
Review on Inflammation Markers in Chronic Kidney Disease


Abstract
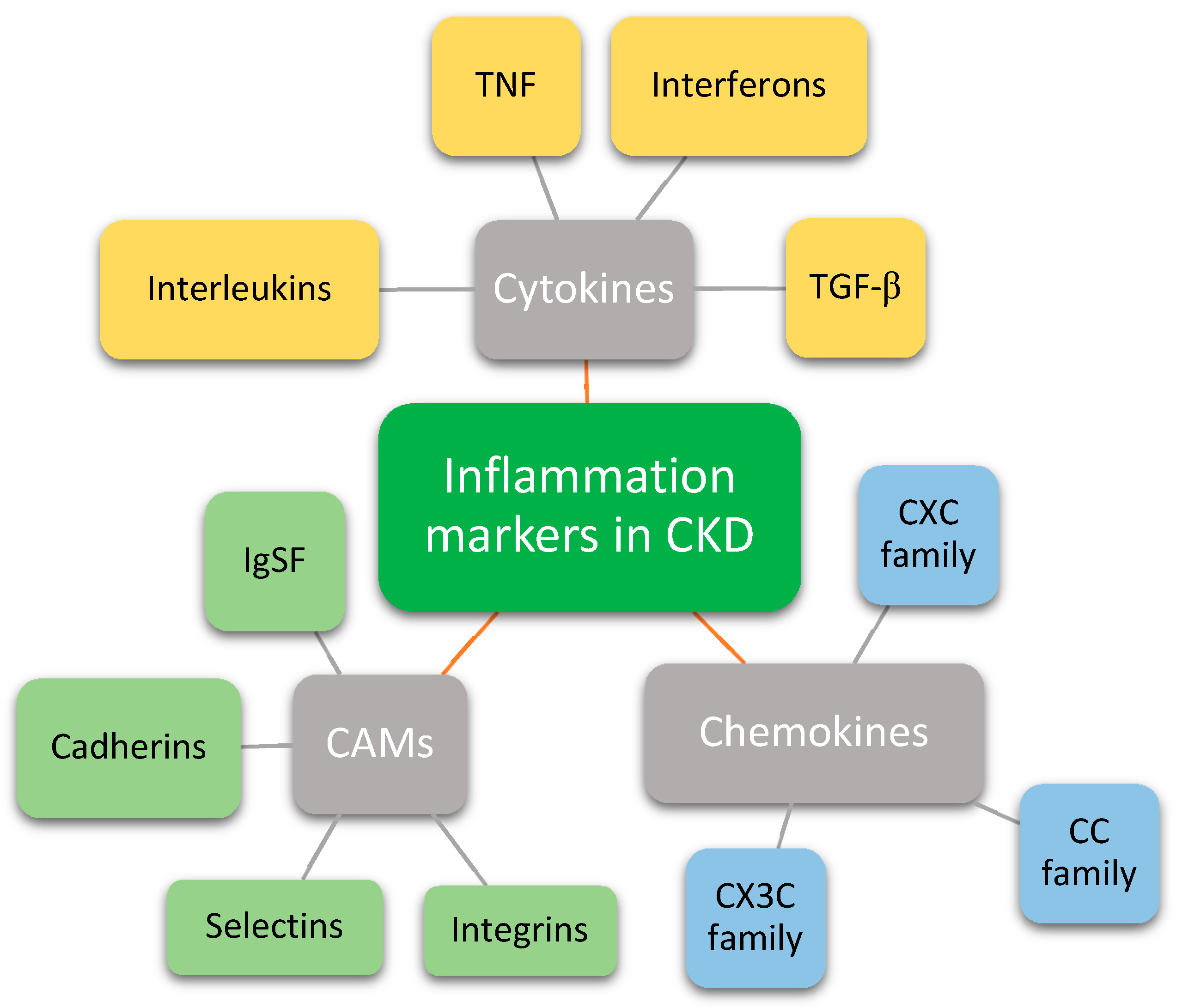
:1. Introduction
2. Cytokines
2.1. Interleukins
2.1.1. Interleukin 6
2.1.2. Interleukin 1 and 18
2.1.3. Inflammasome
2.1.4. Other Interleukins
2.2. Tumor Necrosis Factor
2.3. Interferons
2.4. Transforming Growth Factor Beta
3. Chemokines
3.1. CXC Subfamily
3.2. CC Subfamily
3.3. CX3C Subfamily
4. Cell Adhesion Molecules
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2013, 3, 5–14. [Google Scholar]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Global, regional, and national burden of chronic kidney disease, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [CrossRef] [Green Version]
- Mihai, S.; Codrici, E.; Popescu, I.D.; Enciu, A.M.; Albulescu, L.; Necula, L.G.; Mambet, C.; Anton, G.; Tanase, C. Inflammation-Related Mechanisms in Chronic Kidney Disease Prediction, Progression, and Outcome. J. Immunol. Res. 2018, 2018, 2180373. [Google Scholar] [CrossRef]
- Hojs, R.; Ekart, R.; Bevc, S.; Hojs, N. Markers of Inflammation and Oxidative Stress in the Development and Progression of Renal Disease in Diabetic Patients. Nephron 2016, 133, 159–162. [Google Scholar] [CrossRef]
- Germolec, D.R.; Shipkowski, K.A.; Frawley, R.P.; Evans, E. Markers of Inflammation. Methods Mol. Biol. 2018, 1803, 57–79. [Google Scholar] [PubMed]
- Yeung, Y.T.; Aziz, F.; Guerrero-Castilla, A.; Arguelles, S. Signaling Pathways in Inflammation and Anti-inflammatory Therapies. Curr. Pharm. Des. 2018, 24, 1449–1484. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. Interleukin-6 Family Cytokines. Cold Spring Harb. Perspect. Biol. 2018, 10, a028415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazdi, A.S.; Ghoreschi, K. The Interleukin-1 Family. Adv. Exp. Med. Biol. 2016, 941, 21–29. [Google Scholar]
- Hasegawa, G.; Nakano, K.; Sawada, M.; Uno, K.; Shibayama, Y.; Ienaga, K.; Kondo, M. Possible role of tumor necrosis factor and interleukin-1 in the development of diabetic nephropathy. Kidney Int. 1991, 40, 1007–1012. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Li, B.; Sun, A.; Guo, F. Interleukin-10 Family Cytokines Immunobiology and Structure. Adv. Exp. Med. Biol. 2019, 1172, 79–96. [Google Scholar]
- Tait Wojno, E.D.; Hunter, C.A.; Stumhofer, J.S. The Immunobiology of the Interleukin-12 Family: Room for Discovery. Immunity 2019, 50, 851–870. [Google Scholar] [CrossRef] [PubMed]
- Monin, L.; Gaffen, S.L. Interleukin 17 Family Cytokines: Signaling Mechanisms, Biological Activities, and Therapeutic Implications. Cold Spring Harb. Perspect. Biol. 2018, 10, a028522. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amdur, R.L.; Feldman, H.I.; Gupta, J.; Yang, W.; Kanetsky, P.; Shlipak, M.; Rahman, M.; Lash, J.P.; Townsend, R.R.; Ojo, A.; et al. Inflammation and Progression of CKD: The CRIC Study. Clin. J. Am. Soc. Nephrol. 2016, 11, 1546–1556. [Google Scholar] [CrossRef] [Green Version]
- Desjardins, M.P.; Sidibé, A.; Fortier, C.; Mac-Way, F.; Marquis, K.; De Serres, S.; Larivière, R.; Agharazii, M. Association of interleukin-6 with aortic stiffness in end-stage renal disease. J. Am. Soc. Hypertens. 2018, 12, 5–13. [Google Scholar] [CrossRef]
- Hassan, M.O.; Duarte, R.; Dickens, C.; Dix-Peek, T.; Naidoo, S.; Vachiat, A.; Grinter, S.; Manga, P.; Naicker, S. Interleukin-6 gene polymorhisms and interleukin-6 levels are associated with atherosclerosis in CKD patients. Clin. Nephrol. 2020, 93, 82–86. [Google Scholar] [CrossRef]
- Feng, Y.; Tang, Y.; Zhou, H.; Xie, K. A meta-analysis on correlation between interleukin-6 -174G/C polymorphism and end-stage renal disease. Ren. Fail. 2017, 39, 350–356. [Google Scholar] [CrossRef] [Green Version]
- Durlacher-Betzer, K.; Hassan, A.; Levi, R.; Axelrod, J.; Silver, J.; Naveh-Many, T. Interleukin-6 contributes to the increase in fibroblast growth factor 23 expression in acute and chronic kidney disease. Kidney Int. 2018, 94, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Akchurin, O.; Patino, E.; Dalal, V.; Meza, K.; Bhatia, D.; Brovender, S.; Zhu, Y.S.; Cunningham-Rundles, S.; Perelstein, E.; Kumar, J.; et al. Interleukin-6 Contributes to the Development of Anemia in Juvenile CKD. Kidney Int. Rep. 2019, 4, 470–483. [Google Scholar] [CrossRef] [Green Version]
- Won, H.S.; Kim, H.G.; Yun, Y.S.; Jeon, E.K.; Ko, Y.H.; Kim, Y.S.; Kim, Y.O.; Yoon, S.A. IL-6 is an independent risk factor for resistance to erythropoiesis-stimulating agents in hemodialysis patients without iron deficiency. Hemodial. Int. 2012, 16, 31–37. [Google Scholar] [CrossRef]
- Hashmat, S.; Rudemiller, N.; Lund, H.; Abais-Battad, J.M.; Van Why, S.; Mattson, D.L. Interleukin-6 inhibition attenuates hypertension and associated renal damage in Dahl salt-sensitive rats. Am. J. Physiol. Renal Physiol. 2016, 311, F555–F561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salimi, S.; Shardell, M.D.; Seliger, S.L.; Bandinelli, S.; Guralnik, J.M.; Ferrucci, L. Inflammation and Trajectory of Renal Function in Community-Dwelling Older Adults. J. Am. Geriatr. Soc. 2018, 66, 804–811. [Google Scholar] [CrossRef]
