
Altered Calcium Influx Pathways in Cancer-Associated Fibroblasts

 , , , and
, , , and 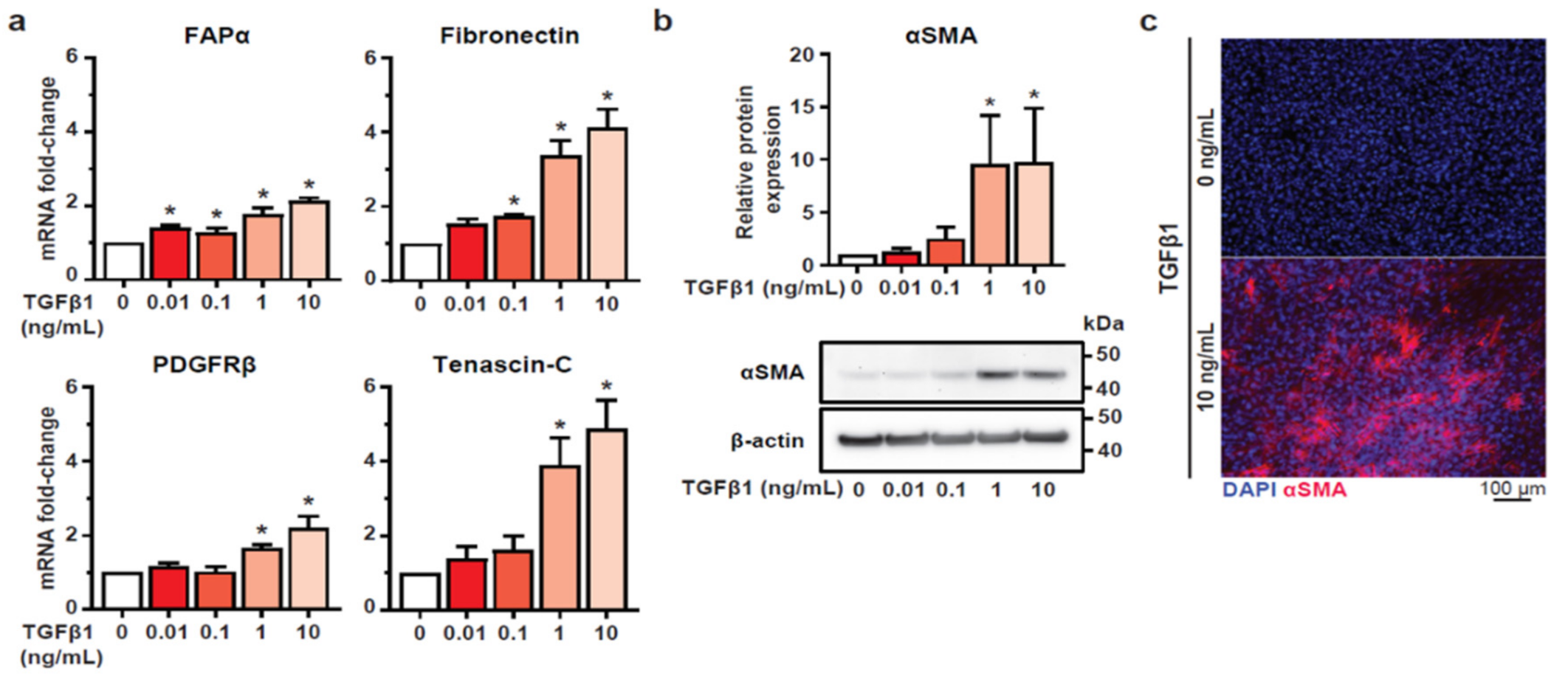{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. siRNA Transfection
2.3. Real-Time Quantitative RT-PCR
2.4. Immunostaining
2.5. Immunoblotting
2.6. Measurement of Intracellular Free Calcium
2.7. Statistical Analysis
3. Results
3.1. TGFβ1 Stimulation Induces a CAF-Like Phenotype in HMF3S Cells
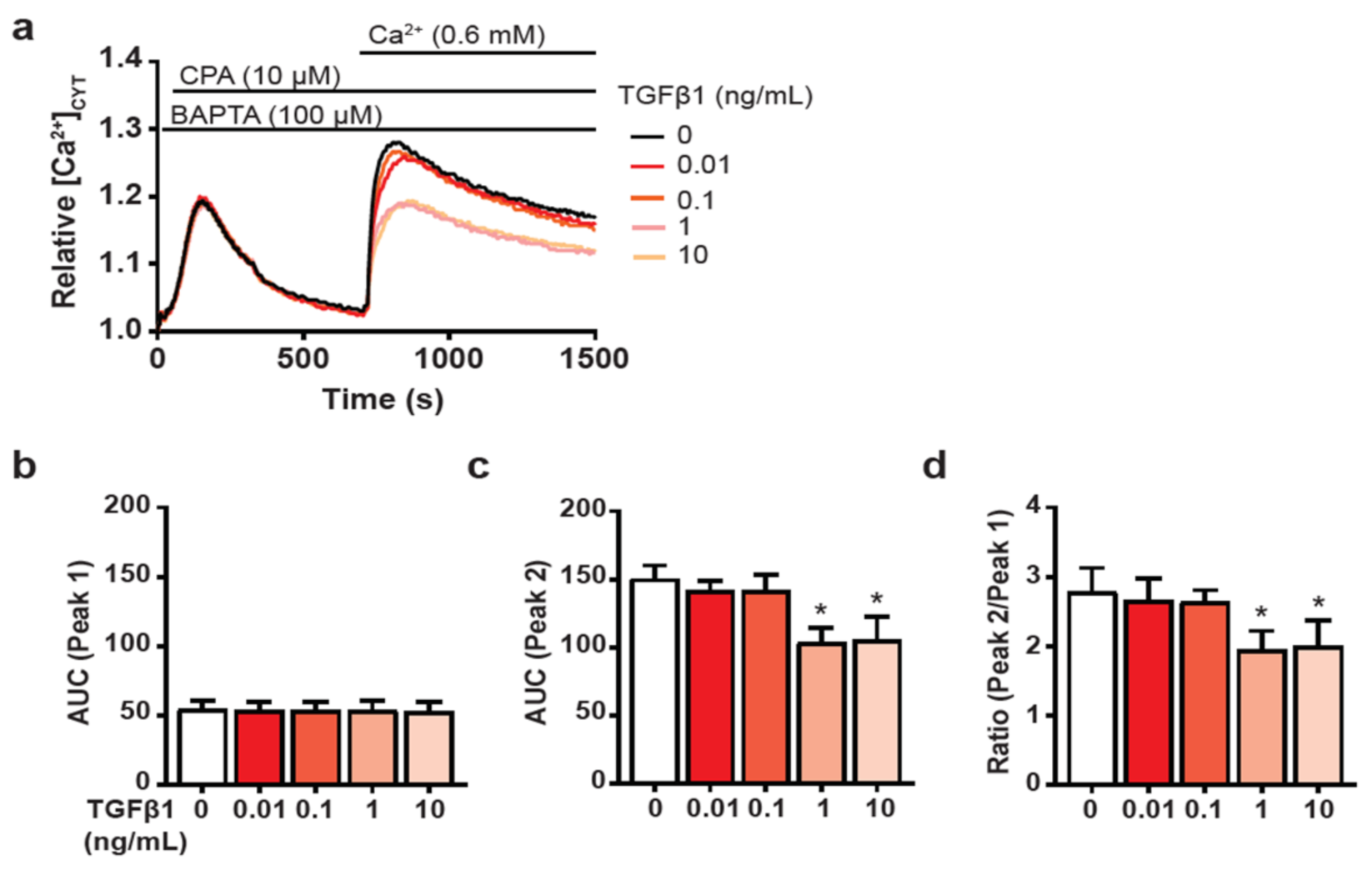
3.2. TGFβ1-Mediated CAF Induction Impairs SOCE in HMF3S Cells
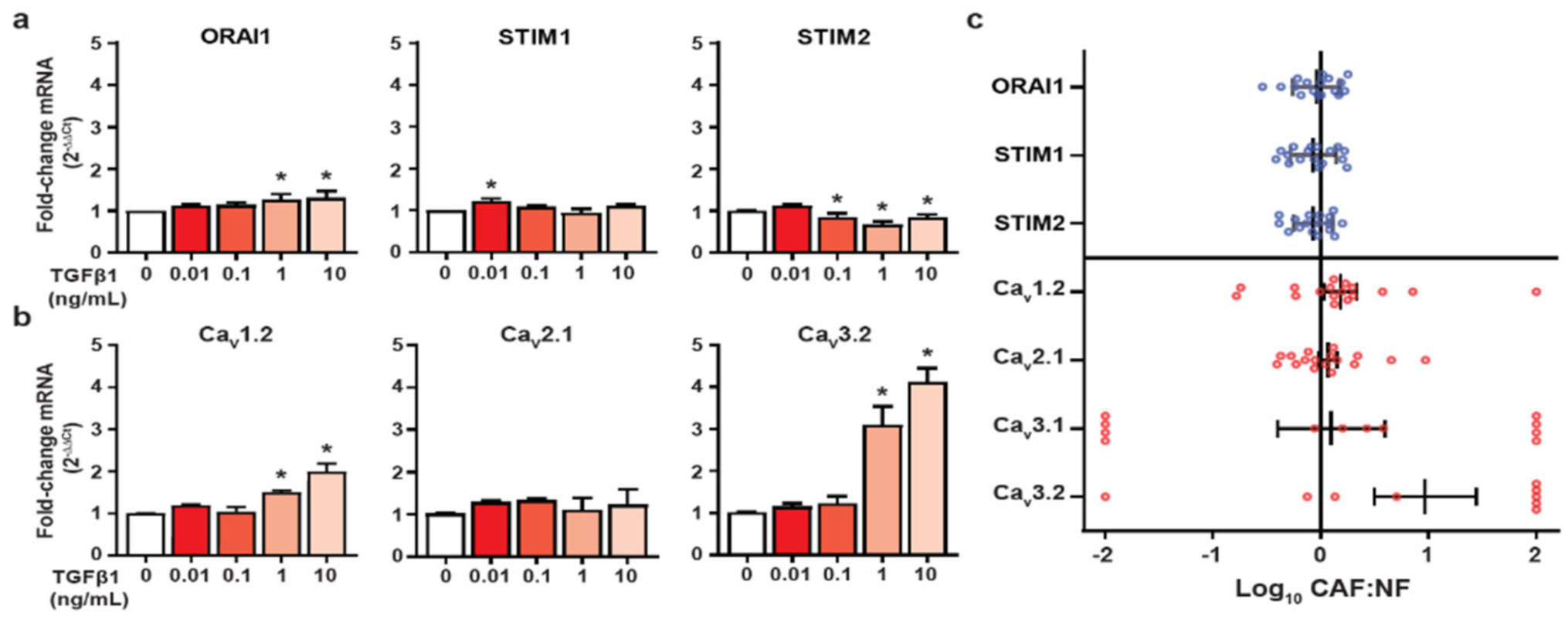
3.3. HMF3S Activation Remodels Calcium Channel and Channel Regulator Expression
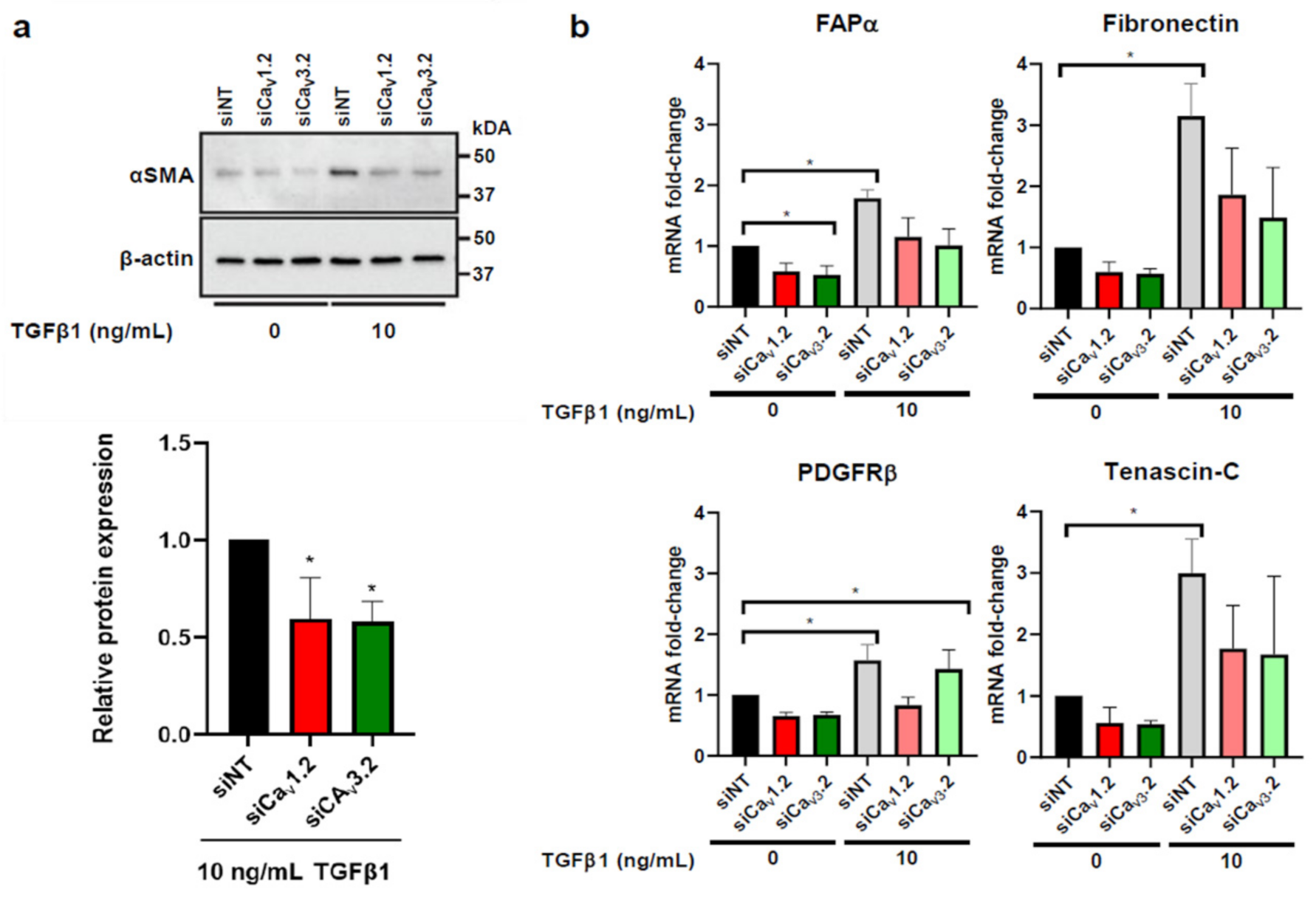
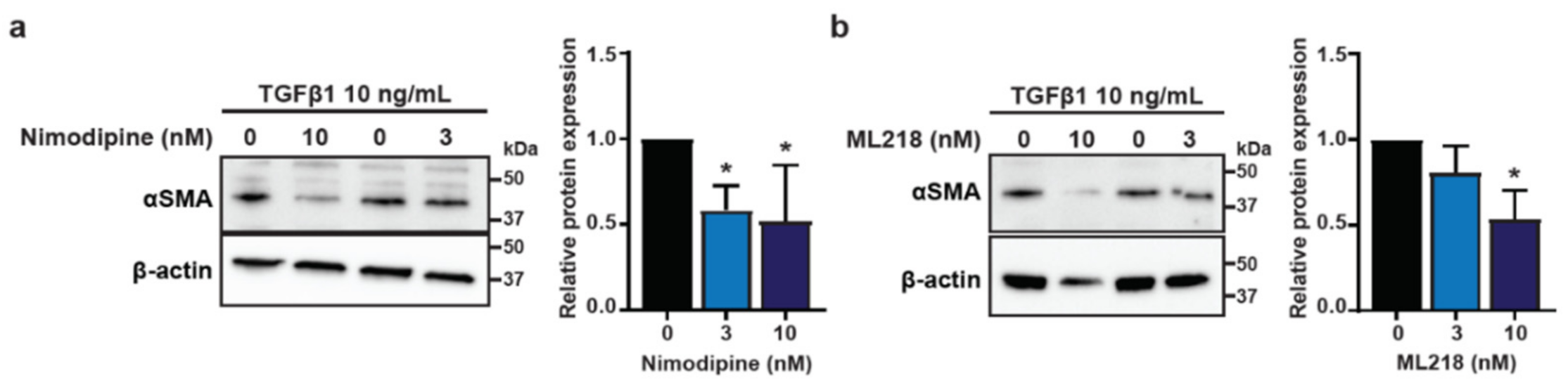
3.4. Voltage-Gated Calcium Channels Are Regulators of CAF Activation in HMF3S Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [Green Version]
- Buchsbaum, R.J.; Oh, S.Y. Breast cancer-associated fibroblasts: Where we are and where we need to go. Cancers 2016, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Melling, G.E.; Flannery, S.E.; Abidin, S.A.; Clemmens, H.; Prajapati, P.; Hinsley, E.E.; Hunt, S.; Catto, J.W.F.; Della Coletta, R.; Mellone, M.; et al. A miRNA-145/TGF-beta1 negative feedback loop regulates the cancer-associated fibroblast phenotype. Carcinogenesis 2018, 39, 798–807. [Google Scholar] [CrossRef]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Hsu, S.C.; Xia, W.; Cao, X.; Shih, J.Y.; Wei, Y.; Abbruzzese, J.L.; Hortobagyi, G.N.; Hung, M.C. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007, 67, 9066–9076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.-H.; Wu, M.-Z.; Chiou, S.-H.; Chen, P.-M.; Chang, S.-Y.; Liu, C.-J.; Teng, S.-C.; Wu, K.-J. Direct regulation of TWIST by HIF-1α promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, X.; Feng, B.; Liu, N.; Wu, Q.; Han, Y.; Nie, Y.; Wu, K.; Shi, Y.; Fan, D. STIM1, a direct target of microRNA-185, promotes tumor metastasis and is associated with poor prognosis in colorectal cancer. Oncogene 2015, 34, 4808–4820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, F.; Azimi, I.; Faville, R.A.; Peters, A.A.; Jalink, K.; Putney, J.W.; Goodhill, G.