
Adipocyte-Specific ACKR3 Regulates Lipid Levels in Adipose Tissue



Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Tissue Homogenization
2.3. Lipid Measurement
2.4. LPL Activity
2.5. HPLC
2.6. RNA Isolation
2.7. cDNA Synthesis
2.8. Droplet Digital PCR
2.9. Western Blot
2.10. Flow Cytometry
2.11. ELISA Assays
2.12. Statistics
3. Results
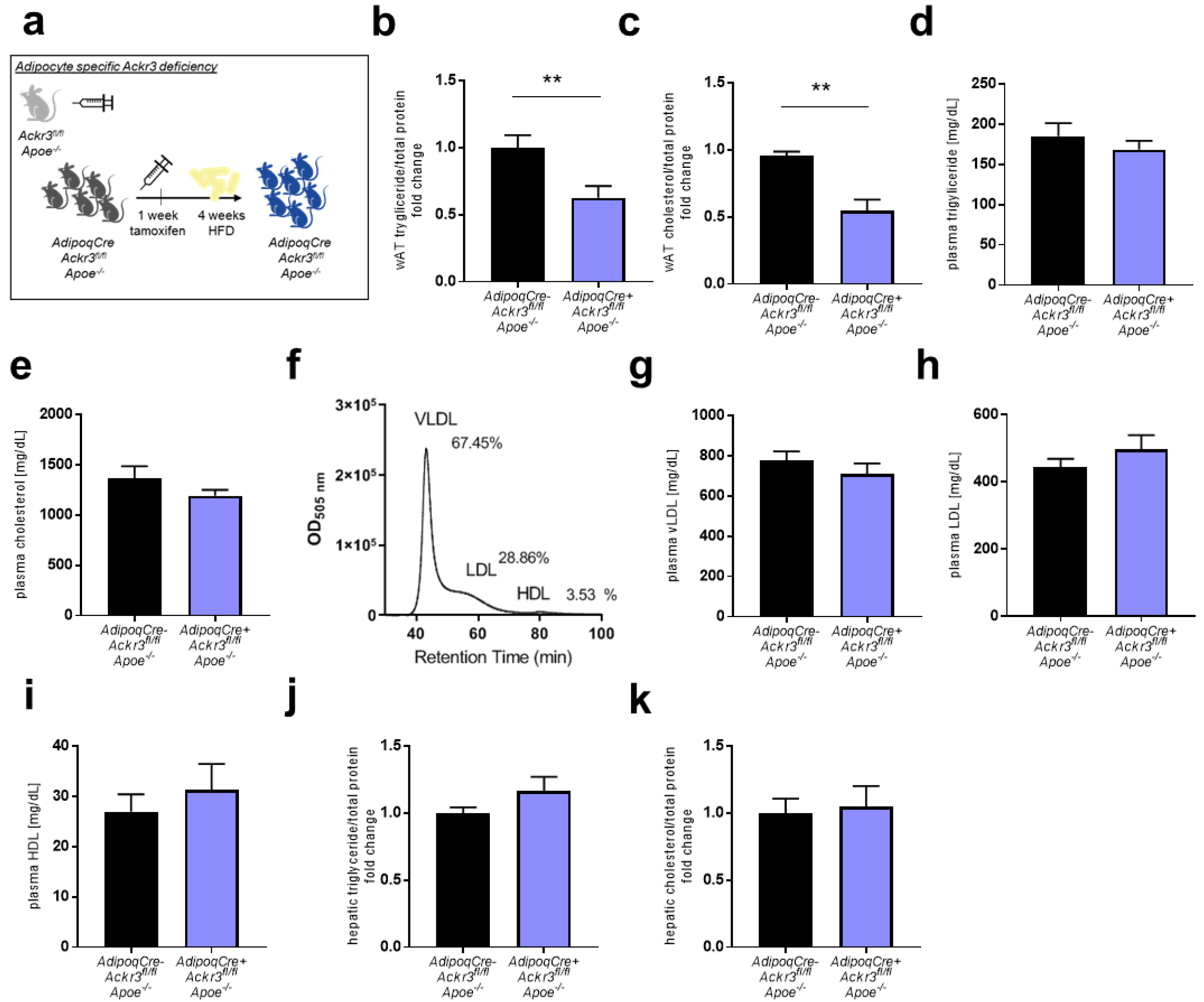
3.1. Adipocyte-Specific Ackr3 Deficiency Decreases AT Lipid Content in Western Diet Fed Mice
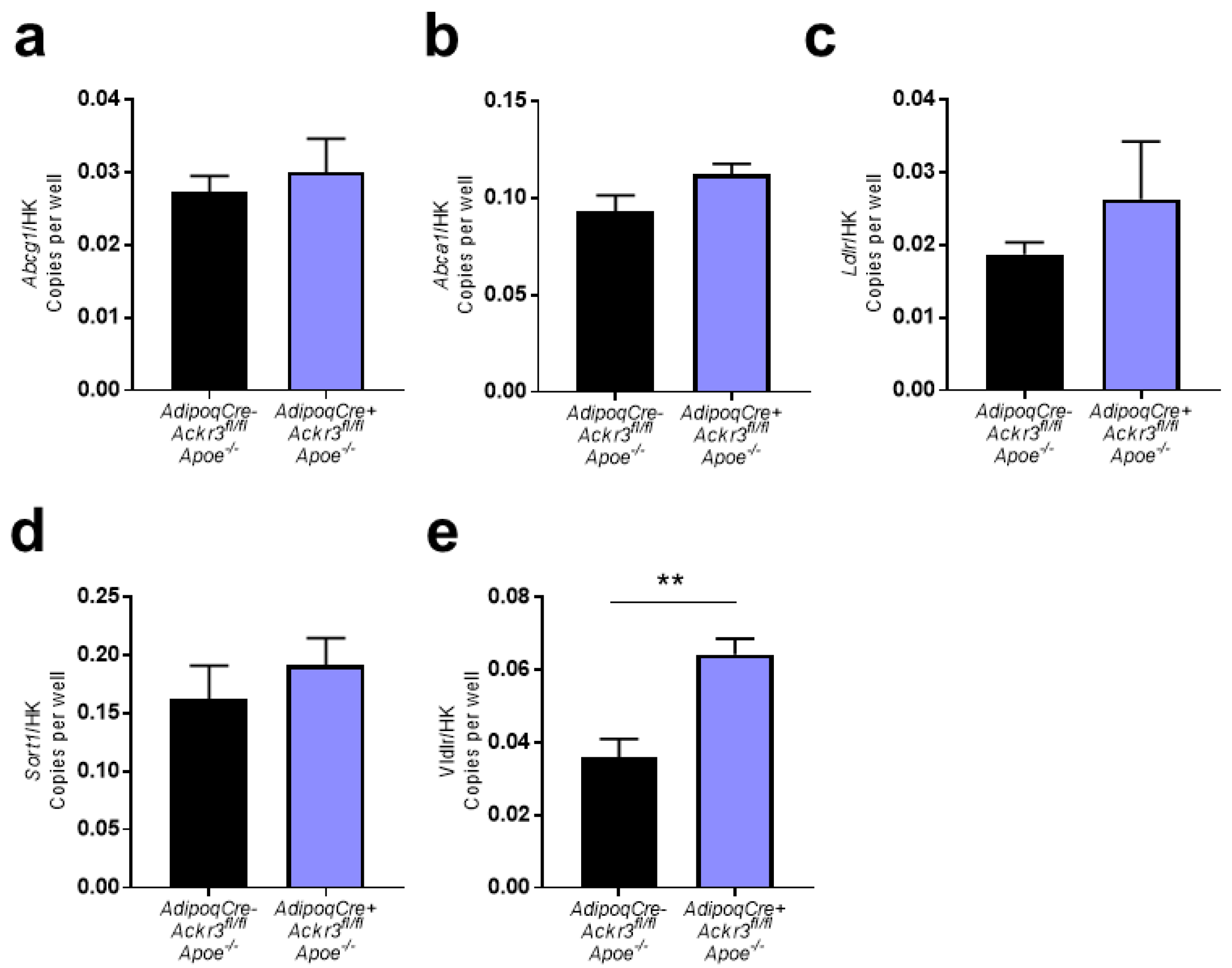
3.2. Expression of Lipid Receptors in wAT Samples
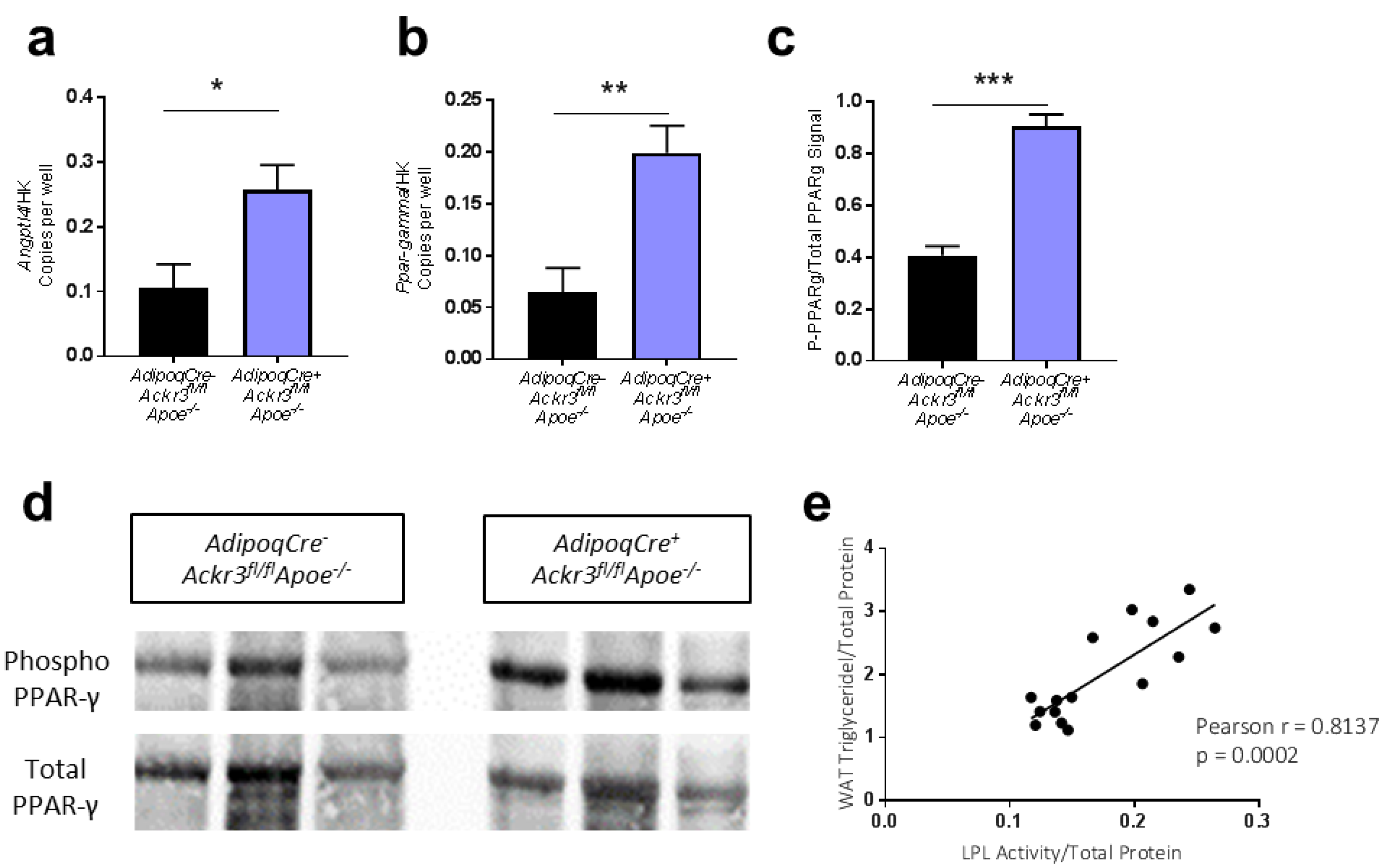
3.3. Lack of ACKR3 in AT Increases Angptl4 and PPAR-γ Expression
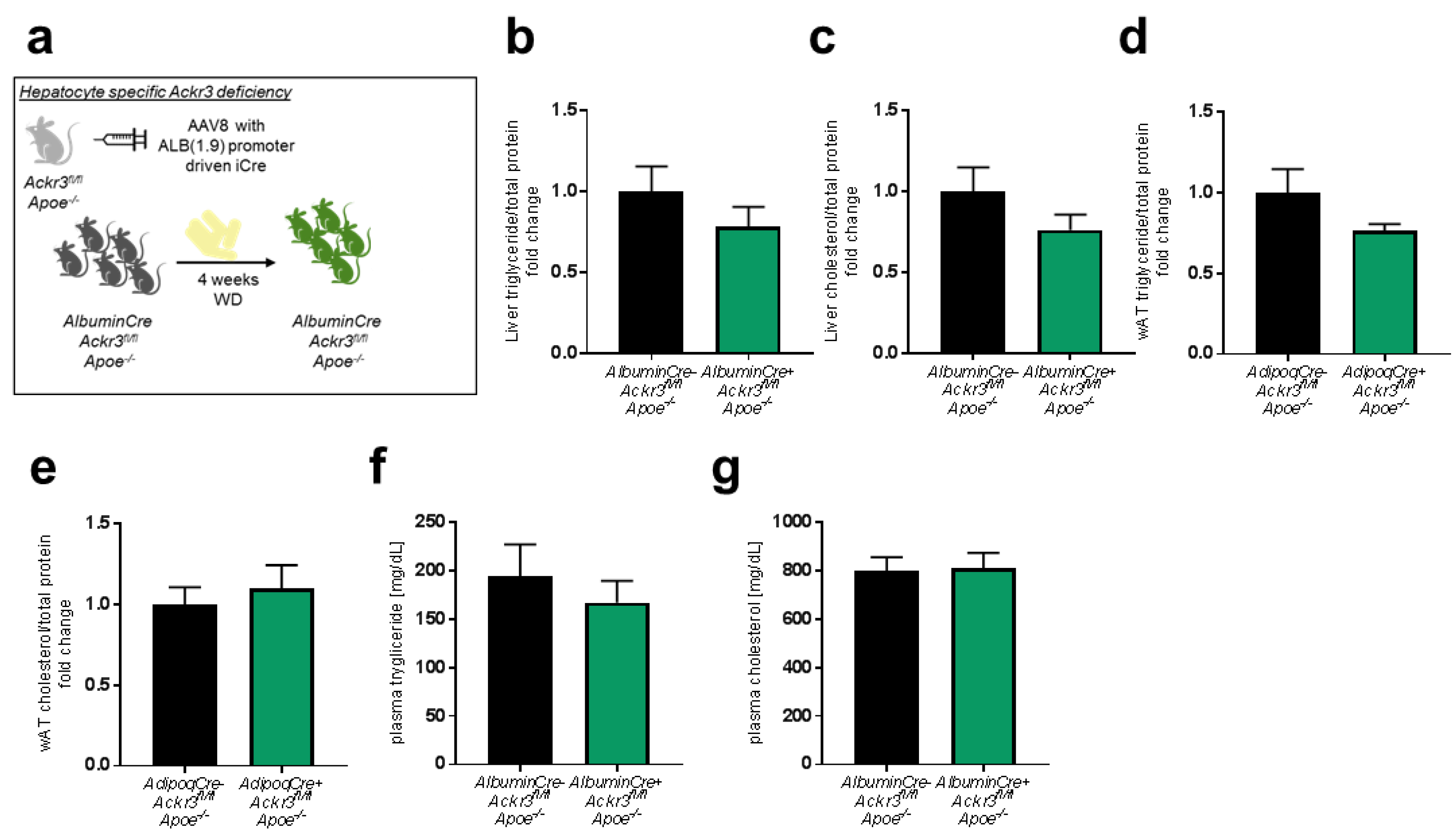
3.4. Hepatic Ackr3 Deficiency Does Not Impact Local or Systemic Lipid Levels
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Frayn, K. Adipose tissue as a buffer for daily lipid flux. Diabetologia 2002, 45, 1201–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.; Liu, M. Adipose tissue in control of metabolism. J. Endocrinol. 2016, 231, R77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matafome, P.; Seiça, R. Function and Dysfunction of Adipose Tissue. Obes. Brain Funct. 2017, 19, 3–31. [Google Scholar] [CrossRef]
- Oda, E. The Metabolic Syndrome as a Concept of Adipose Tissue Disease. Hypertens. Res. 2008, 31, 1283–1291. [Google Scholar] [CrossRef] [Green Version]
