
Lithium and Atypical Antipsychotics: The Possible WNT/β Pathway Target in Glaucoma


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pathophysiology of Glaucoma
3. Oxidative Stress, Inflammation and Glutamate in Glaucoma
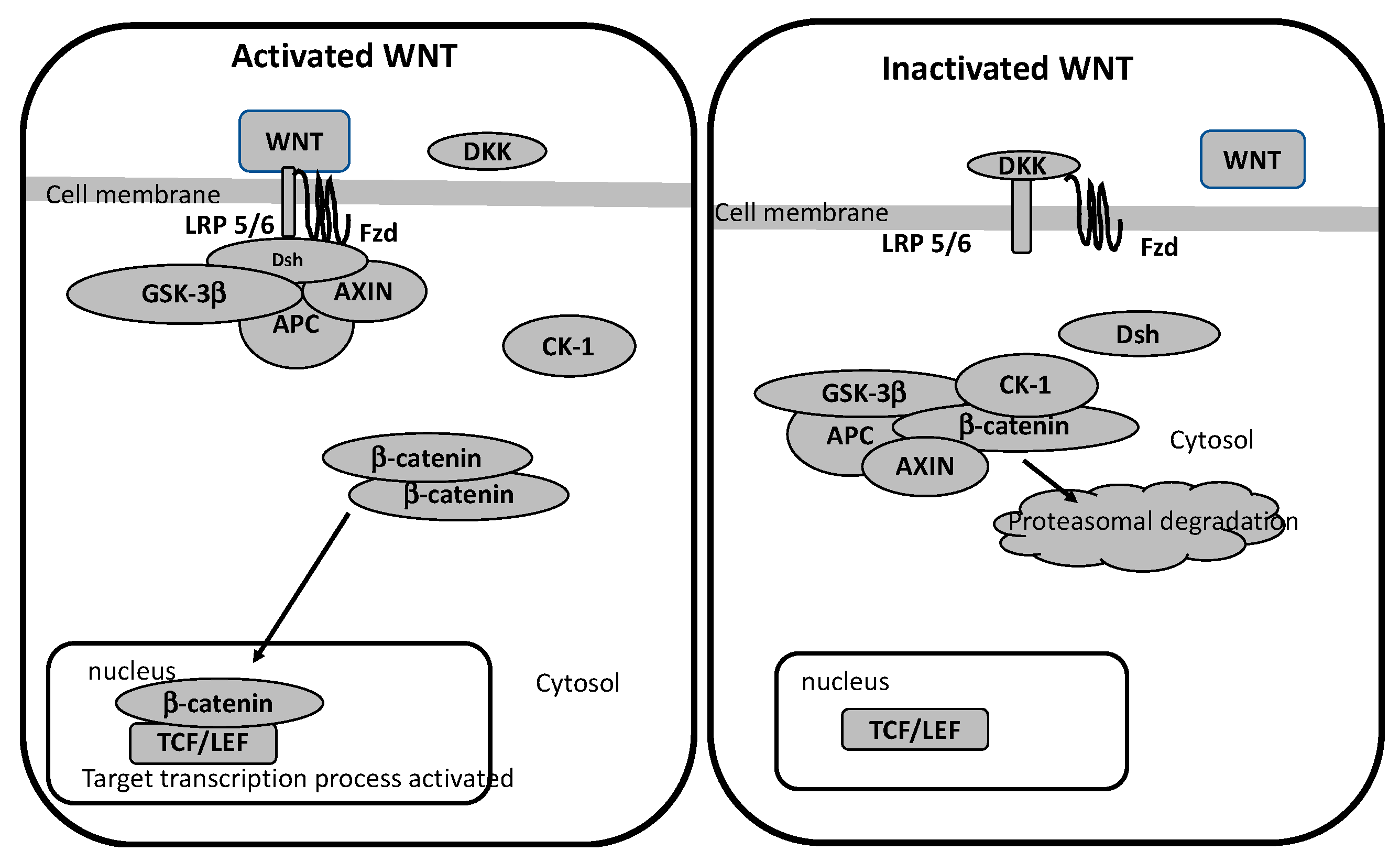
4. WNT/β-Catenin Pathway
WNT/β-Catenin Pathway in Glaucoma
5. Lithium and AAPs in Glaucoma
5.1. Lithium in Glaucoma
5.2. AAPs in Glaucoma
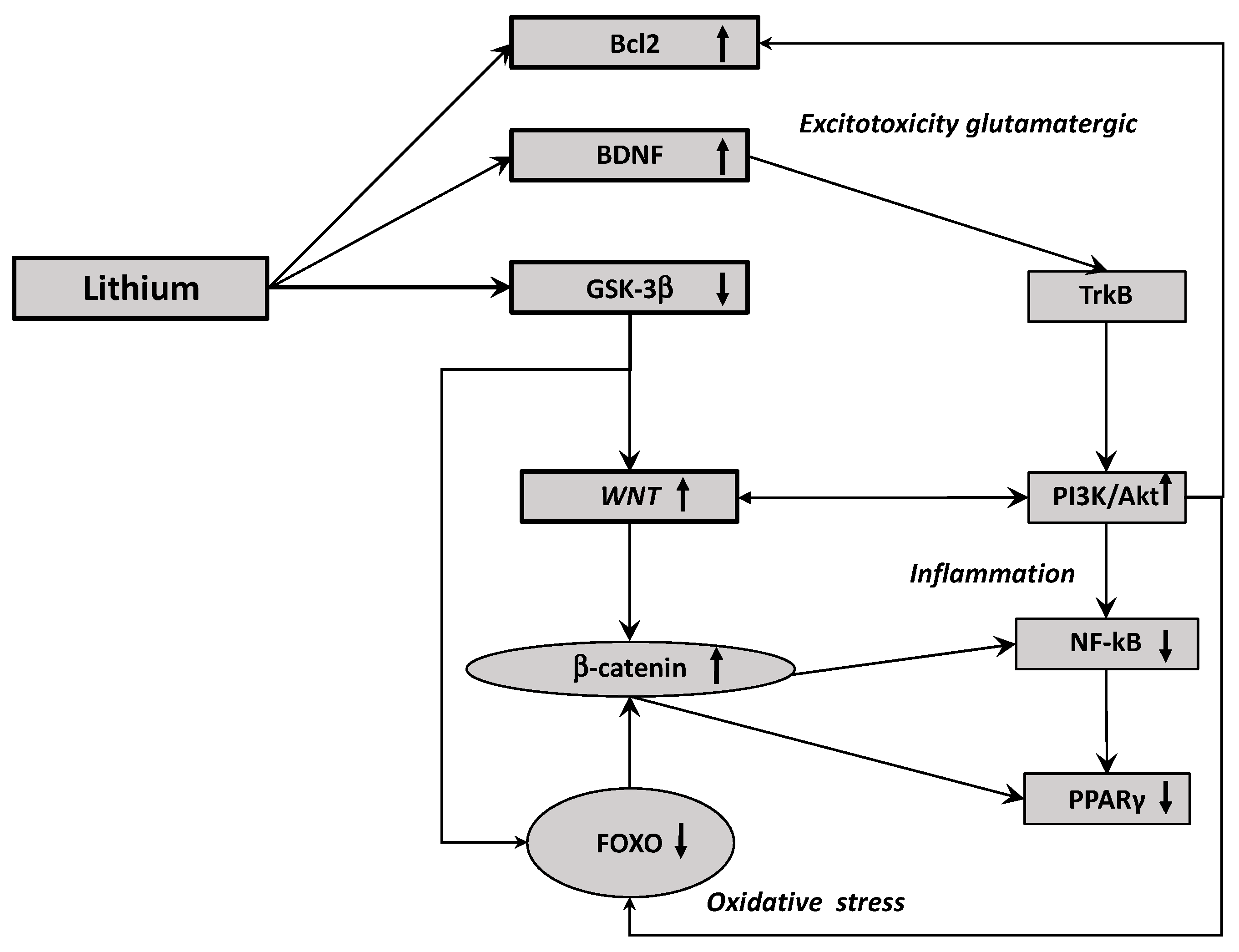
6. Activation of the Canonical WNT Pathway by Lithium: A Potential Therapeutic Strategy
7. Lithium and the Different Altered Pathways Involved in Glaucoma
7.1. Lithium and Inflammation
7.2. Lithium and Glutamatergic Pathway
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tham, Y.-C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.-Y. Global Prevalence of Glaucoma and Projections of Glaucoma Burden through 2040: A Systematic Review and Meta-Analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Harasymowycz, P.; Birt, C.; Gooi, P.; Heckler, L.; Hutnik, C.; Jinapriya, D.; Shuba, L.; Yan, D.; Day, R. Medical Management of Glaucoma in the 21st Century from a Canadian Perspective. J. Ophthalmol. 2016, 2016, 6509809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, J.D.; Khawaja, A.P.; Weizer, J.S. Glaucoma in Adults-Screening, Diagnosis, and Management: A Review. JAMA 2021, 325, 164–174. [Google Scholar] [CrossRef]
- Esporcatte, B.L.B.; Tavares, I.M. Normal-Tension Glaucoma: An Update. Arq. Bras. Oftalmol. 2016, 79, 270–276. [Google Scholar] [CrossRef] [Green Version]
- Allison, K.; Patel, D.; Alabi, O. Epidemiology of Glaucoma: The Past, Present, and Predictions for the Future. Cureus 2020, 12, e11686. [Google Scholar] [CrossRef]
- Grzybowski, A.; Och, M.; Kanclerz, P.; Leffler, C.; Moraes, C.G.D. Primary Open Angle Glaucoma and Vascular Risk Factors: A Review of Population Based Studies from 1990 to 2019. J. Clin. Med. 2020, 9, 761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shields, M.B. Normal-Tension Glaucoma: Is It Different from Primary Open-Angle Glaucoma? Curr. Opin. Ophthalmol. 2008, 19, 85–88. [Google Scholar] [CrossRef]
- Saccà, S.C.; Gandolfi, S.; Bagnis, A.; Manni, G.; Damonte, G.; Traverso, C.E.; Izzotti, A. From DNA Damage to Functional Changes of the Trabecular Meshwork in Aging and Glaucoma. Ageing Res. Rev. 2016, 29, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Lähteenvuo, M.; Tanskanen, A.; Taipale, H.; Hoti, F.; Vattulainen, P.; Vieta, E.; Tiihonen, J. Real-World Effectiveness of Pharmacologic Treatments for the Prevention of Rehospitalization in a Finnish Nationwide Cohort of Patients With Bipolar Disorder. JAMA Psychiatry 2018, 75, 347–355. [Google Scholar] [CrossRef] [Green Version]
- Leeds, P.R.; Yu, F.; Wang, Z.; Chiu, C.-T.; Zhang, Y.; Leng, Y.; Linares, G.R.; Chuang, D.-M. A New Avenue for Lithium: Intervention in Traumatic Brain Injury. ACS Chem. Neurosci. 2014, 5, 422–433. [Google Scholar] [CrossRef]
- Marmol, F. Lithium: Bipolar Disorder and Neurodegenerative Diseases Possible Cellular Mechanisms of the Therapeutic Effects of Lithium. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 1761–1771. [Google Scholar] [CrossRef]
- Young, A.H.; Hammond, J.M. Lithium in Mood Disorders: Increasing Evidence Base, Declining Use? Br. J. Psychiatry 2007, 191, 474–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.A.; Cornelius, V.; Warnock, A.; Bell, A.; Young, A.H. Effectiveness of Mood Stabilizers and Antipsychotics in the Maintenance Phase of Bipolar Disorder: A Systematic Review of Randomized Controlled Trials. Bipolar Disord. 2007, 9, 394–412. [Google Scholar] [CrossRef] [PubMed]
- Baldessarini, R.J.; Tondo, L.; Davis, P.; Pompili, M.; Goodwin, F.K.; Hennen, J. Decreased Risk of Suicides and Attempts during Long-Term Lithium Treatment: A Meta-Analytic Review. Bipolar Disord. 2006, 8, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Pisanu, C.; Melis, C.; Squassina, A. Lithium Pharmacogenetics: Where Do We Stand? Drug Dev. Res. 2016, 77, 368–373. [Google Scholar] [CrossRef]
- Zhu, Z.-F.; Wang, Q.-G.; Han, B.-J.; William, C.P. Neuroprotective Effect and Cognitive Outcome of Chronic Lithium on Traumatic Brain Injury in Mice. Brain Res. Bull. 2010, 83, 272–277. [Google Scholar] [CrossRef]
- Huang, X.; Wu, D.-Y.; Chen, G.; Manji, H.; Chen, D.F. Support of Retinal Ganglion Cell Survival and Axon Regeneration by Lithium through a Bcl-2-Dependent Mechanism. Investig. Ophthalmol. Vis. Sci. 2003, 44, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, J.; Li, F.; Liu, X.; Liu, Z.; Lin, J.; Ge, Y.; Kaminski, J.M.; Summers, J.B.; Wang, Z.; Ge, J.; et al. Lithium Chloride Protects Retinal Neurocytes from Nutrient Deprivation by Promoting DNA Non-Homologous End-Joining. Biochem. Biophys. Res. Commun. 2009, 380, 650–654. [Google Scholar] [CrossRef]
- Villarreal, G.; Chatterjee, A.; Oh, S.S.; Oh, D.-J.; Kang, M.H.; Rhee, D.J. Canonical Wnt Signaling Regulates Extracellular Matrix Expression in the Trabecular Meshwork. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7433–7440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.-B.; Lu, H.-E.; Chen, Y.; Fan, X.-H.; Tong, B. Effect of Lithium Chloride on Endoplasmic Reticulum Stress-Related PERK/ROCK Signaling in a Rat Model of Glaucoma. Pharmazie 2014, 69, 889–893. [Google Scholar]
- Scherk, H.; Pajonk, F.G.; Leucht, S. Second-Generation Antipsychotic Agents in the Treatment of Acute Mania: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Arch. Gen. Psychiatry 2007, 64, 442–455. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liang, J.; Xia, Q.; Zhou, X.; Xie, X. Effects of Lithium Combined with Second-Generation Antipsychotics for the Treatment of Manic Episodes in Patients with Bipolar Disorder: A Naturalistic Study in China. Neuropsychiatr. Dis. Treat. 2020, 16, 2623–2632. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, J.; Murphy, C.; Juster, R. Trabecular Meshwork Cellularity in Primary Open-Angle Glaucoma and Nonglaucomatous Normals. Ophthalmology 1984, 91, 564–579. [Google Scholar] [CrossRef]
- Vernazza, S.; Tirendi, S.; Scarfì, S.; Passalacqua, M.; Oddone, F.; Traverso, C.E.; Rizzato, I.; Bassi, A.M.; Saccà, S.C. 2D- and 3D-Cultures of Human Trabecular Meshwork Cells: A Preliminary Assessment of an in Vitro Model for Glaucoma Study. PLoS ONE 2019, 14, e0221942. [Google Scholar] [CrossRef] [PubMed]
- Vernazza, S.; Tirendi, S.; Bassi, A.M.; Traverso, C.E.; Saccà, S.C. Neuroinflammation in Primary Open-Angle Glaucoma. J. Clin. Med. 2020, 9, 3172. [Google Scholar] [CrossRef]
- Castro, A.; Du, Y. Trabecular Meshwork Regeneration—A Potential Treatment for Glaucoma. Curr. Ophthalmol. Rep. 2019, 7, 80–88. [Google Scholar] [CrossRef]
