G.; Rouhani, H.; Amini, M.; Ostad, S.N.; Khoshayand, M.R.; Atyabi, F.; Tehrani, M.R.; Dinarvand, R. Targeted poly (l-γ-glutamyl glutamine) nanoparticles of docetaxel against folate over-expressed breast cancer cells. Int. J. Pharm. 2014, 467, 123–138. [Google Scholar] [CrossRef]
- Alibolandi, M.; Abnous, K.; Sadeghi, F.; Hosseinkhani, H.; Ramezani, M.; Hadizadeh, F. Folate receptor-targeted multimodal polymersomes for delivery of quantum dots and doxorubicin to breast adenocarcinoma: In vitro and in vivo evaluation. Int. J. Pharm. 2016, 500, 162–178. [Google Scholar] [CrossRef]
- Chiang, W.-H.; Huang, W.-C.; Chang, C.-W.; Shen, M.-Y.; Shih, Z.-F.; Huang, Y.-F.; Lin, S.-C.; Chiu, H.-C. Functionalized polymersomes with outlayered polyelectrolyte gels for potential tumor-targeted delivery of multimodal therapies and MR imaging. J. Control. Release 2013, 168, 280–288. [Google Scholar] [CrossRef]
- Alibolandi, M.; Abnous, K.; Hadizadeh, F.; Taghdisi, S.M.; Alabdollah, F.; Mohammadi, M. Dextran-poly lactide-co-glycolide polymersomes decorated with folate-antennae for targeted delivery of docetaxel to breast adenocarcinima in vitro and in vivo. J. Control. Release 2016, 241, 45–56. [Google Scholar] [CrossRef]
- Ishihara, T.; Takeda, M.; Sakamoto, H.; Kimoto, A.; Kobayashi, C.; Takasaki, N.; Yuki, K.; Tanaka, K.-I.; Takenaga, M.; Igarashi, R.; et al. Accelerated blood clearance phenomenon upon repeated injection of PEG-modified PLA-nanoparticles. Pharm. Res. 2009, 26, 2270–2279. [Google Scholar] [CrossRef]
- Bartlett, J.; Mallon, E.; Cooke, T. The clinical evaluation of HER-2 status: Which test to use? J. Pathol. A J. Pathol. Soc. Great Br. Irel. 2003, 199, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, E.; Kimura, Y.; Oki, E.; Ueda, N.; Futatsugi, M.; Mashino, K. Akt is frequently activated in HER2/neu-positive breast cancers and associated with poor prognosis among hormone-treated patients. Int. J. Cancer 2006, 118, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.K.; Pindiprolu, S.K.S.; Sanapalli, B.K.R.; Yele, V.; Ganesh, G. HER2 targeted biological macromolecule modified liposomes for improved efficacy of capecitabine in breast cancer. Int. J. Biol. Macromol. 2020, 150, 631–636. [Google Scholar] [CrossRef]
- Kim, B.; Shin, J.; Wu, J.; Omstead, D.T.; Kiziltepe, T.; Littlepage, L.E.; Bilgicer, B. Engineering peptide-targeted liposomal nanoparticles optimized for improved selectivity for HER2-positive breast cancer cells to achieve enhanced in vivo efficacy. J. Control. Release 2020, 322, 530–541. [Google Scholar] [CrossRef]
- Stefanick, J.F.; Ashley, J.D.; Kiziltepe, T.; Bilgicer, B. A systematic analysis of peptide linker length and liposomal polyethylene glycol coating on cellular uptake of peptide-targeted liposomes. ACS Nano 2013, 7, 2935–2947. [Google Scholar] [CrossRef]
- Stefanick, J.F.; Ashley, J.D.; Bilgicer, B. Enhanced cellular uptake of peptide-targeted nanoparticles through increased peptide hydrophilicity and optimized ethylene glycol peptide-linker length. ACS Nano 2013, 7, 8115–8127. [Google Scholar] [CrossRef]