- Berg, A.H.; Scherer, P.E. Adipose Tissue, Inflammation, and Cardiovascular Disease. Circ. Res. 2005, 96, 939–949. [Google Scholar] [CrossRef] [Green Version]
- van Greevenbroek, M.M.J.; Schalkwijk, C.G.; Stehouwer, C.D.A. Dysfunctional adipose tissue and low-grade inflammation in the management of the metabolic syndrome: Current practices and future advances. F1000Res 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Benedict, M.; Zhang, X. Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol. 2017, 9, 715–732. [Google Scholar] [CrossRef]
- van der Vorst, E.P.C.; Döring, Y.; Weber, C. Chemokines and their receptors in Atherosclerosis. J. Mol. Med. Berl. 2015, 93, 963–971. [Google Scholar] [CrossRef] [Green Version]
- Raman, D.; Sobolik-Delmaire, T.; Richmond, A. Chemokines in health and disease. Exp. Cell Res. 2011, 317, 575–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, L.; Herlea-Pana, O.; Heuser-Baker, J.; Chen, Y.; Barlic-Dicen, J. Roles of the chemokine system in development of obesity, insulin resistance, and cardiovascular disease. J. Immunol. Res. 2014, 2014, 181450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Chen, W.; Shen, J. CXCR7 Targeting and Its Major Disease Relevance. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, H.; Zhang, H.; Zhu, H. Blocking CXCR7-mediated adipose tissue macrophages chemotaxis attenuates insulin resistance and inflammation in obesity. Biochem. Biophys. Res. Commun. 2016, 479, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, M.; Penfold, M.E.; Koenen, R.R.; Thiemann, A.; Heyll, K.; Akhtar, S.; Koyadan, S.; Wu, Z.; Gremse, F.; et al. Activation of CXCR7 limits atherosclerosis and improves hyperlipidemia by increasing cholesterol uptake in adipose tissue. Circulation 2014, 129, 1244–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukonina, V.; Lookene, A.; Olivecrona, T.; Olivecrona, G. Angiopoietin-like protein 4 converts lipoprotein lipase to inactive monomers and modulates lipase activity in adipose tissue. Proc. Natl. Acad Sci. USA 2006, 103, 17450–17455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Zvi, D.; Barrandon, O.; Hadley, S.; Blum, B.; Peterson, Q.P.; Melton, D.A. Angptl4 links α-cell proliferation following glucagon receptor inhibition with adipose tissue triglyceride metabolism. Proc. Natl. Acad. Sci. USA 2015, 112, 15498–15503. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Zhou, J.; Casimiro, M.C.; Liang, B.; Ojeifo, J.O.; Wang, M.; Hyslop, T.; Wang, C.; Pestell, R.G. Activating peroxisome proliferator-activated receptor gamma mutant promotes tumor growth in vivo by enhancing angiogenesis. Cancer. Res. 2009, 69, 9236–9244. [Google Scholar] [CrossRef] [Green Version]