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G. Induction of epithelial–mesenchymal transition (EMT) in breast cancer cells is calcium signal dependent. Oncogene 2014, 33, 2307–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteith, G.R.; Prevarskaya, N.; Roberts-Thomson, S.J. The calcium–cancer signalling nexus. Nat. Rev. Cancer 2017, 17, 373–380. [Google Scholar] [CrossRef] [Green Version]
- Dang, D.K.; Makena, M.R.; Llongueras, J.P.; Prasad, H.; Ko, M.; Bandral, M.; Rao, R. A Ca2+-ATPase Regulates E-cadherin Biogenesis and Epithelial–Mesenchymal Transition in Breast Cancer Cells. Mol. Cancer Res. 2019, 17, 1735–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Zhang, J.J.; Huang, X.-Y. Orai1 and STIM1 Are Critical for Breast Tumor Cell Migration and Metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, A.; Saha, S. T-Type voltage gated calcium channels: A target in breast cancer? Breast Cancer Res. Treat. 2019, 173, 11–21. [Google Scholar] [CrossRef]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 Clusters and Activates CRAC Channels via Direct Binding of a Cytosolic Domain to Orai1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luik, R.M.; Wang, B.; Prakriya, M.; Wu, M.M.; Lewis, R.S. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nat. Cell Biol. 2008, 454, 538–542. [Google Scholar] [CrossRef]
- Catterall, W.A. Voltage-Gated Calcium Channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef]
- Deliot, N.; Constantin, B. Plasma membrane calcium channels in cancer: Alterations and consequences for cell proliferation and migration. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2512–2522. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.T.; Huang, L.; Pottle, J.E.; Liu, K.; Yang, Y.; Zeng, X.; Keyser, B.M.; Agrawal, K.C.; Hansen, J.B.; Li, M. Selective blockade of T-type Ca2+ channels suppresses human breast cancer cell proliferation. Cancer Lett. 2008, 267, 116–124. [Google Scholar] [CrossRef]