- Alvarado, J.A.; Alvarado, R.G.; Yeh, R.F.; Franse-Carman, L.; Marcellino, G.R.; Brownstein, M.J. A New Insight into the Cellular Regulation of Aqueous Outflow: How Trabecular Meshwork Endothelial Cells Drive a Mechanism That Regulates the Permeability of Schlemm’s Canal Endothelial Cells. Br. J. Ophthalmol. 2005, 89, 1500–1505. [Google Scholar] [CrossRef] [Green Version]
- Salinas-Navarro, M.; Alarcón-Martínez, L.; Valiente-Soriano, F.J.; Jiménez-López, M.; Mayor-Torroglosa, S.; Avilés-Trigueros, M.; Villegas-Pérez, M.P.; Vidal-Sanz, M. Ocular Hypertension Impairs Optic Nerve Axonal Transport Leading to Progressive Retinal Ganglion Cell Degeneration. Exp. Eye Res. 2010, 90, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.-K.; Kim, K.-Y.; Lindsey, J.D.; Angert, M.; Patel, A.; Scott, R.T.; Liu, Q.; Crowston, J.G.; Ellisman, M.H.; Perkins, G.A.; et al. Elevated Hydrostatic Pressure Triggers Release of OPA1 and Cytochrome C, and Induces Apoptotic Cell Death in Differentiated RGC-5 Cells. Mol. Vis. 2009, 15, 120–134. [Google Scholar]
- McMonnies, C.W. Glaucoma History and Risk Factors. J. Optom. 2017, 10, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Saccà, S.C.; Gandolfi, S.; Bagnis, A.; Manni, G.; Damonte, G.; Traverso, C.E.; Izzotti, A. The Outflow Pathway: A Tissue With Morphological and Functional Unity. J. Cell. Physiol. 2016, 231, 1876–1893. [Google Scholar] [CrossRef] [PubMed]
- Izzotti, A.; Saccà, S.C.; Longobardi, M.; Cartiglia, C. Sensitivity of Ocular Anterior Chamber Tissues to Oxidative Damage and Its Relevance to the Pathogenesis of Glaucoma. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5251–5258. [Google Scholar] [CrossRef] [Green Version]
- Saccà, S.C.; Tirendi, S.; Scarfì, S.; Passalacqua, M.; Oddone, F.; Traverso, C.E.; Vernazza, S.; Bassi, A.M. An Advanced in Vitro Model to Assess Glaucoma Onset. ALTEX 2020, 37, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Liton, P.B.; Lin, Y.; Luna, C.; Li, G.; Gonzalez, P.; Epstein, D.L. Cultured Porcine Trabecular Meshwork Cells Display Altered Lysosomal Function When Subjected to Chronic Oxidative Stress. Invest. Ophthalmol Vis. Sci. 2008, 49, 3961–3969. [Google Scholar] [CrossRef]
- Liton, P.B.; Challa, P.; Stinnett, S.; Luna, C.; Epstein, D.L.; Gonzalez, P. Cellular Senescence in the Glaucomatous Outflow Pathway. Exp. Gerontol. 2005, 40, 745–748. [Google Scholar] [CrossRef] [Green Version]
- Chrysostomou, V.; Rezania, F.; Trounce, I.A.; Crowston, J.G. Oxidative Stress and Mitochondrial Dysfunction in Glaucoma. Curr. Opin. Pharm. 2013, 13, 12–15. [Google Scholar] [CrossRef]
- Vohra, R.; Dalgaard, L.M.; Vibaek, J.; Langbøl, M.A.; Bergersen, L.H.; Olsen, N.V.; Hassel, B.; Chaudhry, F.A.; Kolko, M. Potential Metabolic Markers in Glaucoma and Their Regulation in Response to Hypoxia. Acta Ophthalmol. 2019, 97, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Lebrun-Julien, F.; Duplan, L.; Pernet, V.; Osswald, I.; Sapieha, P.; Bourgeois, P.; Dickson, K.; Bowie, D.; Barker, P.A.; Di Polo, A. Excitotoxic Death of Retinal Neurons in Vivo Occurs via a Non-Cell-Autonomous Mechanism. J. Neurosci. 2009, 29, 5536–5545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, P.A.; Marsh-Armstrong, N.; Howell, G.R. Lasker/IRRF Initiative on Astrocytes and Glaucomatous Neurodegeneration Participants Neuroinflammation in Glaucoma: A New Opportunity. Exp. Eye Res. 2017, 157, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Kametani, M.; Chen, D.F. Adaptive Immunity: New Aspects of Pathogenesis Underlying Neurodegeneration in Glaucoma and Optic Neuropathy. Front. Immunol. 2020, 11, 65. [Google Scholar] [CrossRef] [Green Version]
- Eells, J.T. Mitochondrial Dysfunction in the Aging Retina. Biology 2019, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Sreekumar, P.G.; Hinton, D.R.; Kannan, R. The Emerging Role of Senescence in Ocular Disease. Oxid. Med. Cell. Longev. 2020, 2020, 2583601. [Google Scholar] [CrossRef]
- Xu, H.; Chen, M. Targeting the Complement System for the Management of Retinal Inflammatory and Degenerative Diseases. Eur. J. Pharm. 2016, 787, 94–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medzhitov, R. Origin and Physiological Roles of Inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Chen, M.; Luo, C.; Zhao, J.; Devarajan, G.; Xu, H. Immune Regulation in the Aging Retina. Prog. Retin. Eye Res. 2019, 69, 159–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, N.; Arora, P.; Sandhir, R. Perturbed Biochemical Pathways and Associated Oxidative Stress Lead to Vascular Dysfunctions in Diabetic Retinopathy. Oxid. Med. Cell. Longev. 2019, 2019, 8458472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlowska, E.; Szczepanska, J.; Koskela, A.; Kaarniranta, K.; Blasiak, J. Dietary Polyphenols in Age-Related Macular Degeneration: Protection against Oxidative Stress and Beyond. Oxid. Med. Cell. Longev. 2019, 2019, 9682318. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, M.J.; Channon, K.M. Synthesis and Recycling of Tetrahydrobiopterin in Endothelial Function and Vascular Disease. Nitric Oxide 2011, 25, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M. What Controls Aqueous Humour Outflow Resistance? Exp. Eye Res. 2006, 82, 545–557. [Google Scholar] [CrossRef] [Green Version]
- Knepper, P.A.; Goossens, W.; Hvizd, M.; Palmberg, P.F. Glycosaminoglycans of the Human Trabecular Meshwork in Primary Open-Angle Glaucoma. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1360–1367. [Google Scholar]
- Wang, X.; Huai, G.; Wang, H.; Liu, Y.; Qi, P.; Shi, W.; Peng, J.; Yang, H.; Deng, S.; Wang, Y. Mutual Regulation of the Hippo/Wnt/LPA/TGF-β Signaling Pathways and Their Roles in Glaucoma (Review). Int. J. Mol. Med. 2018, 41, 1201–1212. [Google Scholar] [CrossRef]
- Mozaffarieh, M.; Flammer, J. New Insights in the Pathogenesis and Treatment of Normal Tension Glaucoma. Curr. Opin. Pharm. 2013, 13, 43–49. [Google Scholar] [CrossRef]
- Benoist d’Azy, C.; Pereira, B.; Chiambaretta, F.; Dutheil, F. Oxidative and Anti-Oxidative Stress Markers in Chronic Glaucoma: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0166915. [Google Scholar] [CrossRef] [PubMed]
- Gericke, A.; Mann, C.; Zadeh, J.K.; Musayeva, A.; Wolff, I.; Wang, M.; Pfeiffer, N.; Daiber, A.; Li, H.; Xia, N.; et al. Elevated Intraocular Pressure Causes Abnormal Reactivity of Mouse Retinal Arterioles. Oxid. Med. Cell. Longev. 2019, 2019, 9736047. [Google Scholar] [CrossRef] [Green Version]
- Salt, T.E.; Cordeiro, M.F. Glutamate Excitotoxicity in Glaucoma: Throwing the Baby out with the Bathwater? Eye 2006, 20, 730–731; author reply 731–732. [Google Scholar] [CrossRef] [Green Version]