- Qian, T.; Liu, Y.; Dong, Y.; Zhang, L.; Dong, Y.; Sun, Y.; Sun, D. CXCR7 regulates breast tumor metastasis and angiogenesis in vivo and in vitro. Mol. Med. Rep. 2018, 17, 3633–3639. [Google Scholar] [CrossRef]
- Yamada, K.; Maishi, N.; Akiyama, K.; Towfik Alam, M.; Ohga, N.; Kawamoto, T.; Shindoh, M.; Takahashi, N.; Kamiyama, T.; Hida, Y.; et al. CXCL12-CXCR7 axis is important for tumor endothelial cell angiogenic property. Int. J. Cancer 2015, 137, 2825–2836. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Azad, A.K.; Karanika, S.; Basourakos, S.P.; Zuo, X.; Wang, J.; Yang, L.; Yang, G.; Korentzelos, D.; Yin, J.; et al. Enzalutamide and CXCR7 inhibitor combination treatment suppresses cell growth and angiogenic signaling in castration-resistant prostate cancer models. Int. J. Cancer 2018, 142, 2163–2174. [Google Scholar] [CrossRef] [Green Version]
- Sartina, E.; Suguihara, C.; Ramchandran, S.; Nwajei, P.; Rodriguez, M.; Torres, E.; Hehre, D.; Devia, C.; Walters, M.J.; Penfold, M.E.; et al. Antagonism of CXCR7 attenuates chronic hypoxia-induced pulmonary hypertension. Pediatr. Res. 2012, 71, 682–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zabel, B.A.; Wang, Y.; Lewén, S.; Berahovich, R.D.; Penfold, M.E.; Zhang, P.; Powers, J.; Summers, B.C.; Miao, Z.; Zhao, B.; et al. Elucidation of CXCR7-mediated signaling events and inhibition of CXCR4-mediated tumor cell transendothelial migration by CXCR7 ligands. J. Immunol. 