- Bisaillon, J.M.; Motiani, R.K.; González-Cobos, J.C.; Potier, M.; Halligan, K.E.; Alzawahra, W.F.; Barroso, M.; Singer, H.A.; Jourd’Heuil, D.; Trebak, M. Essential role for STIM1/Orai1-mediated calcium influx in PDGF-induced smooth muscle migration. Am. J. Physiol. Physiol. 2010, 298, C993–C1005. [Google Scholar] [CrossRef] [Green Version]
- Kumar, B.; Dreja, K.; Shah, S.S.; Cheong, A.; Xu, S.-Z.; Sukumar, P.; Naylor, J.; Forte, A.; Cipollaro, M.; McHugh, D.; et al. Upregulated TRPC1 Channel in Vascular Injury In Vivo and Its Role in Human Neointimal Hyperplasia. Circ. Res. 2006, 98, 557–563. [Google Scholar] [CrossRef] [Green Version]
- Quignard, J.-F.; Harricane, M.-C.; Ménard, C.; Lory, P.; Nargeot, J.; Capron, L.; Mornet, M.; Richard, S. Transient down-regulation of L-type Ca2+ channel and dystrophin expression after balloon injury in rat aortic cells. Cardiovasc. Res. 2001, 49, 177–188. [Google Scholar] [CrossRef] [Green Version]
- O’Hare, M.J.; Bond, J.; Clarke, C.; Takeuchi, Y.; Atherton, A.J.; Berry, C.; Moody, J.; Silver, A.R.J.; Davies, D.; Alsop, A.E.; et al. Conditional immortalization of freshly isolated human mammary fibroblasts and endothelial cells. Proc. Natl. Acad. Sci. USA 2001, 98, 646–651. [Google Scholar] [CrossRef] [Green Version]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Soon, P.S.H.; Kim, E.; Pon, C.K.; Gill, A.J.; Moore, K.; Spillane, A.J.; E Benn, D.; Baxter, R.C. Breast cancer-associated fibroblasts induce epithelial-to-mesenchymal transition in breast cancer cells. Endocr. Relat. Cancer 2012, 20, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Machaca, K. Ca2+ signaling, genes and the cell cycle. Cell Calcium 2011, 49, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Casey, T.M.; Eneman, J.; Crocker, A.; White, J.; Tessitore, J.; Stanley, M.; Harlow, S.; Bunn, J.Y.; Weaver, D.; Muss, H.; et al. Cancer associated fibroblasts stimulated by transforming growth factor beta1 (TGF-β1) increase invasion rate of tumor cells: A population study. Breast Cancer Res. Treat. 2008, 110, 39–49. [Google Scholar] [CrossRef]
- Stewart, T.A.; Azimi, I.; Brooks, A.J.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Janus kinases and Src family kinases in the regulation of EGF-induced vimentin expression in MDA-MB-468 breast cancer cells. Int. J. Biochem. Cell Biol. 2016, 76, 64–74. [Google Scholar] [CrossRef] [Green Version]
- Azimi, I.; Bong, A.H.; Poo, G.X.H.; Armitage, K.; Lok, D.; Roberts-Thomson, S.J.; Monteith, G.R. Pharmacological inhibition of store-operated calcium entry in MDA-MB-468 basal A breast cancer cells: Consequences on calcium signalling, cell migration and proliferation. Cell. Mol. Life Sci. 2018, 75, 4525–4537. [Google Scholar] [CrossRef]
- Bio Rad. Image Lab Software—User Guide, Version 6.1. Available online: https://www.bio-rad.com/webroot/web/pdf/lsr/literature/10000076953.pdf (accessed on 30 June 2020).
- Azimi, I.; Flanagan, J.U.; Stevenson, R.J.; Inserra, M.; Vetter, I.; Monteith, G.; Denny, W.A. Evaluation of known and novel inhibitors of Orai1-mediated store operated Ca 2+ entry in MDA-MB-231 breast cancer cells using a Fluorescence Imaging Plate Reader assay. Bioorgan. Med. Chem. 2017, 25, 440–449. [Google Scholar] [CrossRef] [Green Version]
- Bassett, J.J.; Bong, A.H.; Janke, E.K.; Robitaille, M.; Roberts-Thomson, S.J.; Peters, A.A.; Monteith, G.R. Assessment of cytosolic free calcium changes during ceramide-induced cell death in MDA-MB-231 breast cancer cells expressing the calcium sensor GCaMP6m. Cell Calcium 2018, 72, 39–50. [Google Scholar] [CrossRef]