- Tezel, G.; Yang, X.; Luo, C.; Peng, Y.; Sun, S.L.; Sun, D. Mechanisms of Immune System Activation in Glaucoma: Oxidative Stress-Stimulated Antigen Presentation by the Retina and Optic Nerve Head Glia. Investig. Ophthalmol. Vis. Sci. 2007, 48, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Wax, M.B. Increased Production of Tumor Necrosis Factor-Alpha by Glial Cells Exposed to Simulated Ischemia or Elevated Hydrostatic Pressure Induces Apoptosis in Cocultured Retinal Ganglion Cells. J. Neurosci. 2000, 20, 8693–8700. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-W.; Karin, M. A Cytokine-Mediated Link between Innate Immunity, Inflammation, and Cancer. J. Clin. Investig. 2007, 117, 1175–1183. [Google Scholar] [CrossRef]
- Sobolewski, C.; Cerella, C.; Dicato, M.; Ghibelli, L.; Diederich, M. The Role of Cyclooxygenase-2 in Cell Proliferation and Cell Death in Human Malignancies. Int. J. Cell Biol. 2010, 2010, 215158. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Ouyang, W.; Huang, C. Inflammation, a Key Event in Cancer Development. Mol. Cancer Res. 2006, 4, 221–233. [Google Scholar] [CrossRef] [Green Version]
- Federico, A.; Morgillo, F.; Tuccillo, C.; Ciardiello, F.; Loguercio, C. Chronic Inflammation and Oxidative Stress in Human Carcinogenesis. Int. J. Cancer 2007, 121, 2381–2386. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Antony, S.; Meitzler, J.L.; Doroshow, J.H. Molecular Mechanisms Underlying Chronic Inflammation-Associated Cancers. Cancer Lett. 2014, 345, 164–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bubici, C.; Papa, S.; Pham, C.G.; Zazzeroni, F.; Franzoso, G. The NF-KappaB-Mediated Control of ROS and JNK Signaling. Histol. Histopathol. 2006, 21, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-H.; Woo, K.J.; Lim, J.H.; Kim, S.; Lee, T.J.; Jung, E.M.; Lee, J.-M.; Park, J.-W.; Kwon, T.K. 8-Hydroxyquinoline Inhibits INOS Expression and Nitric Oxide Production by down-Regulating LPS-Induced Activity of NF-KappaB and C/EBPbeta in Raw 264.7 Cells. Biochem. Biophys. Res. Commun. 2005, 329, 591–597. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative Stress, Inflammation, and Cancer: How Are They Linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duracková, Z. Some Current Insights into Oxidative Stress. Physiol. Res. 2010, 59, 459–469. [Google Scholar] [CrossRef]
- Debnath, T.; Kim, D.H.; Lim, B.O. Natural Products as a Source of Anti-Inflammatory Agents Associated with Inflammatory Bowel Disease. Molecules 2013, 18, 7253–7270. [Google Scholar] [CrossRef]
- Giudice, A.; Montella, M. Activation of the Nrf2-ARE Signaling Pathway: A Promising Strategy in Cancer Prevention. Bioessays 2006, 28, 169–181. [Google Scholar] [CrossRef]
- Lin, M.; Zhai, X.; Wang, G.; Tian, X.; Gao, D.; Shi, L.; Wu, H.; Fan, Q.; Peng, J.; Liu, K.; et al. Salvianolic Acid B Protects against Acetaminophen Hepatotoxicity by Inducing Nrf2 and Phase II Detoxification Gene Expression via Activation of the PI3K and PKC Signaling Pathways. J. Pharmacol.Sci. 2015, 127, 203–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, A.; Lakshmanan, J. The Role of Antioxidants and Other Agents in Alleviating Hyperglycemia Mediated Oxidative Stress and Injury in Liver. Food Funct. 2013, 4, 1148–1184. [Google Scholar] [CrossRef]
- Ting, J.T.; Feng, G. Neurobiology of Obsessive-Compulsive Disorder: Insights into Neural Circuitry Dysfunction through Mouse Genetics. Curr. Opin. Neurobiol. 2011, 21, 842–848. [Google Scholar] [CrossRef]
- Javitt, D.C.; Schoepp, D.; Kalivas, P.W.; Volkow, N.D.; Zarate, C.; Merchant, K.; Bear, M.F.; Umbricht, D.; Hajos, M.; Potter, W.Z.; et al. Translating Glutamate: From Pathophysiology to Treatment. Sci. Transl. Med. 2011, 3, 102mr2. [Google Scholar] [CrossRef] [Green Version]
- Sanacora, G.; Zarate, C.A.; Krystal, J.H.; Manji, H.K. Targeting the Glutamatergic System to Develop Novel, Improved Therapeutics for Mood Disorders. Nat. Rev. Drug Discov. 2008, 7, 426–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daikhin, Y.; Yudkoff, M. Compartmentation of Brain Glutamate Metabolism in Neurons and Glia. J. Nutr. 2000, 130, 1026S–1031S. [Google Scholar] [CrossRef] [PubMed]
- Scimemi, A.; Tian, H.; Diamond, J.S. Neuronal Transporters Regulate Glutamate Clearance, NMDA Receptor Activation, and Synaptic Plasticity in the Hippocampus. J. Neurosci. 2009, 29, 14581–14595. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Hanna, G.L.; Rosenberg, D.R.; Arnold, P.D. The Role of Glutamate Signaling in the Pathogenesis and Treatment of Obsessive-Compulsive Disorder. Pharmacol. Biochem. Behav. 2012, 100, 726–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.-S.; Shutov, L.P.; Gnanasekaran, A.; Lin, Z.; Rysted, J.E.; Ulrich, J.D.; Usachev, Y.M. Nerve Growth Factor (NGF) Regulates Activity of Nuclear Factor of Activated T-Cells (NFAT) in Neurons via the Phosphatidylinositol 3-Kinase (PI3K)-Akt-Glycogen Synthase Kinase 3β (GSK3β) Pathway. J. Biol. Chem. 2014, 289, 31349–31360. [Google Scholar] [CrossRef] [Green Version]
- Lotery, A.J. Glutamate Excitotoxicity in Glaucoma: Truth or Fiction? Eye 2005, 19, 369–370. [Google Scholar] [CrossRef] [Green Version]
- Seki, M.; Lipton, S.A. Targeting Excitotoxic/Free Radical Signaling Pathways for Therapeutic Intervention in Glaucoma. Prog. Brain Res. 2008, 173, 495–510. [Google Scholar] [CrossRef]
- Lee, D.; Shim, M.S.; Kim, K.-Y.; Noh, Y.H.; Kim, H.; Kim, S.Y.; Weinreb, R.N.; Ju, W.-K. Coenzyme Q10 Inhibits Glutamate Excitotoxicity and Oxidative Stress-Mediated Mitochondrial Alteration in a Mouse Model of Glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 993–1005. [Google Scholar] [CrossRef] [Green Version]
- Siliprandi, R.; Canella, R.; Carmignoto, G.; Schiavo, N.; Zanellato, A.; Zanoni, R.; Vantini, G. N-Methyl-D-Aspartate-Induced Neurotoxicity in the Adult Rat Retina. Vis. Neurosci. 1992, 8, 567–573. [Google Scholar] [CrossRef]
- Loh, K.M.; van Amerongen, R.; Nusse, R. Generating Cellular Diversity and Spatial Form: Wnt Signaling and the Evolution of Multicellular Animals. Dev. Cell 2016, 38, 643–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oren, O.; Smith, B.D. Eliminating Cancer Stem Cells by Targeting Embryonic Signaling Pathways. Stem Cell Rev. 2017, 13, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Al-Harthi, L. Wnt/β-Catenin and Its Diverse Physiological Cell Signaling Pathways in Neurodegenerative and Neuropsychiatric Disorders. J. Neuroimmune Pharmacol. 2012, 7, 725–730. [Google Scholar] [CrossRef]
- Marchetti, B.; Pluchino, S. Wnt Your Brain Be Inflamed? Yes, It Wnt! Trends Mol. Med. 2013, 19, 144–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecarpentier, Y.; Claes, V.; Duthoit, G.; Hébert, J.-L. Circadian Rhythms, Wnt/Beta-Catenin Pathway and PPAR Alpha/Gamma Profiles in Diseases with Primary or Secondary Cardiac Dysfunction. Front. Physiol. 2014, 5, 429. [Google Scholar] [CrossRef] [Green Version]
- Lecarpentier, Y.; Vallée, A. Opposite Interplay between PPAR Gamma and Canonical Wnt/Beta-Catenin Pathway in Amyotrophic Lateral Sclerosis. Front. Neurol. 2016, 7, 100. [Google Scholar] [CrossRef] [Green Version]