2009, 183, 3204–3211. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Zhu, Z.; Li, D.; Xu, R.; Wang, T.; Liu, K. Pioglitazone Suppresses CXCR7 Expression To Inhibit Human Macrophage Chemotaxis through Peroxisome Proliferator-Activated Receptor γ. Biochemistry 2015, 54, 6806–6814. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Yang, Y.; Liu, D.; Qi, Y.; Zhang, C.; Zhao, J.; Zhao, S. Activation of PPARγ suppresses proliferation and induces apoptosis of esophageal cancer cells by inhibiting TLR4-dependent MAPK pathway. Oncotarget 2016, 7, 44572–44582. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Fong, K.W.; Gritsina, G.; Zhang, A.; Zhao, J.C.; Kim, J.; Sharp, A.; Yuan, W.; Aversa, C.; Yang, X.J.; et al. Activation of MAPK Signaling by CXCR7 Leads to Enzalutamide Resistance in Prostate Cancer. Cancer Res. 2019, 79, 2580–2592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Han, M.M.; Wang, F.; Xu, L.L.; Yu, H.X.; Yang, P.Y. CXCR7 stimulates MAPK signaling to regulate hepatocellular carcinoma progression. Cell Death Dis. 2014, 5, e1488. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, M.; Leroux, C.; Faulconnier, Y.; Hocquette, J.