- Rovin, B.H.; van Vollenhoven, R.F.; Aranow, C.; Wagner, C.; Gordon, R.; Zhuang, Y.; Belkowski, S.; Hsu, B. A Multicenter, Randomized, Double-Blind, Placebo-Controlled Study to Evaluate the Efficacy and Safety of Treatment With Sirukumab (CNTO 136) in Patients With Active Lupus Nephritis. Arthritis Rheumatol. 2016, 68, 2174–2183. [Google Scholar] [CrossRef] [Green Version]
- Pergola, P.E.; Devalaraja, M.; Fishbane, S.; Chonchol, M.; Mathur, V.S.; Smith, M.T.; Lo, L.; Herzog, K.; Kakkar, R.; Davidson, M.H. Ziltivekimab for Treatment of Anemia of Inflammation in Patients on Hemodialysis: Results from a Phase 1/2 Multicenter, Randomized, Double-Blind, Placebo-Controlled Trial. J. Am. Soc. Nephrol. 2021, 32, 211–222. [Google Scholar] [CrossRef]
- Fukuda, M.; Sawa, N.; Hoshino, J.; Ohashi, K.; Motoaki, M.; Ubara, Y. Tocilizumab preserves renal function in rheumatoid arthritis with AA amyloidosis and end-stage kidney disease: Two case reports. Clin. Nephrol. 2021, 95, 54. [Google Scholar] [CrossRef]
- Chen, W.; Yuan, H.; Cao, W.; Wang, T.; Yu, H.; Fu, Y.; Jiang, B.; Zhou, H.; Guo, H.; Zhao, X. Blocking interleukin-6 trans-signaling protects against renal fibrosis by suppressing STAT3 activation. Theranostics 2019, 9, 3980–3991. [Google Scholar] [CrossRef] [PubMed]
- Palomo, J.; Dietrich, D.; Martin, P.; Palmer, G.; Gabay, C. The interleukin (IL)-1 cytokine family—Balance between agonists and antagonists in inflammatory diseases. Cytokine 2015, 76, 25–37. [Google Scholar] [CrossRef]
- Moorlag, S.; Röring, R.J.; Joosten, L.A.B.; Netea, M.G. The role of the interleukin-1 family in trained immunity. Immunol. Rev. 2018, 281, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Buckley, L.F.; Canada, J.M.; Carbone, S.; Trankle, C.R.; Kadariya, D.; Billingsley, H.; Wohlford, G.F.; Kirkman, D.L.; Abbate, A.; Van Tassel, B.W. Potential role for interleukin-1 in the cardio-renal syndrome. Eur. J. Heart Fail. 2019, 21, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.L.; Hung, A.; Ikizler, T.A.; Farmer-Bailey, H.; Salas-Cruz, N.; Sarkar, S.; Hoofnagle, A.; You, Z.; Chonchol, M. Interleukin-1 inhibition, chronic kidney disease-mineral and bone disorder, and physical function. Clin. Nephrol. 2017, 88, 132–143. [Google Scholar] [CrossRef]
- Nowak, K.L.; Chonchol, M.; Ikizler, T.A.; Farmer-Bailey, H.; Salas, N.; Chaudhry, R.; Wang, W.; Smits, G.; Tengesdal, I.; Dinarello, C.A.; et al. IL-1 Inhibition and Vascular Function in CKD. J. Am. Soc. Nephrol. 2017, 28, 971–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; MacFadyen, J.G.; Glynn, R.J.; Koenig, W.; Libby, P.; Everett, B.M.; Lefkowitz, M.; Thuren, T.; Cornel, J.H. Inhibition of Interleukin-1β by Canakinumab and Cardiovascular Outcomes in Patients With Chronic Kidney Disease. J. Am. Coll. Cardiol. 2018, 71, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- Hung, A.M.; Tsuchida, Y.; Nowak, K.L.; Sarkar, S.; Chonchol, M.; Whitfield, V.; Salas, N.; Dikalova, A.; Yancey, P.G.; Huang, J.; et al. IL-1 Inhibition and Function of the HDL-Containing Fraction of Plasma in Patients with Stages 3 to 5 CKD. Clin. J. Am. Soc. Nephrol. 2019, 14, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Kaplanski, G. Interleukin-18: Biological properties and role in disease pathogenesis. Immunol. Rev. 2018, 281, 138–153. [Google Scholar] [CrossRef] [Green Version]
- Yaribeygi, H.; Atkin, S.L.; Sahebkar, A. Interleukin-18 and diabetic nephropathy: A review. J. Cell. Physiol. 2019, 234, 5674–5682. [Google Scholar] [CrossRef]
- Elsherbiny, N.M.; Al-Gayyar, M.M. The role of IL-18 in type 1 diabetic nephropathy: The problem and future treatment. Cytokine 2016, 81, 15–22. [Google Scholar] [CrossRef]
- Lin, X.; Yuan, J.; Zhao, Y.; Zha, Y. Urine interleukin-18 in prediction of acute kidney injury: A systemic review and meta-analysis. J. Nephrol. 2015, 28, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Rathinam, V.A.; Fitzgerald, K.A. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 2016, 165, 792–800. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wang, Z. Effect and Regulation of the NLRP3 Inflammasome during Renal Fibrosis. Front. Cell Dev. Biol. 2020, 7, 379. [Google Scholar] [CrossRef]
- Mulay, S.R. Multifactorial functions of the inflammasome component NLRP3 in pathogenesis of chronic kidney diseases. Kidney Int. 2019, 96, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Tang, W.; Yi, F. Role of Inflammasome in Chronic Kidney Disease. Adv. Exp. Med. Biol. 2019, 1165, 407–421. [Google Scholar]
- Ermer, T.; Eckardt, K.U.; Aronson, P.S.; Knauf, F. Oxalate, inflammasome, and progression of kidney disease. Curr. Opin. Nephrol. Hypertens. 2016, 25, 363–371. [Google Scholar] [CrossRef]