- Vallée, A.; Lecarpentier, Y. Alzheimer Disease: Crosstalk between the Canonical Wnt/Beta-Catenin Pathway and PPARs Alpha and Gamma. Front. Neurosci. 2016, 10, 459. [Google Scholar] [CrossRef] [Green Version]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Thermodynamics in Neurodegenerative Diseases: Interplay Between Canonical WNT/Beta-Catenin Pathway-PPAR Gamma, Energy Metabolism and Circadian Rhythms. Neuromol. Med. 2018, 20, 174–204. [Google Scholar] [CrossRef]
- Vallée, A.; Vallée, J.-N.; Lecarpentier, Y. Parkinson’s Disease: Potential Actions of Lithium by Targeting the WNT/β-Catenin Pathway, Oxidative Stress, Inflammation and Glutamatergic Pathway. Cells 2021, 10, 230. [Google Scholar] [CrossRef] [PubMed]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of C-MYC as a Target of the APC Pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Shtutman, M.; Zhurinsky, J.; Simcha, I.; Albanese, C.; D’Amico, M.; Pestell, R.; Ben-Ze’ev, A. The Cyclin D1 Gene Is a Target of the Beta-Catenin/LEF-1 Pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 5522–5527. [Google Scholar] [CrossRef] [Green Version]
- Angers, S.; Moon, R.T. Proximal Events in Wnt Signal Transduction. Nat. Rev. Mol. Cell Biol. 2009. [Google Scholar] [CrossRef]
- Sharma, C.; Pradeep, A.; Wong, L.; Rana, A.; Rana, B. Peroxisome Proliferator-Activated Receptor Gamma Activation Can Regulate Beta-Catenin Levels via a Proteasome-Mediated and Adenomatous Polyposis Coli-Independent Pathway. J. Biol. Chem. 2004, 279, 35583–35594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosi, M.C.; Luccarini, I.; Grossi, C.; Fiorentini, A.; Spillantini, M.G.; Prisco, A.; Scali, C.; Gianfriddo, M.; Caricasole, A.; Terstappen, G.C.; et al. Increased Dickkopf-1 Expression in Transgenic Mouse Models of Neurodegenerative Disease. J. Neurochem. 2010, 112, 1539–1551. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/β-Catenin Signaling and Disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inestrosa, N.C.; Montecinos-Oliva, C.; Fuenzalida, M. Wnt Signaling: Role in Alzheimer Disease and Schizophrenia. J. Neuroimmune Pharm. 2012, 7, 788–807. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Interactions between TGF-Β1, Canonical WNT/β-Catenin Pathway and PPAR γ in Radiation-Induced Fibrosis. Oncotarget 2017, 8, 90579–90604. [Google Scholar] [CrossRef] [Green Version]
- Vallée, A.; Lecarpentier, Y.; Vallée, J.-N. Hypothesis of Opposite Interplay Between the Canonical WNT/Beta-Catenin Pathway and PPAR Gamma in Primary Central Nervous System Lymphomas. Curr. Issues Mol. Biol. 2019, 31, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. β-Catenin Is a Target for the Ubiquitin–Proteasome Pathway. Embo J. 1997, 16, 3797–3804. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Pan, W. GSK3: A Multifaceted Kinase in Wnt Signaling. Trends Biochem. Sci. 2010, 35, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Hur, E.-M.; Zhou, F.-Q. GSK3 Signalling in Neural Development. Nat. Rev. Neurosci. 2010, 11, 539–551. [Google Scholar] [CrossRef] [Green Version]
- Ambacher, K.K.; Pitzul, K.B.; Karajgikar, M.; Hamilton, A.; Ferguson, S.S.; Cregan, S.P. The JNK- and AKT/GSK3β- Signaling Pathways Converge to Regulate Puma Induction and Neuronal Apoptosis Induced by Trophic Factor Deprivation. PLoS ONE 2012, 7, e46885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orellana, A.M.M.; Vasconcelos, A.R.; Leite, J.A.; de Sá Lima, L.; Andreotti, D.Z.; Munhoz, C.D.; Kawamoto, E.M.; Scavone, C. Age-Related Neuroinflammation and Changes in AKT-GSK-3β and WNT/β-CATENIN Signaling in Rat Hippocampus. Aging 2015, 7, 1094–1111. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-H.; Zhang, S.-H.; Gao, F.-J.; Lei, Y.; Chen, X.-Y.; Gao, F.; Zhang, S.-J.; Sun, X.-H. RNAi Screening Identifies GSK3β as a Regulator of DRP1 and the Neuroprotection of Lithium Chloride against Elevated Pressure Involved in Downregulation of DRP1. Neurosci. Lett. 2013, 554, 99–104. [Google Scholar] [CrossRef]
- Russo, R.; Adornetto, A.; Cavaliere, F.; Varano, G.P.; Rusciano, D.; Morrone, L.A.; Corasaniti, M.T.; Bagetta, G.; Nucci, C. Intravitreal Injection of Forskolin, Homotaurine, and L-Carnosine Affords Neuroprotection to Retinal Ganglion Cells Following Retinal Ischemic Injury. Mol. Vis. 2015, 21, 718–729. [Google Scholar] [PubMed]
- Giese, K.P. GSK-3: A Key Player in Neurodegeneration and Memory. IUBMB Life 2009, 61, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Vallée, J.-N.; Lecarpentier, Y. PPARγ Agonists: Potential Treatment for Autism Spectrum Disorder by Inhibiting the Canonical WNT/β-Catenin Pathway. Mol. Psychiatry 2018. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Vallée, J.-N. Targeting the Canonical WNT/β-Catenin Pathway in Cancer Treatment Using Non-Steroidal Anti-Inflammatory Drugs. Cells 2019, 8, 726. [Google Scholar] [CrossRef] [Green Version]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. The Influence of Circadian Rhythms and Aerobic Glycolysis in Autism Spectrum Disorder. Transl. Psychiatry 2020, 10, 400. [Google Scholar] [CrossRef]
- Vallée, A.; Vallée, J.-N.; Guillevin, R.; Lecarpentier, Y. Riluzole: A Therapeutic Strategy in Alzheimer’s Disease by Targeting the WNT/β-Catenin Pathway. Aging 2020, 12, 3095–3113. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Vallée, J.-N. Warburg Effect Hypothesis in Autism Spectrum Disorders. Mol. Brain 2018, 11, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, W.; Millar, J.C.; Wang, W.-H.; Silverman, S.M.; Liu, Y.; Wordinger, R.J.; Rubin, J.S.; Pang, I.-H.; Clark, A.F. Existence of the Canonical Wnt Signaling Pathway in the Human Trabecular Meshwork. Investig. Ophthalmol. Vis. Sci. 2012, 53, 7043–7051. [Google Scholar] [CrossRef]
- Wang, W.-H.; McNatt, L.G.; Pang, I.-H.; Millar, J.C.; Hellberg, P.E.; Hellberg, M.H.; Steely, H.T.; Rubin, J.S.; Fingert, J.H.; Sheffield, V.C.; et al. Increased Expression of the WNT Antagonist SFRP-1 in Glaucoma Elevates Intraocular Pressure. J. Clin. Investig. 2008, 118, 1056–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, J.T.; Raghunathan, V.K.; Chang, Y.-R.; Murphy, C.J.; Russell, P. Wnt Inhibition Induces Persistent Increases in Intrinsic Stiffness of Human Trabecular Meshwork Cells. Exp. Eye Res. 2015, 132, 174–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, H.-S.; Lee, H.-S.; Ji, Y.; Rubin, J.S.; Tomarev, S.I. Myocilin Is a Modulator of Wnt Signaling. Mol. Cell. Biol. 2009, 29, 2139–2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tovar-Vidales, T.; Roque, R.; Clark, A.F.; Wordinger, R.J. Tissue Transglutaminase Expression and Activity in Normal and Glaucomatous Human Trabecular Meshwork Cells and Tissues. Investig. Ophthalmol. Vis. Sci. 2008, 49, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Webber, H.C.; Bermudez, J.Y.; Sethi, A.; Clark, A.F.; Mao, W. Crosstalk between TGFβ and Wnt Signaling Pathways in the Human Trabecular Meshwork. Exp. Eye Res. 2016, 148, 97–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webber, H.C.; Bermudez, J.Y.; Millar, J.C.; Mao, W.; Clark, A.F. The Role of Wnt/β-Catenin Signaling and K-Cadherin in the Regulation of Intraocular Pressure. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1454–1466. [Google Scholar] [CrossRef] [Green Version]