- Stefanick, J.F.; Omstead, D.T.; Ashley, J.D.; Deak, P.E.; Mustafaoglu, N.; Kiziltepe, T.; Bilgicer, B. Optimizing design parameters of a peptide targeted liposomal nanoparticle in an in vivo multiple myeloma disease model after initial evaluation in vitro. J. Control. Release 2019, 311, 190–200. [Google Scholar] [CrossRef]
- Mondal, L.; Mukherjee, B.; Das, K.; Bhattacharya, S.; Dutta, D.; Chakraborty, S.; Pal, M.M.; Gaonkar, R.H.; Debnath, M.C. CD-340 functionalized doxorubicin-loaded nanoparticle induces apoptosis and reduces tumor volume along with drug-related cardiotoxicity in mice. Int. J. Nanomed. 2019, 14, 8073. [Google Scholar] [CrossRef] [Green Version]
- Houdaihed, L.; Evans, J.C.; Allen, C. Dual-Targeted Delivery of Nanoparticles Encapsulating Paclitaxel and Everolimus: A Novel Strategy to Overcome Breast Cancer Receptor Heterogeneity. Pharm. Res. 2020, 37, 39. [Google Scholar] [CrossRef]
- Hanstein, B.; Liu, H.; Yancisin, M.C.; Brown, M. Functional analysis of a novel estrogen receptor-β isoform. Mol. Endocrinol. 1999, 13, 129–137. [Google Scholar] [PubMed] [Green Version]
- Rai, S.; Paliwal, R.; Vaidya, B.; Gupta, P.N.; Mahor, S.; Khatri, K.; Goyal, A.K.; Rawat, A.; Vyas, S.P. Estrogen (s) and analogs as a non-immunogenic endogenous ligand in targeted drug/DNA delivery. Curr. Med. Chem. 2007, 14, 2095–2109. [Google Scholar] [PubMed]
- Borrow, A.; Handa, R.J. Estrogen receptors modulation of anxiety-like behavior. In Vitamins and Hormones; Elsevier: Amsterdam, The Netherlands, 2017; Volume 103, pp. 27–52. [Google Scholar]
- Hua, H.; Zhang, H.; Kong, Q.; Jiang, Y. Mechanisms for estrogen receptor expression in human cancer. Exp. Hematol. Oncol. 2018, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Shen, Y.; Li, R. Estrogen synthesis and signaling pathways during aging: From periphery to brain. Trends Mol. Med. 2013, 19, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Omoto, Y.; Iwase, H.; Yamashita, H.; Toyama, T.; Coombes, R.C. Differential expression of estrogen receptor α, β1, and β2 in lobular and ductal breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 1933–1938. [Google Scholar] [CrossRef] [Green Version]
- Eyster, K.M. The Estrogen Receptors: An Overview from Different Perspectives; Estrogen Receptors; Springer: Berlin/Heidelberg, Germany, 2016; pp. 1–10. [Google Scholar]
- Tang, H.; Chen, J.; Wang, L.; Li, Q.; Yang, Y.; Lv, Z.; Bao, H.; Li, Y.; Luan, X.; Li, Y.; et al. Co-delivery of epirubicin and paclitaxel using an estrone-targeted PEGylated liposomal nanoparticle for breast cancer. Int. J. Pharm. 2020, 573, 118806. [Google Scholar] [CrossRef]
- Mamnoon, B.; Feng, L.; Froberg, J.; Choi, Y.; Venkatachalem, S.; Mallik, S. Hypoxia-Responsive, Polymeric Nanocarriers for Targeted Drug Delivery to Estrogen Receptor-Positive Breast Cancer Cell Spheroids. Mol. Pharm. 2020, 17, 4312–4322. [Google Scholar] [CrossRef]