- Andrade-Oliveira, V.; Foresto-Neto, O.; Watanabe, I.K.M.; Zatz, R.; Câmara, N.O.S. Inflammation in Renal Diseases: New and Old Players. Front. Pharmacol. 2019, 10, 1192. [Google Scholar] [CrossRef]
- Kim, S.M.; Kim, Y.G.; Kim, D.J.; Park, S.H.; Jeong, K.H.; Lee, Y.H.; Lim, S.J.; Lee, S.H.; Moon, J.Y. Inflammasome-Independent Role of NLRP3 Mediates Mitochondrial Regulation in Renal Injury. Front. Immunol. 2018, 9, 2563. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.G.; Kim, S.M.; Kim, K.P.; Lee, S.H.; Moon, J.Y. The Role of Inflammasome-Dependent and Inflammasome-Independent NLRP3 in the Kidney. Cells 2019, 8, 1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, H.J.; Suarez-Alvarez, B.; Grigorescu, M.; Foresto-Neto, O.; Steiger, S.; Desai, J.; Marschner, J.A.; Honarpisheh, M.; Shi, C.; Jordan, J.; et al. The macrophage phenotype and inflammasome component NLRP3 contributes to nephrocalcinosis-related chronic kidney disease independent from IL-1-mediated tissue injury. Kidney Int. 2018, 93, 656–669. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, S.M.; Sobey, C.G.; Latz, E.; Mansell, A.; Drummond, G.R. IL-1β and IL-18: Inflammatory markers or mediators of hypertension? Br. J. Pharmacol. 2014, 171, 5589–5602. [Google Scholar] [CrossRef] [PubMed]
- Wada, J.; Makino, H. Innate immunity in diabetes and diabetic nephropathy. Nat. Rev. Nephrol. 2016, 12, 13–26. [Google Scholar] [CrossRef]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.M.; Ling, Y.H.; Huuskes, B.M.; Ferens, D.M.; Saini, N.; Chan, C.T.; Diep, H.; Kett, M.M.; Samuel, C.S.; Kemp-Harper, B.K.; et al. Pharmacological inhibition of the NLRP3 inflammasome reduces blood pressure, renal damage, and dysfunction in salt-sensitive hypertension. Cardiovasc. Res. 2019, 115, 776–787. [Google Scholar] [CrossRef] [Green Version]
- Ke, B.; Shen, W.; Fang, X.; Wu, Q. The NLPR3 inflammasome and obesity-related kidney disease. J. Cell. Mol. Med. 2018, 22, 16–24. [Google Scholar] [CrossRef]
- Wang, Y.J.; Chen, Y.Y.; Hsiao, C.M.; Pan, M.H.; Wang, B.J.; Chen, Y.C.; Ho, C.T.; Huang, K.C.; Chen, R.J. Induction of Autophagy by Pterostilbene Contributes to the Prevention of Renal Fibrosis via Attenuating NLRP3 Inflammasome Activation and Epithelial-Mesenchymal Transition. Front. Cell Dev. Biol. 2020, 8, 436. [Google Scholar] [CrossRef] [PubMed]
- Cortvrindt, C.; Speeckaert, R.; Moerman, A.; Delanghe, J.R.; Speeckaert, M.M. The role of interleukin-17A in the pathogenesis of kidney diseases. Pathology 2017, 49, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Wang, H.; Zhang, L.; Yang, X.; Zhang, M.; Zhu, X.; Ji, X.; Wang, H. Role of interleukin 17 in TGF-β signaling-mediated renal interstitial fibrosis. Cytokine 2018, 106, 80–88. [Google Scholar] [CrossRef]
- Biswas, P.S. IL-17 in Renal Immunity and Autoimmunity. J. Immunol. 2018, 201, 3153–3159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, T.; Luebbe, J.; Paust, H.J.; Panzer, U. Mechanisms and functions of IL-17 signaling in renal autoimmune diseases. Mol. Immunol. 2018, 104, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Koga, T.; Ichinose, K.; Tsokos, G.C. T cells and IL-17 in lupus nephritis. Clin. Immunol. 2017, 185, 95–99. [Google Scholar] [CrossRef]
- Krebs, C.F.; Schmidt, T.; Riedel, J.H.; Panzer, U. T helper type 17 cells in immune-mediated glomerular disease. Nat. Rev. Nephrol. 2017, 13, 647–659. [Google Scholar] [CrossRef]
- Dedong, H.; Feiyan, Z.; Jie, S.; Xiaowei, L.; Shaoyang, W. Analysis of interleukin-17 and interleukin-23 for estimating disease activity and predicting the response to treatment in active lupus nephritis patients. Immunol. Lett. 2019, 210, 33–39. [Google Scholar] [CrossRef]
- Steen, E.H.; Wang, X.; Balaji, S.; Butte, M.J.; Bollyky, P.L.; Keswani, S.G. The Role of the Anti-Inflammatory Cytokine Interleukin-10 in Tissue Fibrosis. Adv. Wound Care 2020, 9, 184–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sziksz, E.; Pap, D.; Lippai, R.; Béres, N.J.; Fekete, A.; Szabó, A.J.; Vannay, A. Fibrosis Related Inflammatory Mediators: Role of the IL-10 Cytokine Family. Mediators Inflamm. 2015, 2015, 764641. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Booz, G.W.; Wang, Y.; Fan, F.; Roman, R.J. Inflammation and renal fibrosis: Recent developments on key signaling molecules as potential therapeutic targets. Eur. J. Pharmacol. 2018, 820, 65–76. [Google Scholar] [CrossRef]
- Wen, Y.; Crowley, S.D. Renal effects of cytokines in hypertension. Curr. Opin. Nephrol. Hypertens. 2018, 27, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Naing, C.; Htet, N.H.; Basavaraj, A.K.; Nalliah, S. An association between IL-10 promoter polymorphisms and diabetic nephropathy: A meta-analysis of case-control studies. J. Diabetes Metab. Disord. 2018, 17, 333–343. [Google Scholar] [CrossRef]