-F.O.; Bocquier, F.O.; Martin, P.; Chilliard, Y. Lipoprotein Lipase Activity and mRNA Are Up-Regulated by Refeeding in Adipose Tissue and Cardiac Muscle of Sheep. J. Nutr. 2000, 130, 749–756. [Google Scholar] [CrossRef] [Green Version]
- Cushing, E.M.; Chi, X.; Sylvers, K.L.; Shetty, S.K.; Potthoff, M.J.; Davies, B.S.J. Angiopoietin-like 4 directs uptake of dietary fat away from adipose during fasting. Mol. Metab. 2017, 6, 809–818. [Google Scholar] [CrossRef]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef] [Green Version]
- Reilly, S.M.; Saltiel, A.R. Adapting to obesity with adipose tissue inflammation. Nat. Rev. Endocrinol. 2017, 13, 633–643. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 4-Week WD | AdipoqCre−Ackr3fl/flApoe−/− | AdipoqCre+Ackr3fl/flApoe−/− | p-Value |
|---|---|---|---|
| Leukocytes [×106/mL] | 2.4 ± 0.2 | 2.1 ± 0.1 | 0.3445 |
| Neutrophils [×105/mL] | 6.5 ± 0.5 | 5.8 ± 0.5 | 0.3701 |
| Classical Monocytes [×105/mL] | 1.9 ± 0.3 | 2.1 ± 0.2 | 0.5925 |
| Non-classical Monocytes [×104/mL] | 7.7 ± 1.3 | 9.3 ± 1.7 | 0.5826 |
| B cells [×105/mL] | 8.7 ± 1.1 | 7.2 ± 0.6 | 0.2358 |
| T cells [×105/mL] | 3.1 ± 0.3 | 2.5 ± 0.2 | 0.1476 |
| Plasma IL-6 [pg/mL] | 6.0 ± 1.5 | 3.5 ± 1.0 | 0.3845 |
| Plasma TNF-α [pg/mL] | 11.6 ± 1.7 | 10.4 ± 0.6 | 0.9157 |