- Manji, H.K.; Moore, G.J.; Rajkowska, G.; Chen, G. Neuroplasticity and Cellular Resilience in Mood Disorders. Mol. Psychiatry 2000, 5, 578–593. [Google Scholar] [CrossRef] [Green Version]
- Phiel, C.J.; Klein, P.S. Molecular Targets of Lithium Action. Annu. Rev. Pharm. Toxicol. 2001, 41, 789–813. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.W.; Chuang, D.M. Long Term Lithium Treatment Suppresses P53 and Bax Expression but Increases Bcl-2 Expression. A Prominent Role in Neuroprotection against Excitotoxicity. J. Biol. Chem. 1999, 274, 6039–6042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donaldson, I.M.; Cuningham, J. Persisting Neurologic Sequelae of Lithium Carbonate Therapy. Arch. Neurol. 1983, 40, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Horton, S.; Tuerk, A.; Cook, D.; Cook, J.; Dhurjati, P. Maximum Recommended Dosage of Lithium for Pregnant Women Based on a PBPK Model for Lithium Absorption. Adv. Bioinform. 2012, 2012, 352729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, M.; Stegmayr, B.; Salander Renberg, E.; Werneke, U. Lithium Intoxication: Incidence, Clinical Course and Renal Function—A Population-Based Retrospective Cohort Study. J. Psychopharmacol. 2016, 30, 1008–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erden, A.; Karagöz, H.; Başak, M.; Karahan, S.; Cetinkaya, A.; Avci, D.; Bugǧday, I. Lithium Intoxication and Nephrogenic Diabetes Insipidus: A Case Report and Review of Literature. Int. J. Gen. Med. 2013, 6, 535–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canan, F.; Kaya, A.; Bulur, S.; Albayrak, E.S.; Ordu, S.; Ataoglu, A. Lithium Intoxication Related Multiple Temporary Ecg Changes: A Case Report. Cases J. 2008, 1, 156. [Google Scholar] [CrossRef] [Green Version]
- Mohandas, E.; Rajmohan, V. Lithium Use in Special Populations. Indian J. Psychiatry 2007, 49, 211–218. [Google Scholar] [CrossRef]
- Kibirige, D.; Luzinda, K.; Ssekitoleko, R. Spectrum of Lithium Induced Thyroid Abnormalities: A Current Perspective. Thyroid Res. 2013, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Abou-Saleh, M.T.; Coppen, A. The Efficacy of Low-Dose Lithium: Clinical, Psychological and Biological Correlates. J. Psychiatr. Res. 1989, 23, 157–162. [Google Scholar] [CrossRef]
- Zhang, H.; Tao, J.; Zhang, S.; Lv, X. LncRNA MEG3 Reduces Hippocampal Neuron Apoptosis via the PI3K/AKT/MTOR Pathway in a Rat Model of Temporal Lobe Epilepsy. Neuropsychiatr. Dis. Treat. 2020, 16, 2519–2528. [Google Scholar] [CrossRef]
- Chen, D.F.; Schneider, G.E.; Martinou, J.C.; Tonegawa, S. Bcl-2 Promotes Regeneration of Severed Axons in Mammalian CNS. Nature 1997, 385, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.-T.; Wang, Z.; Hunsberger, J.G.; Chuang, D.-M. Therapeutic Potential of Mood Stabilizers Lithium and Valproic Acid: Beyond Bipolar Disorder. Pharm. Rev. 2013, 65, 105–142. [Google Scholar] [CrossRef] [Green Version]
- Estaquier, J.; Arnoult, D. Inhibiting Drp1-Mediated Mitochondrial Fission Selectively Prevents the Release of Cytochrome c during Apoptosis. Cell Death Differ. 2007, 14, 1086–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuettauf, F.; Rejdak, R.; Thaler, S.; Bolz, S.; Lehaci, C.; Mankowska, A.; Zarnowski, T.; Junemann, A.; Zagorski, Z.; Zrenner, E.; et al. Citicoline and Lithium Rescue Retinal Ganglion Cells Following Partial Optic Nerve Crush in the Rat. Exp. Eye Res. 2006, 83, 1128–1134. [Google Scholar] [CrossRef]
- Li, X.; Bijur, G.N.; Jope, R.S. Glycogen Synthase Kinase-3beta, Mood Stabilizers, and Neuroprotection. Bipolar Disord. 2002, 4, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Khasawneh, F.T.; Shankar, G.S. Minimizing Cardiovascular Adverse Effects of Atypical Antipsychotic Drugs in Patients with Schizophrenia. Cardiol. Res. Pr. 2014, 2014, 273060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Civantos Calzada, B.; Aleixandre de Artiñano, A. Alpha-Adrenoceptor Subtypes. Pharm. Res. 2001, 44, 195–208. [Google Scholar] [CrossRef]
- Ronaldson, K.J. Cardiovascular Disease in Clozapine-Treated Patients: Evidence, Mechanisms and Management. CNS Drugs 2017, 31, 777–795. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Seki, M.; Sato, Y.; Nagamine, T. Quetiapine-Induced Bradycardia and Hypotension in the Elderly—A Case Report. Innov. Clin. Neurosci. 2016, 13, 34–36. [Google Scholar] [PubMed]
- Wilson, M.P.; Nordstrom, K.; Hopper, A.; Porter, A.; Castillo, E.M.; Vilke, G.M. Risperidone in the Emergency Setting Is Associated with More Hypotension in Elderly Patients. J. Emerg. Med. 2017, 53, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Citrome, L. Iloperidone: Chemistry, Pharmacodynamics, Pharmacokinetics and Metabolism, Clinical Efficacy, Safety and Tolerability, Regulatory Affairs, and an Opinion. Expert Opin. Drug Metab. Toxicol. 2010, 6, 1551–1564. [Google Scholar] [CrossRef]
- Tonin, F.S.; Wiens, A.; Fernandez-Llimos, F.; Pontarolo, R. Iloperidone in the Treatment of Schizophrenia: An Evidence-Based Review of Its Place in Therapy. Core Evid. 2016, 11, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kee Choi, Y.; Tarazi, F.I. Long-Term Effects of Iloperidone on Cerebral Dopamine Receptor Subtypes. Synapse 2018, e22039. [Google Scholar] [CrossRef]
- Langedijk, J.; Mantel-Teeuwisse, A.K.; Slijkerman, D.S.; Schutjens, M.-H.D.B. Drug Repositioning and Repurposing: Terminology and Definitions in Literature. Drug Discov. Today 2015, 20, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.V.; Patel, E.P.; Vyas, B.A.; Lodha, S.R.; Kalyankar, G.G. Repurposing of Iloperidone: Antihypertensive and Ocular Hypotensive Activity in Animals. Eur. J. Pharm. Sci. 2020, 143, 105173. [Google Scholar] [CrossRef]
- Al-Amin, M.M.; Nasir Uddin, M.M.; Mahmud Reza, H. Effects of Antipsychotics on the Inflammatory Response System of Patients with Schizophrenia in Peripheral Blood Mononuclear Cell Cultures. Clin. Psychopharmacol. Neurosci. 2013, 11, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Caruso, G.; Grasso, M.; Fidilio, A.; Tascedda, F.; Drago, F.; Caraci, F. Antioxidant Properties of Second-Generation Antipsychotics: Focus on Microglia. Pharmaceuticals 2020, 13, 457. [Google Scholar] [CrossRef] [PubMed]