- Williams, A.; Wang, E.C.; Thurner, L.; Liu, C.J. Review: Novel Insights Into Tumor Necrosis Factor Receptor, Death Receptor 3, and Progranulin Pathways in Arthritis and Bone Remodeling. Arthritis Rheumatol. 2016, 68, 2845–2856. [Google Scholar] [CrossRef]
- Vanamee, É.S.; Faustman, D.L. Structural principles of tumor necrosis factor superfamily signaling. Sci. Signal. 2018, 11, eaao4910. [Google Scholar] [CrossRef] [Green Version]
- Wallach, D. The Tumor Necrosis Factor Family: Family Conventions and Private Idiosyncrasies. Cold Spring Harb. Perspect. Biol. 2018, 10, a028431. [Google Scholar] [CrossRef] [PubMed]
- Egli-Spichtig, D.; Imenez Silva, P.H.; Glaudemans, B.; Gehring, N.; Bettoni, C.; Zhang, M.Y.H.; Pastor-Arroyo, E.M.; Schőnenberger, D.; Rajski, M.; Hoogewijs, D.; et al. Tumor necrosis factor stimulates fibroblast growth factor 23 levels in chronic kidney disease and non-renal inflammation. Kidney Int. 2019, 96, 890–905. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, T.T.; Chen, J.; Qiu, W. Elevated expression levels of serum insulin-like growth factor-1, tumor necrosis factor-α and vascular endothelial growth factor 165 might exacerbate type 2 diabetic nephropathy. J. Diabetes Investig. 2017, 8, 108–114. [Google Scholar] [CrossRef] [Green Version]
- Carlsson, A.C.; Östgren, C.J.; Nystrom, F.H.; Länne, T.; Jennersjö, P.; Larsson, A.; Ärnlöv, J. Association of soluble tumor necrosis factor receptors 1 and 2 with nephropathy, cardiovascular events, and total mortality in type 2 diabetes. Cardiovasc. Diabetol. 2016, 15, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, Y.J.; An, J.N.; Kim, C.T.; Yang, S.H.; Lee, H.; Kim, D.K.; Joo, K.W.; Paik, J.H.; Kang, S.W.; Park, J.T.; et al. Circulating Tumor Necrosis Factor α Receptors Predict the Outcomes of Human IgA Nephropathy: A Prospective Cohort Study. PLoS ONE 2015, 10, e0132826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, A.S.; You, H.; Gao, T.; Cooper, T.K.; Nedospasov, S.A.; Vacher, J.; Wilkinson, P.F.; Farrell, F.X.; Reeves, W.B. Macrophage-derived tumor necrosis factor-α mediates diabetic renal injury. Kidney Int. 2015, 88, 722–733. [Google Scholar] [CrossRef] [Green Version]
- Park, M.; Maristany, D.; Huang, D.; Shlipak, M.G.; Whooley, M. Associations of tumor necrosis factor alpha receptor type 1 with kidney function decline, cardiovascular events, and mortality risk in persons with coronary artery disease: Data from the Heart and Soul Study. Atherosclerosis 2017, 263, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wu, W.; Zhang, X.; Huang, Y.; Wen, Y.; Li, X.; Gao, R. Serum levels of tumor necrosis factor alpha in patients with IgA nephropathy are closely associated with disease severity. BMC Nephrol. 2018, 19, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhagat Singh, A.K.; Jeyaruban, A.S.; Wilson, G.J.; Ranganathan, D. Adalimumab-induced IgA nephropathy. BMJ Case Rep. 2019, 12, e226442. [Google Scholar] [CrossRef]
- Kim, S.K.; Choe, J.Y.; Kwak, S.G.; Bae, J.; Park, S.H.; Lee, H. Effect of tumour necrosis factor-alpha inhibitors on renal function in patients with rheumatoid arthritis from the KOBIO registry from 2012 to 2016. Clin. Exp. Rheumatol. 2018, 36, 1022–1030. [Google Scholar]
- Mohamed, H.E.; Asker, M.E.; Keshawy, M.M.; Hasan, R.A.; Mahmoud, Y.K. Inhibition of tumor necrosis factor-α enhanced the antifibrotic effect of empagliflozin in an animal model with renal insulin resistance. Mol. Cell. Biochem. 2020, 466, 45–54. [Google Scholar] [CrossRef]
- Youssef, D.M.; El-Shal, A.S.; Hussein, S.; Salah, K.; Ahmed, A. Tumor necrosis factor alpha gene polymorphisms and haplotypes in Egyptian children with nephrotic syndrome. Cytokine 2018, 102, 76–82. [Google Scholar] [CrossRef]
- Ramírez-Bello, J.; Cadena-Sandoval, D.; Mendoza-Rincón, J.F.; Barbosa-Cobos, R.E.; Sánchez-Muñoz, F.; Amezcua-Guerra, L.M.; Sierra-Martínez, M.; Jiménez-Morales, S. Tumor necrosis factor gene polymorphisms are associated with systemic lupus erythematosus susceptibility or lupus nephritis in Mexican patients. Immunol. Res. 2018, 66, 348–354. [Google Scholar]
- Negishi, H.; Taniguchi, T.; Yanai, H. The Interferon (IFN) Class of Cytokines and the IFN Regulatory Factor (IRF) Transcription Factor Family. Cold Spring Harb. Perspect. Biol. 2018, 10, a028423. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.J.; Lichtnekert, J.; Allam, R. Interferon-alpha and -beta in kidney inflammation. Kidney Int. 2010, 77, 848–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grzegorzewska, A.E.; Swiderska, M.K.; Warchol, W. Interferon-λ3 as a Predictor of Survival in Hemodialysis Patients. Curr. Mol. Med. 2018, 18, 207–215. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, D.; Zhang, Q.; Liu, W. Interferon-alpha for IgM nephropathy? Clin. Nephrol. 2014, 82, 133–137. [Google Scholar] [CrossRef]