| Plasma CXCL12 [pg/mL] | 452.1 ± 50.2 | 386.3 ± 66.08 | 0.4360 |
| Mouse weight [g] | 24.5 ± 0.5 | 25.6 ± 0.7 | 0.2575 |
| wAT weight [mg] | 341.6 ± 62.71 | 384.6 ± 72.18 | 0.6633 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gencer, S.; Döring, Y.; Jansen, Y.; Bayasgalan, S.; Schengel, O.; Müller, M.; Peters, L.J.F.; Weber, C.; van der Vorst, E.P.C. Adipocyte-Specific ACKR3 Regulates Lipid Levels in Adipose Tissue. Biomedicines 2021, 9, 394. https://doi.org/10.3390/biomedicines9040394
Gencer S, Döring Y, Jansen Y, Bayasgalan S, Schengel O, Müller M, Peters LJF, Weber C, van der Vorst EPC. Adipocyte-Specific ACKR3 Regulates Lipid Levels in Adipose Tissue. Biomedicines. 2021; 9(4):394. https://doi.org/10.3390/biomedicines9040394
Chicago/Turabian StyleGencer, Selin, Yvonne Döring, Yvonne Jansen, Soyolmaa Bayasgalan, Olga Schengel, Madeleine Müller, Linsey J. F. Peters, Christian Weber, and Emiel P. C. van der Vorst. 2021. "Adipocyte-Specific ACKR3 Regulates Lipid Levels in Adipose Tissue" Biomedicines 9, no. 4: 394. https://doi.org/10.3390/biomedicines9040394
APA StyleGencer, S., Döring, Y., Jansen, Y., Bayasgalan, S., Schengel, O., Müller, M., Peters, L. J. F., Weber, C., & van der Vorst, E. P. C. (2021). Adipocyte-Specific ACKR3 Regulates Lipid Levels in Adipose Tissue. Biomedicines, 9(4), 394. https://doi.org/10.3390/biomedicines9040394