- Noto, C.; Ota, V.K.; Gouvea, E.S.; Rizzo, L.B.; Spindola, L.M.N.; Honda, P.H.S.; Cordeiro, Q.; Belangero, S.I.; Bressan, R.A.; Gadelha, A.; et al. Effects of Risperidone on Cytokine Profile in Drug-Naïve First-Episode Psychosis. Int. J. Neuropsychopharmacol. 2014, 18. [Google Scholar] [CrossRef] [PubMed]
- Tendilla-Beltrán, H.; Meneses-Prado, S.; Vázquez-Roque, R.A.; Tapia-Rodríguez, M.; Vázquez-Hernández, A.J.; Coatl-Cuaya, H.; Martín-Hernández, D.; MacDowell, K.S.; Garcés-Ramírez, L.; Leza, J.C.; et al. Risperidone Ameliorates Prefrontal Cortex Neural Atrophy and Oxidative/Nitrosative Stress in Brain and Peripheral Blood of Rats with Neonatal Ventral Hippocampus Lesion. J. Neurosci. 2019, 39, 8584–8599. [Google Scholar] [CrossRef]
- Casquero-Veiga, M.; García-García, D.; MacDowell, K.S.; Pérez-Caballero, L.; Torres-Sánchez, S.; Fraguas, D.; Berrocoso, E.; Leza, J.C.; Arango, C.; Desco, M.; et al. Risperidone Administered during Adolescence Induced Metabolic, Anatomical and Inflammatory/Oxidative Changes in Adult Brain: A PET and MRI Study in the Maternal Immune Stimulation Animal Model. Eur. Neuropsychopharmacol. 2019, 29, 880–896. [Google Scholar] [CrossRef]
- Aringhieri, S.; Carli, M.; Kolachalam, S.; Verdesca, V.; Cini, E.; Rossi, M.; McCormick, P.J.; Corsini, G.U.; Maggio, R.; Scarselli, M. Molecular Targets of Atypical Antipsychotics: From Mechanism of Action to Clinical Differences. Pharmacol. Ther. 2018, 192, 20–41. [Google Scholar] [CrossRef] [PubMed]
- Ribaudo, G.; Bortoli, M.; Pavan, C.; Zagotto, G.; Orian, L. Antioxidant Potential of Psychotropic Drugs: From Clinical Evidence to In Vitro and In Vivo Assessment and toward a New Challenge for in Silico Molecular Design. Antioxidants 2020, 9, 714. [Google Scholar] [CrossRef] [PubMed]
- Carli, M.; Aringhieri, S.; Kolachalam, S.; Longoni, B.; Grenno, G.; Rossi, M.; Gemignani, A.; Fornai, F.; Maggio, R.; Scarselli, M. Is Adult Hippocampal Neurogenesis Really Relevant for the Treatment of Psychiatric Disorders? Curr. Neuropharmacol. 2020. [Google Scholar] [CrossRef]
- Chitranshi, N.; Dheer, Y.; Abbasi, M.; You, Y.; Graham, S.L.; Gupta, V. Glaucoma Pathogenesis and Neurotrophins: Focus on the Molecular and Genetic Basis for Therapeutic Prospects. Curr. Neuropharmacol. 2018, 16, 1018–1035. [Google Scholar] [CrossRef]
- Quigley, H.A.; McKinnon, S.J.; Zack, D.J.; Pease, M.E.; Kerrigan-Baumrind, L.A.; Kerrigan, D.F.; Mitchell, R.S. Retrograde Axonal Transport of BDNF in Retinal Ganglion Cells Is Blocked by Acute IOP Elevation in Rats. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3460–3466. [Google Scholar]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in Neuronal Development and Function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.-H.; Dwyer, D.S. Second-Generation Antipsychotic Drugs, Olanzapine, Quetiapine, and Clozapine Enhance Neurite Outgrowth in PC12 Cells via PI3K/AKT, ERK, and Pertussis Toxin-Sensitive Pathways. J. Mol. Neurosci. 2005, 27, 43–64. [Google Scholar] [CrossRef]
- Aringhieri, S.; Kolachalam, S.; Gerace, C.; Carli, M.; Verdesca, V.; Brunacci, M.G.; Rossi, C.; Ippolito, C.; Solini, A.; Corsini, G.U.; et al. Clozapine as the Most Efficacious Antipsychotic for Activating ERK 1/2 Kinases: Role of 5-HT2A Receptor Agonism. Eur. Neuropsychopharmacol. 2017, 27, 383–398. [Google Scholar] [CrossRef]
- Morais, M.; Patrício, P.; Mateus-Pinheiro, A.; Alves, N.D.; Machado-Santos, A.R.; Correia, J.S.; Pereira, J.; Pinto, L.; Sousa, N.; Bessa, J.M. The Modulation of Adult Neuroplasticity Is Involved in the Mood-Improving Actions of Atypical Antipsychotics in an Animal Model of Depression. Transl. Psychiatry 2017, 7, e1146. [Google Scholar] [CrossRef] [Green Version]
- Chikama, K.; Yamada, H.; Tsukamoto, T.; Kajitani, K.; Nakabeppu, Y.; Uchimura, N. Chronic Atypical Antipsychotics, but Not Haloperidol, Increase Neurogenesis in the Hippocampus of Adult Mouse. Brain Res. 2017, 1676, 77–82. [Google Scholar] [CrossRef]
- Richa, S.; Yazbek, J.-C. Ocular Adverse Effects of Common Psychotropic Agents: A Review. CNS Drugs 2010, 24, 501–526. [Google Scholar] [CrossRef]
- Tripathi, R.C.; Tripathi, B.J.; Haggerty, C. Drug-Induced Glaucomas: Mechanism and Management. Drug Saf. 2003, 26, 749–767. [Google Scholar] [CrossRef] [PubMed]
- Faiq, M.A.; Wollstein, G.; Schuman, J.S.; Chan, K.C. Cholinergic Nervous System and Glaucoma: From Basic Science to Clinical Applications. Prog. Retin. Eye Res. 2019, 72, 100767. [Google Scholar] [CrossRef]
- Almasieh, M.; Zhou, Y.; Kelly, M.E.; Casanova, C.; Di Polo, A. Structural and Functional Neuroprotection in Glaucoma: Role of Galantamine-Mediated Activation of Muscarinic Acetylcholine Receptors. Cell Death Dis. 2010, 1, e27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chew, M.L.; Mulsant, B.H.; Pollock, B.G.; Lehman, M.E.; Greenspan, A.; Kirshner, M.A.; Bies, R.R.; Kapur, S.; Gharabawi, G. A Model of Anticholinergic Activity of Atypical Antipsychotic Medications. Schizophr. Res. 2006, 88, 63–72. [Google Scholar] [CrossRef]
- Stroup, T.S.; Gray, N. Management of Common Adverse Effects of Antipsychotic Medications. World Psychiatry 2018, 17, 341–356. [Google Scholar] [CrossRef]
- Libro, R.; Bramanti, P.; Mazzon, E. The Role of the Wnt Canonical Signaling in Neurodegenerative Diseases. Life Sci. 2016, 158, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Alda, M.; Priller, J.; Young, L.T. International Group For The Study Of Lithium Treated Patients (IGSLI) Implications of the Neuroprotective Effects of Lithium for the Treatment of Bipolar and Neurodegenerative Disorders. Pharmacopsychiatry 2003, 36 (Suppl. 3), S250–S254. [Google Scholar] [CrossRef]
- Rowe, M.K.; Chuang, D.-M. Lithium Neuroprotection: Molecular Mechanisms and Clinical Implications. Expert Rev. Mol. Med. 2004, 6, 1–18. [Google Scholar] [CrossRef]
- Rowe, M.K.; Wiest, C.; Chuang, D.-M. GSK-3 Is a Viable Potential Target for Therapeutic Intervention in Bipolar Disorder. Neurosci. Biobehav. Rev. 2007, 31, 920–931. [Google Scholar] [CrossRef] [Green Version]
- Alural, B.; Ozerdem, A.; Allmer, J.; Genc, K.; Genc, S. Lithium Protects against Paraquat Neurotoxicity by NRF2 Activation and MiR-34a Inhibition in SH-SY5Y Cells. Front. Cell. Neurosci. 2015, 9, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaulieu, J.-M.; Gainetdinov, R.R.; Caron, M.G. The Akt-GSK-3 Signaling Cascade in the Actions of Dopamine. Trends Pharmacol. Sci. 2007, 28, 166–172. [Google Scholar] [CrossRef]
- Gould, T.D.; Chen, G.; Manji, H.K. In Vivo Evidence in the Brain for Lithium Inhibition of Glycogen Synthase Kinase-3. Neuropsychopharmacology 2004, 29, 32–38. [Google Scholar] [CrossRef]
- O’Brien, W.T.; Harper, A.D.; Jové, F.; Woodgett, J.R.; Maretto, S.; Piccolo, S.; Klein, P.S. Glycogen Synthase Kinase-3beta Haploinsufficiency Mimics the Behavioral and Molecular Effects of Lithium. J. Neurosci. 2004, 24, 6791–6798. [Google Scholar] [CrossRef] [Green Version]