- Zheng, N.; Wang, B.; Fan, J.; Luo, N.; Kong, Q.; Ye, H.; Zhang, J.; Ming, H.; Yu, X. Increased Abundance of Plasmacytoid Dendritic Cells and Interferon-Alpha Induces Plasma Cell Differentiation in Patients of IgA Nephropathy. Mediators Inflamm. 2017, 2017, 4532409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Wei, L.; Liu, X.; Wang, L.; Niu, D.; Jin, T.; Yao, G.; Wang, M.; Yu, Q.; Fu, R. Association Between IFN-γ Gene Polymorphisms and IgA Nephropathy in a Chinese Han Population. Kidney Blood Press. Res. 2017, 42, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Nassar, G.M.; Pedro, P.; Remmers, R.E.; Mohanty, L.B.; Smith, W. Reversible renal failure in a patient with the hypereosinophilia syndrome during therapy with alpha interferon. Am. J. Kidney Dis. 1998, 31, 121–126. [Google Scholar] [CrossRef]
- Ozturk, M.; Basoglu, F.; Yilmaz, M.; Ozagari, A.A.; Baybas, S. Interferon β associated nephropathy in a Multiple Sclerosis patient: A case and review. Mult. Scler. Relat. Disord. 2016, 9, 50–53. [Google Scholar] [CrossRef]
- Migliorini, A.; Angelotti, M.L.; Mulay, S.R.; Kulkarni, O.O.; Demleitner, J.; Dietrich, A.; Sagrinati, C.; Ballerini, L.; Peired, A.; Shankland, S.J.; et al. The antiviral cytokines IFN-α and IFN-β modulate parietal epithelial cells and promote podocyte loss: Implications for IFN toxicity, viral glomerulonephritis, and glomerular regeneration. Am. J. Pathol. 2013, 183, 431–440. [Google Scholar] [CrossRef]
- Poosti, F.; Bansal, R.; Yazdani, S.; Prakash, J.; Post, E.; Klok, P.; van der Born, J.; de Borst, M.H.; van Goor, H.; Poelstra, K.; et al. Selective delivery of IFN-γ to renal interstitial myofibroblasts: A novel strategy for the treatment of renal fibrosis. FASEB J. 2015, 29, 1029–1042. [Google Scholar] [CrossRef]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Alexander, P.B.; Wang, X.F. TGF-β Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb. Perspect. Biol. 2017, 9, a022145. [Google Scholar] [CrossRef] [Green Version]
- Lodyga, M.; Hinz, B. TGF-β1—A truly transforming growth factor in fibrosis and immunity. Semin Cell Dev. Biol. 2020, 101, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Sutariya, B.; Jhonsa, D.; Saraf, M.N. TGF-β: The connecting link between nephropathy and fibrosis. Immunopharmacol. Immunotoxicol. 2016, 38, 39–49. [Google Scholar] [CrossRef]
- Isaka, Y. Targeting TGF-β Signaling in Kidney Fibrosis. Int. J. Mol. Sci. 2018, 19, 2532. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.B.; Qu, X.; Caruana, G.; Li, J. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation 2016, 92, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Higgins, S.P.; Tang, Y.; Higgins, C.E.; Mian, B.; Zhang, W.; Czekay, R.P.; Samarakoon, R.; Conti, D.J.; Higgins, P.J. TGF-β1/p53 signaling in renal fibrogenesis. Cell. Signal. 2018, 43, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mou, X.; Zhou, D.Y.; Ma, J.R.; Liu, Y.H.; Chen, H.P.; Hu, Y.B.; Shou, C.M.; Chen, J.W.; Liu, W.H.; Ma, G.L. Serum TGF-β1 as a Biomarker for Type 2 Diabetic Nephropathy: A Meta-Analysis of Randomized Controlled Trials. PLoS ONE 2016, 11, e0149513. [Google Scholar] [CrossRef] [PubMed]
- Horikoshi, S.; Fukuda, N.; Tsunemi, A.; Okamura, M.; Otsuki, M.; Endo, M.; Abe, M. Contribution of TGF-β1 and Effects of Gene Silencer Pyrrole-Imidazole Polyamides Targeting TGF-β1 in Diabetic Nephropathy. Molecules 2020, 25, 950. [Google Scholar] [CrossRef] [Green Version]
- Voelker, J.; Berg, P.H.; Sheetz, M.; Duffin, K.; Shen, T.; Moser, B.; Greene, T.; Blumenthal, S.S.; Rychlik, I.; Yagil, Y.; et al. Anti-TGF-β1 Antibody Therapy in Patients with Diabetic Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 953–962. [Google Scholar] [CrossRef] [Green Version]
- Feng, M.; Tang, P.M.; Huang, X.R.; Sun, S.F.; You, Y.K.; Xiao, J.; Lv, L.L.; Xu, A.P.; Lan, H.Y. TGF-β Mediates Renal Fibrosis via the Smad3-Erbb4-IR Long Noncoding RNA Axis. Mol. Ther. 2018, 26, 148–161. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Luo, M.L.; Song, E.; Zhou, Z.; Ma, T.; Wang, J.; Jia, N.; Wang, G.; Nie, S.; Liu, Y.; et al. Long noncoding RNA lnc-TSI inhibits renal fibrogenesis by negatively regulating the TGF-β/Smad3 pathway. Sci. Transl. Med. 2018, 10, eaat2039. [Google Scholar] [CrossRef] [Green Version]
- You, Y.K.; Luo, Q.; Wu, W.F.; Zhang, J.J.; Zhu, H.J.; Lao, L.; Lan, H.Y.; Chen, H.Y.; Cheng, Y.X. Petchiether A attenuates obstructive nephropathy by suppressing TGF-β/Smad3 and NF-κB signalling. J. Cell. Mol. Med. 2019, 23, 5576–5587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.K.; Lee, J.H.; Ryoo, I.G.; Lee, S.H.; Ku, S.K.; Kwak, M.K. Bardoxolone ameliorates TGF-β1-associated renal fibrosis through Nrf2/Smad7 elevation. Free Radic. Biol. Med. 2019, 138, 33–42. [Google Scholar] [CrossRef]
- Vinader, V.; Afarinkia, K. A beginner’s guide to chemokines. Future Med. Chem. 2012, 4, 845–852. [Google Scholar] [CrossRef]
- Miller, M.C.; Mayo, K.H. Chemokines from a Structural Perspective. Int. J. Mol. Sci. 2017, 18, 2088. [Google Scholar] [CrossRef] [Green Version]