- Chen, K.-L.; Pan, F.; Jiang, H.; Chen, J.-F.; Pei, L.; Xie, F.-W.; Liang, H.J. Highly enriched CD133+ CD44+ stem-like cells with CD133+ CD44 high metastatic subset in HCT116 colon cancer cells. Clin. Exp. metastasis 2011, 28, 751–763. [Google Scholar] [CrossRef]
- Wang, Z.; Shi, Q.; Wang, Z.; Gu, Y.; Shen, Y.; Sun, M.; Deng, M.; Zhang, H.; Fang, J.; Zhang, S.; et al. Clinicopathologic correlation of cancer stem cell markers CD44, CD24, VEGF and HIF-1α in ductal carcinoma in situ and invasive ductal carcinoma of breast: An immunohistochemistry-based pilot study. Pathol. Res. Pract. 2011, 207, 505–513. [Google Scholar] [CrossRef]
- Orian-Rousseau, V. CD44, a therapeutic target for metastasising tumours. Eur. J. Cancer 2010, 46, 1271–1277. [Google Scholar] [CrossRef]
- Yang, Y.; Long, Y.; Wang, Y.; Ren, K.; Li, M.; Zhang, Z.; Xiang, B.; He, Q. Enhanced anti-tumor and anti-metastasis therapy for triple negative breast cancer by CD44 receptor-targeted hybrid self-delivery micelles. Int. J. Pharm. 2020, 577, 119085. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, M.; Tian, L.; Qiu, Y.; Yu, Q.; Wang, X.; Guo, R.; He, Q. Facile strategy by hyaluronic acid functional carbon dot-doxorubicin nanoparticles for CD44 targeted drug delivery and enhanced breast cancer therapy. Int. J. Pharm. 2020, 578, 119122. [Google Scholar] [CrossRef] [PubMed]
- Sang, M.; Han, L.; Luo, R.; Qu, W.; Zheng, F.; Zhang, K.; Liu, F.; Xue, J.; Liu, W.; Feng, F.; et al. CD44 targeted redox-triggered self-assembly with magnetic enhanced EPR effects for effective amplification of gambogic acid to treat triple-negative breast cancer. Biomater. Sci. 2020, 8, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Hamori, M.; Yoshimatsu, S.; Hukuchi, Y.; Shimizu, Y.; Fukushima, K.; Sugioka, N.; Nishimura, A.; Shibata, N. Preparation and pharmaceutical evaluation of nano-fiber matrix supported drug delivery system using the solvent-based electrospinning method. Int. J. Pharm. 2014, 464, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Schieber, C.; Bestetti, A.; Lim, J.P.; Ryan, A.D.; Nguyen, T.L.; Eldridge, R.; White, A.R.; Gleeson, P.A.; Donnelly, P.S.; Williams, S.J.; et al. Conjugation of Transferrin to Azide-Modified CdSe/ZnS Core–Shell Quantum Dots using Cyclooctyne Click Chemistry. Angew. Chem. Int. Ed. 2012, 51, 10523–10527. [Google Scholar] [CrossRef]
- Anabousi, S.; Bakowsky, U.; Schneider, M.; Huwer, H.; Lehr, C.-M.; Ehrhardt, C. In vitro assessment of transferrin-conjugated liposomes as drug delivery systems for inhalation therapy of lung cancer. Eur. J. Pharm. Sci. 2006, 29, 367–374. [Google Scholar] [CrossRef]
- Vandewalle, B.; Granier, A.; Peyrat, J.; Bonneterre, J.; Lefebvre, J. Transferrin receptors in cultured breast cancer cells. J. Cancer Res. Clin. Oncol. 1985, 110, 71–76. [Google Scholar] [CrossRef]
- Cui, T.; Zhang, S.; Sun, H. Co-delivery of doxorubicin and pH-sensitive curcumin prodrug by transferrin-targeted nanoparticles for breast cancer treatment. Oncol. Rep. 2017, 37, 1253–1260. [Google Scholar] [CrossRef]