- Yang, L.; Huang, J.; Ren, X.; Gorska, A.E.; Chytil, A.; Aakre, M.; Carbone, D.P.; Matrisian, L.M.; Richmond, A.; Lin, P.C.; et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 2008, 13, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Dvořánková, B.; Szabo, P.; Lacina, L.; Gál, P.; Uhrova, J.; Zima, T.; Kaltner, H.; André, S.; Gabius, H.-J.; Syková, E.; et al. Human Galectins Induce Conversion of Dermal Fibroblasts into Myofibroblasts and Production of Extracellular Matrix: Potential Application in Tissue Engineering and Wound Repair. Cells Tissues Organs 2011, 194, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef] [Green Version]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef]
- Lewis, R.S. The molecular choreography of a store-operated calcium channel. Nature 2007, 446, 284–287. [Google Scholar] [CrossRef]
- Parekh, A.B.; Penner, R. Store depletion and calcium influx. Physiol. Rev. 1997, 77, 901–930. [Google Scholar] [CrossRef]
- Tang, L.; El-Din, T.G.; Swanson, T.M.; Pryde, D.C.; Scheuer, T.; Zheng, L.T.N.; Catterall, W.A. Structural basis for inhibition of a voltage-gated Ca2+ channel by Ca2+ antagonist drugs. Nat. Cell Biol. 2016, 537, 117–121. [Google Scholar] [CrossRef]
- Xiang, Z.; Thompson, A.D.; Brogan, J.T.; Schulte, M.L.; Melancon, B.J.; Mi, D.; Lewis, L.M.; Zou, B.; Yang, L.; Morrison, R.; et al. The Discovery and Characterization of ML218: A Novel, Centrally Active T-Type Calcium Channel Inhibitor with Robust Effects in STN Neurons and in a Rodent Model of Parkinson’s Disease. ACS Chem. Neurosci. 2011, 2, 730–742. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.S.; Spitzer, N.C. Calcium Signaling in Neuronal Development. Cold Spring Harb. Perspect. Biol. 2011, 3, a004259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrol, H.P.; Church, L.D.; Bacon, P.A.; Young, S.P. Intracellular calcium signalling patterns reflect the differentiation status of human T cells. Clin. Exp. Immunol. 