- Mehta, N.N.; Matthews, G.J.; Krishnamoorthy, P.; Shah, R.; McLaughlin, C.; Patel, P.; Budoff, M.; Chen, J.; Wolman, M.; Go, A.; et al. Higher plasma CXCL12 levels predict incident myocardial infarction and death in chronic kidney disease: Findings from the Chronic Renal Insufficiency Cohort study. Eur. Heart J. 2014, 35, 2115–2122. [Google Scholar] [CrossRef] [Green Version]
- Klimczak-Tomaniak, D.; Pilecki, T.; Żochowska, D.; Sieńko, D.; Janiszewski, M.; Pączek, L.; Kuch, M. CXCL12 in Patients with Chronic Kidney Disease and Healthy Controls: Relationships to Ambulatory 24-Hour Blood Pressure and Echocardiographic Measures. Cardiorenal. Med. 2018, 8, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.H.; Advani, S.L.; Thai, K.; Kabir, M.G.; Sood, M.M.; Gibson, I.W.; Yuen, D.A.; Connely, K.A.; Marsden, P.A.; Kelly, D.J.; et al. SDF-1/CXCR4 signaling preserves microvascular integrity and renal function in chronic kidney disease. PLoS ONE 2014, 9, e92227. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, V.; Bosquetti, B.; Gonçalves, S.M.; Bucharles, S.G.; Rempel, L.; Maciel, R.A.; de Oliveira, S.G.; Pecoits Filho, R.; Marwues Stinghen, A.E. Uremic serum inhibits in vitro expression of chemokine SDF-1: Impact of uremic toxicity on endothelial injury. J. Bras. Nefrol. 2014, 36, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Bouabdallah, J.; Zibara, K.; Issa, H.; Lenglet, G.; Kchour, G.; Caus, T.; Six, I.; Chourkroun, G.; Kamel, S.; Bennis, Y. Endothelial cells exposed to phosphate and indoxyl sulphate promote vascular calcification through interleukin-8 secretion. Nephrol. Dial. Transplant. 2019, 34, 1125–1134. [Google Scholar] [CrossRef]
- Alsharidah, A.S.; Alzogaibi, M.A.; Bayoumy, N.M.; Alghonaim, M. Neutrophil chemokines levels in different stages of nephrotic syndrome. Saudi J. Kidney Dis Transpl. 2017, 28, 1256–1263. [Google Scholar] [CrossRef]
- Lin, Z.; Gong, Q.; Zhou, Z.; Zhang, W.; Liao, S.; Liu, Y.; Yan, X.; Pan, X.; Lin, S.; Li, X. Increased plasma CXCL16 levels in patients with chronic kidney diseases. Eur. J. Clin. Investig. 2011, 41, 836–845. [Google Scholar] [CrossRef]
- Zhao, L.; Wu, F.; Jin, L.; Lu, T.; Yang, L.; Pan, X.; Shao, C.; Li, X.; Lin, Z. Serum CXCL16 as a novel marker of renal injury in type 2 diabetes mellitus. PLoS ONE 2014, 9, e87786. [Google Scholar] [CrossRef]
- Scurt, F.G.; Menne, J.; Brandt, S.; Bernhardt, A.; Mertens, P.R.; Haller, H.; Chatzikyrkou, C.; ROADMAP Steering Committee. Systemic Inflammation Precedes Microalbuminuria in Diabetes. Kidney Int. Rep. 2019, 4, 1373–1386. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.H.; Kim, K.P.; Park, S.H.; Kim, D.J.; Kim, Y.G.; Moon, J.Y.; Jung, S.W.; Kim, J.S.; Jeong, K.H.; Lee, S.Y.; et al. Urinary chemokine C-X-C motif ligand 16 and endostatin as predictors of tubulointerstitial fibrosis in patients with advanced diabetic kidney disease. Nephrol. Dial. Transplant. 2021, 36, 295–305. [Google Scholar] [CrossRef]
- Tok, A.; Seyithanoğlu, M.; Ozer, A.; Erkayıran, U.; Karaküçük, S.; Çelebi, A. The serum level of soluble CXCL16 is increased in preeclampsia and associated with hepatic/renal damage. J. Matern. Fetal Neonatal Med. 2019, 1–6. [Google Scholar] [CrossRef]
- Abujam, B.; Cheekatla, S.; Aggarwal, A. Urinary CXCL-10/IP-10 and MCP-1 as markers to assess activity of lupus nephritis. Lupus 2013, 22, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Marie, M.A.; Abu Khalil, R.E.; Habib, H.M. Urinary CXCL10: A marker of nephritis in lupus patients. Reumatismo 2014, 65, 292–297. [Google Scholar] [CrossRef] [Green Version]
- Gregg, L.P.; Tio, M.C.; Li, X.; Adams-Huet, B.; de Lemos, J.A.; Hedayati, S.S. Association of Monocyte Chemoattractant Protein-1 with Death and Atherosclerotic Events in Chronic Kidney Disease. Am. J. Nephrol. 2018, 47, 395–405. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira Junior, W.V.; Silva, A.P.F.; de Figueiredo, R.C.; Gomes, K.B.; Simões, E.S.A.C.; Dusse, L.M.S.A.; Rios, D.R. Association between dyslipidemia and CCL2 in patients undergoing hemodialysis. Cytokine 2020, 125, 154858. [Google Scholar] [CrossRef]
- Zhang, W.R.; Craven, T.E.; Malhotra, R.; Cheung, A.K.; Chonchol, M.; Drawz, P.; Sarnak, M.J.; Parikh, C.R.; Shlipak, M.G.; Ix, J.H.; et al. Kidney Damage Biomarkers and Incident Chronic Kidney Disease During Blood Pressure Reduction: A Case-Control Study. Ann. Intern. Med. 2018, 169, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Satirapoj, B. Tubulointerstitial Biomarkers for Diabetic Nephropathy. J. Diabetes Res. 2018, 2018, 2852398. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Wu, L. Association between MCP-1 2518 A>G gene polymorphism and chronic kidney disease. Int. Urol. Nephrol. 2018, 50, 2245–2253. [Google Scholar] [CrossRef]