- Gao, W.; Ye, G.; Duan, X.; Yang, X.; Yang, V.C. Transferrin receptor-targeted pH-sensitive micellar system for diminution of drug resistance and targetable delivery in multidrug-resistant breast cancer. Int. J. Nanomed. 2017, 12, 1047. [Google Scholar] [CrossRef] [Green Version]
- Pavalko, F.M.; Otey, C.A. Role of adhesion molecule cytoplasmic domains in mediating interactions with the cytoskeleton. Proc. Soc. Exp. Biol. Med. 1994, 205, 282–293. [Google Scholar] [CrossRef]
- Haass, N.K.; Smalley, K.S.; Li, L.; Herlyn, M. Adhesion, migration and communication in melanocytes and melanoma. Pigment Cell Res. 2005, 18, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Chen, X. Integrin targeted delivery of chemotherapeutics. Theranostics 2011, 1, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, P.C.; Strömblad, S.; Klemke, R.; Visscher, D.; Sarkar, F.H.; Cheresh, D.A. Antiintegrin alpha v beta 3 blocks human breast cancer growth and angiogenesis in human skin. J. Clin. Investig. 1995, 96, 1815–1822. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.A.; Majeti, B.K.; Barnes, L.A.; Makale, M.; Weis, S.M.; Lutu-Fuga, K.; Wrasidlo, W.; Cheresh, D.A. Nanoparticle-mediated drug delivery to tumor vasculature suppresses metastasis. Proc. Natl. Acad. Sci. USA 2008, 105, 9343–9348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, P.C.; Strömblad, S.; Sanders, L.C.; von Schalscha, T.L.; Aimes, R.T.; Stetler-Stevenson, W.G.; Quigley, J.P.; Cheresh, D.A. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin αvβ3. Cell 1996, 85, 683–693. [Google Scholar] [CrossRef] [Green Version]
- Yadav, A.S.; Radharani, N.N.V.; Gorain, M.; Bulbule, A.; Shetti, D.; Roy, G.; Baby, T.; Kundu, G.C. RGD functionalized chitosan nanoparticle mediated targeted delivery of raloxifene selectively suppresses angiogenesis and tumor growth in breast cancer. Nanoscale 2020, 12, 10664–10684. [Google Scholar] [CrossRef]
- Covarrubias, G.; He, F.; Raghunathan, S.; Turan, O.; Peiris, P.M.; Schiemann, W.P.; Karathanasis, E. Effective treatment of cancer metastasis using a dual-ligand nanoparticle. PLoS ONE 2019, 14, e0220474. [Google Scholar] [CrossRef] [Green Version]
- Taheri, A.; Dinarvand, R.; Nouri, F.S.; Khorramizadeh, M.R.; Borougeni, A.T.; Mansoori, P.; Atyabi, F. Use of biotin targeted methotrexate–human serum albumin conjugated nanoparticles to enhance methotrexate antitumor efficacy. Int. J. Nanomed. 2011, 6, 1863. [Google Scholar]
- Patra, P.; Mitra, S.; Gupta, A.D.; Pradhan, S.; Bhattacharya, S.; Ahir, M.; Mukherjee, S.; Sarkar, S.; Roy, S.; Chattopadhyay, S.; et al. Simple synthesis of biocompatible biotinylated porous hexagonal ZnO nanodisc for targeted doxorubicin delivery against breast cancer cell: In vitro and in vivo cytotoxic potential. Colloids Surf. B Biointerfaces 2015, 133, 88–98. [Google Scholar] [CrossRef]
- Yuan, Z.; Wang, H.; Hu, Z.; Huang, Y.; Yao, F.; Sun, S.; Wu, B. Quercetin inhibits proliferation and drug resistance in KB/VCR oral cancer cells and enhances its sensitivity to vincristine. Nutr. Cancer 2015, 67, 126–136. [Google Scholar] [CrossRef]