- Jope, R.S.; Johnson, G.V.W. The Glamour and Gloom of Glycogen Synthase Kinase-3. Trends Biochem. Sci. 2004, 29, 95–102. [Google Scholar] [CrossRef]
- Gould, T.D.; Einat, H.; O’Donnell, K.C.; Picchini, A.M.; Schloesser, R.J.; Manji, H.K. Beta-Catenin Overexpression in the Mouse Brain Phenocopies Lithium-Sensitive Behaviors. Neuropsychopharmacology 2007, 32, 2173–2183. [Google Scholar] [CrossRef] [Green Version]
- Gould, T.D.; O’Donnell, K.C.; Picchini, A.M.; Dow, E.R.; Chen, G.; Manji, H.K. Generation and Behavioral Characterization of Beta-Catenin Forebrain-Specific Conditional Knock-out Mice. Behav. Brain Res. 2008, 189, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Gould, T.D.; Quiroz, J.A.; Singh, J.; Zarate, C.A.; Manji, H.K. Emerging Experimental Therapeutics for Bipolar Disorder: Insights from the Molecular and Cellular Actions of Current Mood Stabilizers. Mol. Psychiatry 2004, 9, 734–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barthel, A.; Schmoll, D.; Unterman, T.G. FoxO Proteins in Insulin Action and Metabolism. Trends Endocrinol. Metab. 2005, 16, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Ambrogini, E.; Han, L.; Manolagas, S.C.; Jilka, R.L. Increased Lipid Oxidation Causes Oxidative Stress, Increased Peroxisome Proliferator-Activated Receptor-Gamma Expression, and Diminished pro-Osteogenic Wnt Signaling in the Skeleton. J. Biol. Chem. 2009, 284, 27438–27448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essers, M.A.G.; de Vries-Smits, L.M.M.; Barker, N.; Polderman, P.E.; Burgering, B.M.T.; Korswagen, H.C. Functional Interaction between Beta-Catenin and FOXO in Oxidative Stress Signaling. Science 2005, 308, 1181–1184. [Google Scholar] [CrossRef] [PubMed]
- Hoogeboom, D.; Essers, M.A.G.; Polderman, P.E.; Voets, E.; Smits, L.M.M.; Burgering, B.M.T. Interaction of FOXO with Beta-Catenin Inhibits Beta-Catenin/T Cell Factor Activity. J. Biol. Chem. 2008, 283, 9224–9230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Z.; Liu, L.; Zhang, R.; Li, X. Lithium Reduces FoxO3a Transcriptional Activity by Decreasing Its Intracellular Content. Biol. Psychiatry 2007, 62, 1423–1430. [Google Scholar] [CrossRef]
- Shao, L.; Young, L.T.; Wang, J.-F. Chronic Treatment with Mood Stabilizers Lithium and Valproate Prevents Excitotoxicity by Inhibiting Oxidative Stress in Rat Cerebral Cortical Cells. Biol. Psychiatry 2005, 58, 879–884. [Google Scholar] [CrossRef]
- De Vasconcellos, A.P.S.; Nieto, F.B.; Crema, L.M.; Diehl, L.A.; de Almeida, L.M.; Prediger, M.E.; da Rocha, E.R.; Dalmaz, C. Chronic Lithium Treatment Has Antioxidant Properties but Does Not Prevent Oxidative Damage Induced by Chronic Variate Stress. Neurochem. Res. 2006, 31, 1141–1151. [Google Scholar] [CrossRef]
- Cui, J.; Shao, L.; Young, L.T.; Wang, J.-F. Role of Glutathione in Neuroprotective Effects of Mood Stabilizing Drugs Lithium and Valproate. Neuroscience 2007, 144, 1447–1453. [Google Scholar] [CrossRef]
- Frey, B.N.; Andreazza, A.C.; Kunz, M.; Gomes, F.A.; Quevedo, J.; Salvador, M.; Gonçalves, C.A.; Kapczinski, F. Increased Oxidative Stress and DNA Damage in Bipolar Disorder: A Twin-Case Report. Prog. Neuropsychopharmacol. Biol. Psychiatry 2007, 31, 283–285. [Google Scholar] [CrossRef]
- Machado-Vieira, R.; Andreazza, A.C.; Viale, C.I.; Zanatto, V.; Cereser, V.; da Silva Vargas, R.; Kapczinski, F.; Portela, L.V.; Souza, D.O.; Salvador, M.; et al. Oxidative Stress Parameters in Unmedicated and Treated Bipolar Subjects during Initial Manic Episode: A Possible Role for Lithium Antioxidant Effects. Neurosci. Lett. 2007, 421, 33–36. [Google Scholar] [CrossRef]
- Kim, Y.-H.; Rane, A.; Lussier, S.; Andersen, J.K. Lithium Protects against Oxidative Stress-Mediated Cell Death in α-Synuclein-Overexpressing in Vitro and in Vivo Models of Parkinson’s Disease. J. Neurosci. Res. 2011, 89, 1666–1675. [Google Scholar] [CrossRef] [Green Version]
- Halleskog, C.; Mulder, J.; Dahlström, J.; Mackie, K.; Hortobágyi, T.; Tanila, H.; Kumar Puli, L.; Färber, K.; Harkany, T.; Schulte, G. WNT Signaling in Activated Microglia Is Proinflammatory. Glia 2011, 59, 119–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- L’episcopo, F.; Serapide, M.F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Pluchino, S.; Marchetti, B. A Wnt1 Regulated Frizzled-1/β-Catenin Signaling Pathway as a Candidate Regulatory Circuit Controlling Mesencephalic Dopaminergic Neuron-Astrocyte Crosstalk: Therapeutical Relevance for Neuron Survival and Neuroprotection. Mol. Neurodegener. 2011, 6, 49. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Hottiger, M.O. Crosstalk between Wnt/β-Catenin and NF-ΚB Signaling Pathway during Inflammation. Front. Immunol. 2016, 7, 378. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB System. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Miller, S.A.; Wang, H.-Y.; Xia, W.; Wen, Y.; Zhou, B.P.; Li, Y.; Lin, S.-Y.; Hung, M.-C. Beta-Catenin Interacts with and Inhibits NF-Kappa B in Human Colon and Breast Cancer. Cancer Cell 2002, 2, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Liao, Y.; Ma, K.; Wang, Y.; Zhang, G.; Yang, R.; Deng, J. PI3K Is Required for the Physical Interaction and Functional Inhibition of NF-ΚB by β-Catenin in Colorectal Cancer Cells. Biochem. Biophys. Res. Commun. 2013, 434, 760–766. [Google Scholar] [CrossRef]
- Martin, M.; Rehani, K.; Jope, R.S.; Michalek, S.M. Toll-like Receptor-Mediated Cytokine Production Is Differentially Regulated by Glycogen Synthase Kinase 3. Nat. Immunol. 2005, 6, 777–784. [Google Scholar] [CrossRef]
- Manicassamy, S.; Reizis, B.; Ravindran, R.; Nakaya, H.; Salazar-Gonzalez, R.M.; Wang, Y.-C.; Pulendran, B. Activation of Beta-Catenin in Dendritic Cells Regulates Immunity versus Tolerance in the Intestine. Science 2010, 329, 849–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.H.; Song, J.S.; Yu, J.M.; Yu, S.S.; Choi, S.J.; Kim, D.H.; Jung, J.S. Differential Effect of NF-KappaB Activity on Beta-Catenin/Tcf Pathway in Various Cancer Cells. FEBS Lett. 2008, 582, 616–622. [Google Scholar] [CrossRef] [Green Version]
- Fliniaux, I.; Mikkola, M.L.; Lefebvre, S.; Thesleff, I. Identification of Dkk4 as a Target of Eda-A1/Edar Pathway Reveals an Unexpected Role of Ectodysplasin as Inhibitor of Wnt Signalling in Ectodermal Placodes. Dev. Biol. 2008, 320, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez de la Concepción, M.L.; Yubero, P.; Iglesias, R.; Giralt, M.; Villarroya, F. Lithium Inhibits Brown Adipocyte Differentiation. FEBS Lett. 2005, 579, 1670–1674. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-H.; Olson, P.; Evans, R.M. Minireview: Lipid Metabolism, Metabolic Diseases, and Peroxisome Proliferator-Activated Receptors. Endocrinology 2003, 144, 2201–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marx, N.; Duez, H.; Fruchart, J.-C.; Staels, B. Peroxisome Proliferator-Activated Receptors and Atherogenesis: Regulators of Gene Expression in Vascular Cells. Circ. Res. 2004, 94, 1168–1178. [Google Scholar] [CrossRef]