- Vianna, H.R.; Soares, C.M.; Silveira, K.D.; Elmiro, G.S.; Mendes, P.M.; de Sousa Tavares, M.; Teixeira, M.M.; Miranda, D.M.; Silva, A.C.S.E. Cytokines in chronic kidney disease: Potential link of MCP-1 and dyslipidemia in glomerular diseases. Pediatr. Nephrol. 2013, 28, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Musiał, K.; Bargenda, A.; Drożdż, D.; Zwolińska, D. New Markers of Inflammation and Tubular Damage in Children with Chronic Kidney Disease. Dis Markers. 2017, 2017, 9389432. [Google Scholar] [CrossRef] [Green Version]
- Brix, S.R.; Stege, G.; Disteldorf, E.; Hoxha, E.; Krebs, C.; Krohn, S.; Otto, B.; Klätschke, K.; Herden, E.; Heymann, F.; et al. CC Chemokine Ligand 18 in ANCA-Associated Crescentic GN. J. Am. Soc. Nephrol. 2015, 26, 2105–2117. [Google Scholar] [CrossRef] [Green Version]
- Wagrowska-Danilewicz, M.; Danilewicz, M.; Stasikowska, O. CC chemokines and chemokine receptors in IgA nephropathy (IgAN) and in non-IgA mesangial proliferative glomerulonephritis (MesProGN). the immunohistochemical comparative study. Pol. J. Pathol. 2005, 56, 121–126. [Google Scholar]
- Mansour, S.G.; Puthumana, J.; Coca, S.G.; Gentry, M.; Parikh, C.R. Biomarkers for the detection of renal fibrosis and prediction of renal outcomes: A systematic review. BMC Nephrol. 2017, 18, 72. [Google Scholar] [CrossRef] [Green Version]
- Stroo, I.; Claessen, N.; Teske, G.J.; Butter, L.M.; Florquin, S.; Leemans, J.C. Deficiency for the chemokine monocyte chemoattractant protein-1 aggravates tubular damage after renal ischemia/reperfusion injury. PLoS ONE 2015, 10, e0123203. [Google Scholar] [CrossRef] [Green Version]
- Rudemiller, N.P.; Patel, M.B.; Zhang, J.D.; Jeffs, A.D.; Karlovich, N.S.; Griffiths, R.; Kan, M.J.; Buckley, A.F.; Gunn, M.D.; Crowley, S.D. C-C Motif Chemokine 5 Attenuates Angiotensin II-Dependent Kidney Injury by Limiting Renal Macrophage Infiltration. Am. J. Pathol. 2016, 186, 2846–2856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haller, H.; Bertram, A.; Nadrowitz, F.; Menne, J. Monocyte chemoattractant protein-1 and the kidney. Curr. Opin. Nephrol. Hypertens. 2016, 25, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Q.; Cheng, K.; Ming, Y. CX3CL1/CX3CR1 Axis, as the Therapeutic Potential in Renal Diseases: Friend or Foe? Curr. Gene Ther. 2017, 17, 442–452. [Google Scholar] [CrossRef] [Green Version]
- Fu, D.; Senouthai, S.; Wang, J.; You, Y. FKN Facilitates HK-2 Cell EMT and Tubulointerstitial Lesions via the Wnt/β-Catenin Pathway in a Murine Model of Lupus Nephritis. Front. Immunol. 2019, 10, 784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senouthai, S.; Wang, J.; Fu, D.; You, Y. Fractalkine is Involved in Lipopolysaccharide-Induced Podocyte Injury through the Wnt/β-Catenin Pathway in an Acute Kidney Injury Mouse Model. Inflammation 2019, 42, 1287–1300. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; Guo, S.M.; Li, Y.Q.; Yang, Y.; Li, M.L.; Han, M.; He, X.F.; Ge, S.W.; Xu, G. Plasma fractalkine levels are associated with renal inflammation and outcomes in immunoglobulin A nephropathy. Nephrol. Dial. Transplant. 2019, 34, 1549–1558. [Google Scholar] [CrossRef]
- You, Y.; Qin, Y.; Lin, X.; Yang, F.; Wang, J.; Yuan, F.; Sooranna, S.R.; Pinhu, L. Upregulated fractalkine levels in Chinese patients with lupus nephritis. Cytokine 2018, 104, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Nordlohne, J.; Ge, S.; Hertel, B.; Melk, A.; Rong, S.; Haller, H.; von Vietinghoff, S. T Cell CX3CR1 Mediates Excess Atherosclerotic Inflammation in Renal Impairment. J. Am Soc. Nephrol. 2016, 27, 1753–1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagci, B.; Bagci, G.; Huzmeli, C.; Sezgin, I.; Ozdemir, O. Associations of fractalkine receptor (CX3CR1) and CCR5 gene variants with hypertension, diabetes and atherosclerosis in chronic renal failure patients undergoing hemodialysis. Int. Urol. Nephrol. 2016, 48, 1163–1170. [Google Scholar] [CrossRef]
- Harjunpää, H.; Llort Asens, M.; Guenther, C.; Fagerholm, S.C. Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front. Immunol. 2019, 10, 1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khodabandehlou, K.; Masehi-Lano, J.J.; Poon, C.; Wang, J.; Chung, E.J. Targeting cell adhesion molecules with nanoparticles using in vivo and flow-based in vitro models of atherosclerosis. Exp. Biol. Med. 2017, 242, 799–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Hamm, L.L.; Mohler, E.R.; Hudaihed, A.; Arora, R.; Chen, C.S.; Liu, Y.; Browne, G.; Mills, K.T.; Kleinpeter, M.A.; et al. Interrelationship of Multiple Endothelial Dysfunction Biomarkers with Chronic Kidney Disease. PLoS ONE 2015, 10, e0132047. [Google Scholar]