2008, 153, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.L.; Rios, E.; Durães, C.; Ribeiro, R.; Machado, J.C.; Mantovani, A.; Barbosa, M.A.; Carneiro, F.M.; Oliveira, M.J. The Two Faces of Tumor-Associated Macrophages and Their Clinical Significance in Colorectal Cancer. Front. Immunol. 2019, 10, 1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vancauwenberghe, E.; Noyer, L.; Derouiche, S.; Lemonnier, L.; Gosset, P.; Sadofsky, L.R.; Mariot, P.; Warnier, M.; Bokhobza, A.; Slomianny, C.; et al. Activation of mutated TRPA1 ion channel by resveratrol in human prostate cancer associated fibroblasts (CAF). Mol. Carcinog. 2017, 56, 1851–1867. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.S.; Yeung, T.-L.; Yip, K.-P.; Wong, K.-K.; Ho, S.Y.; Mangala, L.S.; Sood, A.K.; Lopez-Berestein, G.; Sheng, J.; Wong, S.T.; et al. Cancer-associated fibroblasts regulate endothelial adhesion protein LPP to promote ovarian cancer chemoresistance. J. Clin. Investig. 2017, 128, 589–606. [Google Scholar] [CrossRef] [Green Version]
- Tao, L.; Huang, G.; Wang, R.; Pan, Y.; He, Z.; Chu, X.; Song, H.; Chen, L. Cancer-associated fibroblasts treated with cisplatin facilitates chemoresistance of lung adenocarcinoma through IL-11/IL-11R/STAT3 signaling pathway. Sci. Rep. 2016, 6, 38408. [Google Scholar] [CrossRef]
- Zhang, H.; Deng, T.; Liu, R.; Ning, T.; Yang, H.; Liu, D.; Zhang, Q.; Lin, D.; Ge, S.; Bai, M.; et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol. Cancer 2020, 19, 43. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-Y.; Lai, M.-D.; Phan, N.N.; Sun, Z.; Lin, Y.-C. Meta-Analysis of Public Microarray Datasets Reveals Voltage-Gated Calcium Gene Signatures in Clinical Cancer Patients. PLoS ONE 2015, 10, e0125766. [Google Scholar] [CrossRef] [Green Version]
- Stadler, S.; Nguyen, C.H.; Schachner, H.; Milovanovic, D.; Holzner, S.; Brenner, S.; Eichsteininger, J.; Stadler, M.; Senfter, D.; Krenn, L.; et al. Colon cancer cell-derived 12(S)-HETE induces the retraction of cancer-associated fibroblast via MLC2, RHO/ROCK and Ca2+ signalling. Cell. Mol. Life Sci. 2017, 74, 1907–1921. [Google Scholar] [CrossRef] [Green Version]