- Ferreira, R.J.; Ferreira, M.J.U.; dos Santos, D.J. Reversing cancer multidrug resistance: Insights into the efflux by ABC transports from in silico studies. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2015, 5, 27–55. [Google Scholar] [CrossRef]
- Lv, L.; Liu, C.; Chen, C.; Yu, X.; Chen, G.; Shi, Y.; Qin, F.; Ou, J.; Qiu, K.; Li, G.; et al. Quercetin and doxorubicin co-encapsulated biotin receptor-targeting nanoparticles for minimizing drug resistance in breast cancer. Oncotarget 2016, 7, 32184. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Reyes, M.; Maya-Núñez, G.; Pérez-Solis, M.A.; López-Muñoz, E.; Guillén, N.; Olivo-Marin, J.-C.; Aguilar-Rojas, A. Treatment of Breast Cancer with Gonadotropin-Releasing Hormone Analogs. Front. Oncol. 2019, 9, 943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghanghoria, R.; Kesharwani, P.; Tekade, R.K.; Jain, N.K. Targeting luteinizing hormone-releasing hormone: A potential therapeutics to treat gynecological and other cancers. J. Control. Release 2018, 269, 277–301. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Wang, Q.; Jiang, J.; Luo, Y.; Sun, Z. A conjugate of methotrexate and an analog of luteinizing hormone releasing hormone shows increased efficacy against prostate cancer. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Tang, Z.; Zhang, Y.; Lv, S.; Yu, H.; Zhang, D.; Hong, H.; Chen, X. LHRH-peptide conjugated dextran nanoparticles for targeted delivery of cisplatin to breast cancer. J. Mater. Chem. B 2014, 2, 3490–3499. [Google Scholar] [CrossRef]
- Li, M.; Tang, Z.; Zhang, Y.; Lv, S.; Li, Q.; Chen, X. Targeted delivery of cisplatin by LHRH-peptide conjugated dextran nanoparticles suppresses breast cancer growth and metastasis. Acta Biomater. 2015, 18, 132–143. [Google Scholar] [CrossRef]
- Zhang, L.; Ren, Y.; Wang, Y.; He, Y.; Feng, W.; Song, C. Pharmacokinetics, distribution and anti-tumor efficacy of liposomal mitoxantrone modified with a luteinizing hormone-releasing hormone receptor-specific peptide. Int. J. Nanomed. 2018, 13, 1097. [Google Scholar] [CrossRef] [Green Version]
- Trynda-Lemiesz, L. Paclitaxel–HSA interaction. Binding sites on HSA molecule. Bioorg. Med. Chem. 2004, 12, 3269–3275. [Google Scholar] [CrossRef]
- Hua, S.; De Matos, M.B.; Metselaar, J.M.; Storm, G. Current trends and challenges in the clinical translation of nanoparticulate nanomedicines: Pathways for translational development and commercialization. Front. Pharmacol. 2018, 9, 790. [Google Scholar] [CrossRef]
- Xu, J.; Wong, D.H.; Byrne, J.D.; Chen, K.; Bowerman, C.; DeSimone, J.M. Future of the particle replication in nonwetting templates (PRINT) technology. Angew. Chem. Int. Ed. 2013, 52, 6580–6589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, N. Challenges in development of nanoparticle-based therapeutics. AAPS J. 2012, 14, 282–295. [Google Scholar] [PubMed] [Green Version]