- Favretto, G.; Cunha, R.S.D.; Dalboni, M.A.; Oliveira, R.B.; Barreto, F.C.; Massy, Z.A.; Marques Stinghen, A.E. Endothelial Microparticles in Uremia: Biomarkers and Potential Therapeutic Targets. Toxins 2019, 11, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bevc, S.; Sabic, S.; Hojs, R. Atherosclerosis in hemodialysis patients--the role of microinflammation. Ren. Fail. 2008, 30, 1012–1016. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.M.; Thijs, L.; Zhang, Z.Y.; Yang, W.Y.; Huang, Q.F.; Wei, F.F.; Kuznetsova, T.; Jennings, A.M.; Delles, C.; Lennox, R.; et al. Glomerular function in relation to circulating adhesion molecules and inflammation markers in a general population. Nephrol. Dial. Transplant. 2018, 33, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zhang, M.; Sun, C.; Wang, H.; Tang, T.; Xia, Y.; Shao, Q.; Liu, J.; Jiang, C. Soluble Vascular Cell Adhesion Molecule-1 Is Associated With Disease Activity in Adult-Onset Minimal Change Disease. Am. J. Med. Sci. 2019, 357, 311–315. [Google Scholar] [CrossRef]
- Tycová, I.; Hrubá, P.; Maixnerová, D.; Girmanová, E.; Mrázová, P.; Straňavová, L.; Zachoval, R.; Merta, M.; Slatinská, J.; Kollár, M.; et al. Molecular profiling in IgA nephropathy and focal and segmental glomerulosclerosis. Physiol. Res. 2018, 67, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Lau, Y.C.; Xiong, Q.; Blann, A.D.; Lip, G.Y. Relationship between renal function and circulating microparticles, soluble P-selectin and E-selectin levels in atrial fibrillation. J. Thromb. Thrombolysis. 2017, 43, 18–23. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.J.; Yeoh, L.Y.; Sum, C.F.; Tavintharan, S.; Ng, X.W.; Liu, S.; Lee, S.B.M.; Tang, W.E.; Lim, S.C.; SMART2D Study. Vascular cell adhesion molecule-1, but not intercellular adhesion molecule-1, is associated with diabetic kidney disease in Asians with type 2 diabetes. J. Diabetes Complicat. 2015, 29, 707–712. [Google Scholar] [CrossRef]
- Karimi, Z.; Kahe, F.; Jamil, A.; Marszalek, J.; Ghanbari, A.; Afarideh, M.; Khajeh, E.; Noshad, S.; Esteghamati, A.; Chi, G. Intercellular adhesion molecule-1 in diabetic patients with and without microalbuminuria. Diabetes Metab. Syndr. 2018, 12, 365–368. [Google Scholar] [CrossRef]
- El-Dawla, N.M.Q.; Sallam, A.M.; El-Hefnawy, M.H.; El-Mesallamy, H.O. E-cadherin and periostin in early detection and progression of diabetic nephropathy: Epithelial-to-mesenchymal transition. Clin. Exp. Nephrol. 2019, 23, 1050–1057. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ling, Y.; Yang, F.; Deng, J.; Rong, L.; Jiang, M.; Jiang, X. The mTOR/p70S6K1 signaling pathway in renal fibrosis of children with immunoglobulin A nephropathy. J. Renin Angiotensin Aldosterone Syst. 2017, 18, 1470320317717831. [Google Scholar] [CrossRef] [Green Version]
- Sawada, K.; Toyoda, M.; Kaneyama, N.; Shiraiwa, S.; Moriya, H.; Miyatake, H.; Tanaka, E.; Yamamoto, N.; Miyauchi, M.; Kimura, M.; et al. Upregulation of α3β1-Integrin in Podocytes in Early-Stage Diabetic Nephropathy. J. Diabetes Res. 2016, 2016, 9265074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Zhang, J.; Haimbach, R.; Zhu, W.; Mayer-Ezell, R.; Garcia-Calvo, M.; Smith, E.; Price, O.; Kan, Y.; Zycband, E.; et al. An integrin antagonist (MK-0429) decreases proteinuria and renal fibrosis in the ZSF1 rat diabetic nephropathy model. Pharmacol. Res. Perspect. 2017, 5, e00354. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Inflammation Marker | Original Article | Review/Meta-Analysis/Case Report |
|---|---|---|
| IL 6 | [15,16,17,19,20,22,23,25,27,30,34,145] | [8,18,36] |
| IL 1 | [15,16,20,23,30,31,32,33,34,45,47,51] | [9,28,29,35,36,39,41,42,43,46,49,50,64] |
| IL 18 | [23,51,122] | [9,28,35,36,37,38,39,41,42,46,49] |
| IL 17 | [51,55,60] | [13,54,56,57,58,59,64] |
| IL 10 | [20] | [11,61,62,63,64,65] |
| Inflammasome | [33,34,45,47,51,53] | [39,40,41,42,43,44,46,49,50,52] |
| TNF | [15,16,20,23,34,51,69,70,71,72,73,74,75,76,77,78,79,80,145] | [36,63,64,66,67,68] |
| IFN | [83,85,86,90] | [63,64,81,84,87,88] |
| TGF | [15,20,47,53,55,99,100,101,102,103,104,115,138,145,154] | [41,63,64,91,92,93,94,95,96,97,98,129] |
| CXC | [108,111,112,115,116,117] | [106] |
| CC | [51,120,121,122,126,127,130,131,139,145,147] | [36,59,63,106,123,124,129,132] |
| CX3C | [134,135,136,137,138,139] | [106,133] |
| CAMs | [51,142,145,146,147,148,149,150,151,152,153,154] | [93,140,141,143] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petreski, T.; Piko, N.; Ekart, R.; Hojs, R.; Bevc, S. Review on Inflammation Markers in Chronic Kidney Disease. Biomedicines 2021, 9, 182. https://doi.org/10.3390/biomedicines9020182
Petreski T, Piko N, Ekart R, Hojs R, Bevc S. Review on Inflammation Markers in Chronic Kidney Disease. Biomedicines. 2021; 9(2):182. https://doi.org/10.3390/biomedicines9020182
Chicago/Turabian StylePetreski, Tadej, Nejc Piko, Robert Ekart, Radovan Hojs, and Sebastjan Bevc. 2021. "Review on Inflammation Markers in Chronic Kidney Disease" Biomedicines 9, no. 2: 182. https://doi.org/10.3390/biomedicines9020182
APA StylePetreski, T., Piko, N., Ekart, R., Hojs, R., & Bevc, S. (2021). Review on Inflammation Markers in Chronic Kidney Disease. Biomedicines, 9(2), 182. https://doi.org/10.3390/biomedicines9020182