- Coleman, D.T.; Gray, A.L.; Stephens, C.A.; Scott, M.L.; Cardelli, J.A. Repurposed drug screen identifies cardiac glycosides as inhibitors of TGF-β-induced cancer-associated fibroblast differentiation. Oncotarget 2016, 7, 32200–32209. [Google Scholar] [CrossRef]
- Altamirano, J.; Li, Y.; DeSantiago, J.; Piacentino, V.; Houser, S.R.; Bers, D.M. The inotropic effect of cardioactive glycosides in ventricular myocytes requires Na+-Ca2+ exchanger function. J. Physiol. 2006, 575, 845–854. [Google Scholar] [CrossRef]
- Tobar, N.; Toyos, M.; Urra, C.; Méndez, N.; Arancibia, R.; Smith, P.C.; Martínez, J. c-Jun N terminal kinase modulates NOX-4 derived ROS production and myofibroblasts differentiation in human breast stromal cells. BMC Cancer 2014, 14, 640. [Google Scholar] [CrossRef] [Green Version]
- Hodeify, R.; Selvaraj, S.; Wen, J.; Arredouani, A.; Hubrack, S.; Dib, M.; Al-Thani, S.N.; McGraw, T.; Machaca, K. A STIM1-dependent “trafficking trap” mechanism regulates Orai1 plasma membrane residence and Ca2+ influx levels. Cell Sci. 2015, 128, 3143–3154. [Google Scholar] [CrossRef] [Green Version]
- Sobradillo, D.; Hernández-Morales, M.; Ubierna, D.; Moyer, M.P.; Núñez, L.; Villalobos, C. A Reciprocal Shift in Transient Receptor Potential Channel 1 (TRPC1) and Stromal Interaction Molecule 2 (STIM2) Contributes to Ca2+ Remodeling and Cancer Hallmarks in Colorectal Carcinoma Cells. J. Biol. Chem. 2014, 289, 28765–28782. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Huang, X.-Y. Ca2+ Influx through L-type Ca2+ Channels Controls the Trailing Tail Contraction in Growth Factor-induced Fibroblast Cell Migration. J. Biol. Chem. 2005, 280, 27130–27137. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Jia, Z.; Kong, J.; Zhang, F.; Fang, S.; Li, X.; Li, W.; Yang, X.; Luo, Y.; Lin, B.; et al. Carcinoma-Associated Fibroblasts Lead the Invasion of Salivary Gland Adenoid Cystic Carcinoma Cells by Creating an Invasive Track. PLoS ONE 2016, 11, e0150247. [Google Scholar]