- Parker, N.; Turk, M.J.; Westrick, E.; Lewis, J.D.; Low, P.S.; Leamon, C.P. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal. Biochem. 2005, 338, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Hultdin, J.; Van Guelpen, B.; Bergh, A.; Hallmans, G.; Stattin, P. Plasma folate, vitamin B12, and homocysteine and prostate cancer risk: A prospective study. Int. J. Cancer 2005, 113, 819–824. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Constituents (1) of Nanoformulations and Their Sizes (2) | Name of Drug (1), Its Amount Used (2) and Encapsulation (3a) or Loading (3b) Efficiency | Cell Lines (1) and No. of Cells Used (2) to Induce Tumors in Animals (3) (Mice/Rats) | Days Required to Form Tumors (1) and Their Sizes (2) Prior to Administration | Route of Delivery (Intraven-Ous (i.v.)/Intratum-Oral/Peritoneal (i.p.)/Oral) | Amount of Drug Administered at One Time (1) and Frequency of Administration (2) and Interval Time (3) | Total Period of Tumor Measurement since 1st Administration | Fold Reduction in Tumor Size (1 a) or Mass (1b) Compared to Free Drug at the End | Decrease in Body Weight for Drug-Free Particles (1) and Drug-Loaded Particles (2) |
|---|---|---|---|---|---|---|---|---|
| (1) EGF peptide- PMBN (2) ~200 nm [75] | (1) Paclitaxel (2) 15 mg/kg and (3a) NR (3b) NR | (1) A431 and H69 (2) 1.0 × 106 (3) mice | (1) NR (not reported) (2) 100 mm3 | i.p. | (1) 15 mg/kg (2) 5 (3) 1 day | 21 days | (1a) 2-fold (1b) NR | (2) No (2) No |
| (1) EGF peptides-polymeric micelle (2) 20–100 nm [72] | (1) Aminoflavone (2) 7 mg/kg and (3a) NR (3b) 16.7% | (1) MDA-MB-468 (2) 1.0 × 106 (3) mice | (1) 56 days (2) 500 mm3 | i.v. | (1) 7 mg/kg (2) 5 (3) 4 days | 46 days | (1a) 7-fold (1b) NR | (2) NR (2) NR |
| (1) EGF peptides-PLGA-PEG (2) 210 nm [88] | (1) Curcumin (2) 5 mg/kg and (3a) 92.3 ± 2.7 (3b) NR | (1) MCF-7 cells (2) 1.0 × 107 (3) mice | (1) 7 days (2) NR | i.v. | (1) 5 mg/kg (2) 20 (3) 1 days | 21 days | (1a) 7-fold (1b) 2-fold | (1) NR (2) NR |
| (1) Folic acid-liposome (2) ~190 nm [99] | (1) Celastrol (Cs) and Irinotecan (IR) (2) NR and (3a) 90% for both (3b) 28.5 ± 0.8% (Cs) and 14.7 ± 0.5% (Ir) | (1) MDA-MB-231 (2) 1.0 × 107 (3) mice | (1) NR (2) 100 mm3 | i.v. | (1) NR (2) 4 (3) 3 days | 25 days | (1a) 3-fold (1b) 2-fold | (1) Yes (2) No |
| (1) Folic acid-lipid–polymer hybrid nanoparticles (NPs) (2) 279.9 ± 8.7 nm, [103] | (1) paclitaxel. (2) 20 mg/kg (3a) 91.16% ± 1.12% (3b) 27.36% ± 0.91% | (1) EMT6 (2) 2.0 × 107 (3) mice | (1) NR (2) 100 mm3 | Intratum-oral | (1) 20 mg/kg (2) 5 (3) 2 days | 16 days | (1a) no significant reduction (1b) NR | (1) Yes (2) No |
| (1) Folic acid-Gold nanorods-liposome (2) 154 nm, [104] | (1) Doxorubicin (2) 2.5 mg/kg and (3a) 54.73 ± 2.13% (3b) NR | (1) 4T1 (2) 1.0 × 106 (3) mice | (1) 10 days (2) 100 mm3 | i.v. | (1) 2.5 mg/kg (2) NR (3) NR | 15 days | (1a) 2.9-fold (1b) NR | (1) No (2) No |
| (1) Folic acid-PGG NPs (2) 131.96 ± 5.34 nm, [106] | (1) Docetaxel (2) 10 mg/kg and (3a) 67.83 ± 3.29(%) (3b) NR | (1) 4T1 (2) 1.0 × 105 (3) mice | (1) NR (2) 100–200 mm3 | i.v. | (1) 10 mg/kg (2) 4 (3) 7 days | 28 days | (1a) 2.6-fold (1b) NR | (1) Yes (2) N0 |