- Cunard, R.; Ricote, M.; DiCampli, D.; Archer, D.C.; Kahn, D.A.; Glass, C.K.; Kelly, C.J. Regulation of Cytokine Expression by Ligands of Peroxisome Proliferator Activated Receptors. J. Immunol. 2002, 168, 2795–2802. [Google Scholar] [CrossRef] [Green Version]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The Peroxisome Proliferator-Activated Receptor-Gamma Is a Negative Regulator of Macrophage Activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef]
- Giannini, S.; Serio, M.; Galli, A. Pleiotropic Effects of Thiazolidinediones: Taking a Look beyond Antidiabetic Activity. J. Endocrinol. Investig. 2004, 27, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y. Crosstalk Between Peroxisome Proliferator-Activated Receptor Gamma and the Canonical WNT/β-Catenin Pathway in Chronic Inflammation and Oxidative Stress During Carcinogenesis. Front. Immunol. 2018, 9, 745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Thermodynamics in Gliomas: Interactions between the Canonical WNT/Beta-Catenin Pathway and PPAR Gamma. Front. Physiol. 2017, 8, 352. [Google Scholar] [CrossRef] [Green Version]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Demyelination in Multiple Sclerosis: Reprogramming Energy Metabolism and Potential PPARγ Agonist Treatment Approaches. Int. J. Mol. Sci. 2018, 19, 1212. [Google Scholar] [CrossRef] [Green Version]
- Park, K.S.; Lee, R.D.; Kang, S.-K.; Han, S.Y.; Park, K.L.; Yang, K.H.; Song, Y.S.; Park, H.J.; Lee, Y.M.; Yun, Y.P.; et al. Neuronal Differentiation of Embryonic Midbrain Cells by Upregulation of Peroxisome Proliferator-Activated Receptor-Gamma via the JNK-Dependent Pathway. Exp. Cell Res. 2004, 297, 424–433. [Google Scholar] [CrossRef]
- Grimes, C.A.; Jope, R.S. The Multifaceted Roles of Glycogen Synthase Kinase 3beta in Cellular Signaling. Prog. Neurobiol. 2001, 65, 391–426. [Google Scholar] [CrossRef]
- Jeon, M.; Rahman, N.; Kim, Y.-S. Wnt/β-Catenin Signaling Plays a Distinct Role in Methyl Gallate-Mediated Inhibition of Adipogenesis. Biochem. Biophys. Res. Commun. 2016, 479, 22–27. [Google Scholar] [CrossRef]
- Gustafson, B.; Eliasson, B.; Smith, U. Thiazolidinediones Increase the Wingless-Type MMTV Integration Site Family (WNT) Inhibitor Dickkopf-1 in Adipocytes: A Link with Osteogenesis. Diabetologia 2010, 53, 536–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, A.; Haldar, S. The Relationship between BcI2, Bax and P53: Consequences for Cell Cycle Progression and Cell Death. Mol. Hum. Reprod. 1998, 4, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Rajkowska, G.; Du, F.; Seraji-Bozorgzad, N.; Manji, H.K. Enhancement of Hippocampal Neurogenesis by Lithium. J. Neurochem. 2000, 75, 1729–1734. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.H.; Arraf, Z. Prevention of MPTP (N-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine) Dopaminergic Neurotoxicity in Mice by Chronic Lithium: Involvements of Bcl-2 and Bax. Neuropharmacology 2004, 46, 1130–1140. [Google Scholar] [CrossRef]
- Ruvolo, P.P.; Deng, X.; May, W.S. Phosphorylation of Bcl2 and Regulation of Apoptosis. Leukemia 2001, 15, 515–522. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-L.; Lin, C.-F.; Chiang, C.-W.; Jan, M.-S.; Lin, Y.-S. Lithium Inhibits Ceramide- and Etoposide-Induced Protein Phosphatase 2A Methylation, Bcl-2 Dephosphorylation, Caspase-2 Activation, and Apoptosis. Mol. Pharmacol. 2006, 70, 510–517. [Google Scholar] [CrossRef]
- Chen, R.-W.; Qin, Z.-H.; Ren, M.; Kanai, H.; Chalecka-Franaszek, E.; Leeds, P.; Chuang, D.-M. Regulation of C-Jun N-Terminal Kinase, P38 Kinase and AP-1 DNA Binding in Cultured Brain Neurons: Roles in Glutamate Excitotoxicity and Lithium Neuroprotection. J. Neurochem. 2003, 84, 566–575. [Google Scholar] [CrossRef]
- Abousaab, A.; Lang, F. Up-Regulation of Excitatory Amino Acid Transporters EAAT3 and EAAT4 by Lithium Sensitive Glycogen Synthase Kinase GSK3ß. Cell. Physiol. Biochem. 2016, 40, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.F.; Hokin, L.E. Lithium Acutely Inhibits and Chronically Up-Regulates and Stabilizes Glutamate Uptake by Presynaptic Nerve Endings in Mouse Cerebral Cortex. Proc. Natl Acad Sci USA 1998, 95, 8363–8368. [Google Scholar] [CrossRef] [Green Version]
- Mayer, M.L.; Miller, R.J. Excitatory Amino Acid Receptors, Second Messengers and Regulation of Intracellular Ca2+ in Mammalian Neurons. Trends Pharm. Sci. 1990, 11, 254–260. [Google Scholar] [CrossRef]
- Yoshii, A.; Constantine-Paton, M. Postsynaptic BDNF-TrkB Signaling in Synapse Maturation, Plasticity, and Disease. Dev. Neurobiol. 2010, 70, 304–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheuing, L.; Chiu, C.-T.; Liao, H.-M.; Chuang, D.-M. Antidepressant Mechanism of Ketamine: Perspective from Preclinical Studies. Front. Neurosci. 2015, 9, 249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, C.-T.; Scheuing, L.; Liu, G.; Liao, H.-M.; Linares, G.R.; Lin, D.; Chuang, D.-M. The Mood Stabilizer Lithium Potentiates the Antidepressant-like Effects and Ameliorates Oxidative Stress Induced by Acute Ketamine in a Mouse Model of Stress. Int. J. Neuropsychopharmacol. 2014, 18. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.-T.; Chuang, D.-M. Molecular Actions and Therapeutic Potential of Lithium in Preclinical and Clinical Studies of CNS Disorders. Pharmacol. Ther. 2010, 128, 281–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Y.; Xie, K.; Dong, H.; Zhang, H.; Wang, F.; Li, Y.; Xiong, L. Hyperbaric Oxygen Preconditioning Protects Cortical Neurons against Oxygen-Glucose Deprivation Injury: Role of Peroxisome Proliferator-Activated Receptor-Gamma. Brain Res. 2012, 1452, 140–150. [Google Scholar] [CrossRef]
- Heneka, M.T.; Landreth, G.E. PPARs in the Brain. Biochim. Biophys. Acta 2007, 1771, 1031–1045. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallée, A.; Vallée, J.-N.; Lecarpentier, Y. Lithium and Atypical Antipsychotics: The Possible WNT/β Pathway Target in Glaucoma. Biomedicines 2021, 9, 473. https://doi.org/10.3390/biomedicines9050473
Vallée A, Vallée J-N, Lecarpentier Y. Lithium and Atypical Antipsychotics: The Possible WNT/β Pathway Target in Glaucoma. Biomedicines. 2021; 9(5):473. https://doi.org/10.3390/biomedicines9050473
Chicago/Turabian StyleVallée, Alexandre, Jean-Noël Vallée, and Yves Lecarpentier. 2021. "Lithium and Atypical Antipsychotics: The Possible WNT/β Pathway Target in Glaucoma" Biomedicines 9, no. 5: 473. https://doi.org/10.3390/biomedicines9050473
APA StyleVallée, A., Vallée, J. -N., & Lecarpentier, Y. (2021). Lithium and Atypical Antipsychotics: The Possible WNT/β Pathway Target in Glaucoma. Biomedicines, 9(5), 473. https://doi.org/10.3390/biomedicines9050473