- Arandkar, S.; Furth, N.; Elisha, Y.; Nataraj, N.B.; van der Kuip, H.; Yarden, Y.; Aulitzky, W.; Ulitsky, I.; Geiger, B.; Oren, M. Altered p53 functionality in cancer-associated fibroblasts contributes to their cancer-supporting features. Proc. Natl. Acad. Sci. USA 2018, 115, 6410–6415. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Kamiya, K.; Kodama, I.; Toyama, J. Cell Cycle-related Changes in the Voltage-gated Ca2+ Currents in Cultured Newborn Rat Ventricular Myocytes. J. Mol. Cell. Cardiol. 1998, 30, 1095–1103. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadras, F.; Stewart, T.A.; Robitaille, M.; Peters, A.A.; Croft, P.K.-d.; Soon, P.S.; Saunus, J.M.; Lakhani, S.R.; Roberts-Thomson, S.J.; Monteith, G.R. Altered Calcium Influx Pathways in Cancer-Associated Fibroblasts. Biomedicines 2021, 9, 680. https://doi.org/10.3390/biomedicines9060680
Sadras F, Stewart TA, Robitaille M, Peters AA, Croft PK-d, Soon PS, Saunus JM, Lakhani SR, Roberts-Thomson SJ, Monteith GR. Altered Calcium Influx Pathways in Cancer-Associated Fibroblasts. Biomedicines. 2021; 9(6):680. https://doi.org/10.3390/biomedicines9060680
Chicago/Turabian StyleSadras, Francisco, Teneale A. Stewart, Mélanie Robitaille, Amelia A. Peters, Priyakshi Kalita-de Croft, Patsy S. Soon, Jodi M. Saunus, Sunil R. Lakhani, Sarah J. Roberts-Thomson, and Gregory R. Monteith. 2021. "Altered Calcium Influx Pathways in Cancer-Associated Fibroblasts" Biomedicines 9, no. 6: 680. https://doi.org/10.3390/biomedicines9060680
APA StyleSadras, F., Stewart, T. A., Robitaille, M., Peters, A. A., Croft, P. K. -d., Soon, P. S., Saunus, J. M., Lakhani, S. R., Roberts-Thomson, S. J., & Monteith, G. R. (2021). Altered Calcium Influx Pathways in Cancer-Associated Fibroblasts. Biomedicines, 9(6), 680. https://doi.org/10.3390/biomedicines9060680