| (1) Folic acid-Quantum dot-PEG-PLGA polymersomes (2) 170.53 ± 1.21 nm, [107] | (1) Doxorubicin (2) 7 mg/kg and (3a) 54.26 ± 1.23% (3b) 10.82 ± 0.87 | (1) 4T1 (2) 2.0 × 105 (3) mice | (1) NR (2) 80–100 mm3 | i.v. | (1) 7 mg/kg (2) 1 (3) 0 days | 21 days | (1a) 5-fold (1b) NR | (1) Yes (2) No |
| (1) Folic acid-Dextran–PLGA polymersomes (2) 178.53 ± 2.5 nm, [109] | (1) Docetaxel (2) 10 mg/kg and (3a) 78.85 ± 3.81% % (3b) 9.32 ± 0.27 | (1) 4T1 (2) 5.0 × 105 (3) mice | (1) 7 days (2) 80–100 mm3 | i.v. | (1) 10 mg/kg (2) 1 (3) 0 days | 21 days | (1a) 4.5-fold (1b) NR | (1) Yes (2) No |
| (1) HER2 peptide- chitosan-liposome (2) 116.18 ± 1.73 nm, [114] | (1) Capecitabine (2) 10 mg/kg and (3a) 82.21 ± 0.62% (3b) NR | (1) MCF-7 (2) 1.5 × 105 (3) mice | (1) 14 days (2) NR | i.v. | (1) 10 mg/kg (2) 9 (3) 2 days | 21 days | (1a) 29-fold (1b) NR | (1) NR (2) NR |
| (1) HER2 peptide-liposome (2) ~80 nm [115] | (1) Doxorubicin (2) 3 mg/kg and (3a) NR (3b) > 98% | (1) MMTV/neu transgenic (2) NR (3) mice | (1) NR (2) ~150–200 mm3 | i.v. | (1) 3 mg/kg (2) 5 (3) 2 days | 25 days | (1a) 3.5-fold (1b) NR | (1) Yes (2) No |
| (1) HER2 (CD-340)-antibody-PLGA (2) 241 nm [119] | (1) Doxorubicin (2) 3 mg/kg and (3a) 88 ± 0.17% (3b) 8 ± 0.1 | (1) SKBR-3 (2) 1.0 × 107 (3) mice | (1) NR (2) 100–150 mm3 | i.v. | (1) 3 mg/kg (2) 3 (3) 2 days | 25 days | (1a) 2-fold (1b) NR | (1) No (2) No |
| (1) Estron ligand-pegylated liposome (2) ~120 nm [128] | (1) Paclitaxel (PTX), epirubicin (EPI) (2) 9.42 mg/kg (PTX), 6 mg/kg (EPI) and (3a) 90.23 ± 0.52 (EPI), 61.85 ± 0.56 (PTX) (3b) 5.07 ± 0.84 (EPI), 4.39 ± 0.67(PTX) | (1) MCF-7 (2) 2.0 × 106 (3) mice | (1) NR (2) ~100–200 mm3 | i.v. | (1) 9.42 mg/kg (PTX), 6 mg/kg(EPI) (2) 5 (3) 2 days | 35 days | (1a) 6-fold (1b) NR | (1) Yes (2) No |
| (1) Hyaluronic acid-hybrid micelle (2) ~125 nm [133] | (1) Doxorubicin (2) 3 mg/kg and (3a) NR (3b) NR | (1) 4T1 (2) 2.5 × 105 (3) mice | (1) NR (2) ~100 mm3 | i.v. | (1) 3 mg/kg (2) 4 (3) 2 days | 20 days | (1a) 4.8-fold (1b) 4.4 fold | (1) Yes (2) No |
| (1) Hyaluronic acid-p-CBA-Carbon dots (2) ~125 nm [134] | (1) Doxorubicin (2) 3 mg/kg and (3a) NR (3b) 18.13% | (1) 4T1 (2) 1.0 × 107 (3) mice | (1) 6 days (2) ~50–100 mm3 | i.v. | (1) 3 mg/kg (2) 5 (3) 2 days | 21 days | (1a) 4-fold (1b) 2.5 fold | (1) Yes (2) No |
| (1) mPEG-HA/CSO-SS-Hex/SPION (2) ~100 nm [135] | (1) Gambogic acid (2) 6 mg/kg and (3a) 85.1% (3b) 23.7% | (1) 4T1 (2) NR (3) mice | (1) 15 days (2) 300–400 mm3 | i.v. | (1) 6 mg/kg (2) 6 (3) 2 days | 13 days | (1a) 10-fold (1b) 4- fold | (1) Yes (2) No |
| (1) Tf-PEG-NPs (2) 89 nm [140] | (1) Doxorubicin (DOX) Curcumin (CUR) (2) 50 mg/kg (DOX), 50 mg/kg(CUR) and (3a) 82.7 ± 4.1% (D-OX), 85.3 ± 3.2%(CUR) (3b) NR, 4.6 ± 0.8% (CUR) | (1) MCF-7 (2) 1.0 × 106 (3) mice | (1) NR (2) NR | i.v. | (1) 50 mg/kg (2) 7 (3) 7 days | 49 days | (1a) 6-fold (1b) NR | (1) No (2) No |
| (1) αvβ3 integrin RGD peptide-chitosan NPs (2) 200 nm [148] | (1) Raloxifene (2) 10 mg/kg and (3a) 50% (3b) NR | (1) 4T1 (2) 3.0 × 105 (3) mice | (1) NR (2) NR | Oral | (1) 10 mg/kg (2) 4 (3) 1 days | 13 days | (1a) 5-fold (1b) 2.8-fold | (1) No (2) No |
| (1) EGF and αvβ3 integrin peptide-liposome (2) 100 nm [149] | (1) Doxorubicin (2) 7.5 mg/kg and (3a) NR (3b) 18.13% | (1) D2. A1 (2) 5.0 × 105 (3) mice | (1) NR (2) NR | i.v. | (1) 7.5 mg/kg (2) 3 (3) 1 days | 49 days | (1a) NR (1b) NR,BLI signalling reduced to 1.33 fold | (1) NR (2) NR |
| (1) Biotinylated porous hexagonal ZnO nanodisc (2) 200 nm [151] | (1) Doxorubicin (2) 50 µg/mL and (3a) NR (3b) 63% | (1) EAC (2) 1.0 × 106 (3) mice | (1) 12 days (2) 150 mm3 | i.v. | (1) 50 µg/mL (2) NR (3) NR | 28 days | (1a) 3.5-fold (1b) 5.3-fold | (1) NR (2) NR |
| (1) Biotinylated polymeric NPs (2) 105.8 ± 1.4 nm [154] | (1) Doxorubicin (DOX), Quercetin (QUT) (2) 5 mg/kg and (3a) 86% (DOX), 91% (QUT) (3b) 3.6% (DOX), 7.9% (QUT) | (1) MCF-7/ADR (2) 3.0 × 107 (3) mice | (1) NR (2) 50 mm3 | i.v. | (1) 5 mg/kg (2) 8 (3) 3 days | 25 days | (1a) 1.95-fold (1b) 7-fold | (1) Yes (2) No |
| (1) LHRH-peptide conjugated dextran NPs (2) ~22 nm [158] | (1) Cisplatin (2) 4 mg/kg, 10 mg/kg and (3a) 85.5% (3b) 7.5% | (1) MCF-7 (2) 1.5 × 106 (3) mice | (1) NR (2) 50 mm3 | i.v. | (1) 4 mg/kg and 10 mg/kg (2) 3 (3) 4 days | 20 days | (1a) 1.1-fold (4 mg/kg), 1.5-fold (10 mg/kg) (1b) NR | (1) Yes (2) No |
| (1) LHRH-peptide conjugated dextran NPs (2) ~22 nm [159] | (1) Cisplatin (2) 5 mg/kg, 10 mg/kg and (3a) 85.5% (3b) 7.5% | (1) 4T1 (2) 1.5 × 106 (3) mice | (1) NR (2) 50 mm3 | i.v. | (1) 5 mg/kg and 10 mg/kg (2) 3 (3) 4 days | 20 days | (1a) 1.4-fold (5 mg/kg), 2.4-fold (10 mg/kg) (1b) 1.3-fold (5 mg/kg), 2.6-fold (10 mg/kg) | (1) Yes (2) No |
| (1) LHRH-peptide conjugated liposome (2) 103.3 ± 0.70 nm, [160] | (1) Mitoxantrone (2) 2.5 mg/kg, and (3a) 98.2% (3b) 9.5% | 1) MCF-7 (2) 4.0 × 106 (3) mice | (1) NR (2) 100 mm3 | i.v. | (1) 2.5 mg/kg and (2) 3 (3) 7 days | 21 days | (1a) 2-fold (1b) NR | (1) Yes (2) No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jahan, S.; Karim, M.E.; Chowdhury, E.H. Nanoparticles Targeting Receptors on Breast Cancer for Efficient Delivery of Chemotherapeutics. Biomedicines 2021, 9, 114. https://doi.org/10.3390/biomedicines9020114
Jahan S, Karim ME, Chowdhury EH. Nanoparticles Targeting Receptors on Breast Cancer for Efficient Delivery of Chemotherapeutics. Biomedicines. 2021; 9(2):114. https://doi.org/10.3390/biomedicines9020114
Chicago/Turabian StyleJahan, Sulltana, Md. Emranul Karim, and Ezharul Hoque Chowdhury. 2021. "Nanoparticles Targeting Receptors on Breast Cancer for Efficient Delivery of Chemotherapeutics" Biomedicines 9, no. 2: 114. https://doi.org/10.3390/biomedicines9020114
APA StyleJahan, S., Karim, M. E., & Chowdhury, E. H. (2021). Nanoparticles Targeting Receptors on Breast Cancer for Efficient Delivery of Chemotherapeutics. Biomedicines, 9(2), 114. https://doi.org/10.3390/biomedicines9020114