
Immunotherapeutic Potential of m6A-Modifiers and MicroRNAs in Controlling Acute Myeloid Leukaemia

 ,
,  ,
,  and
and
Abstract
:1. Introduction
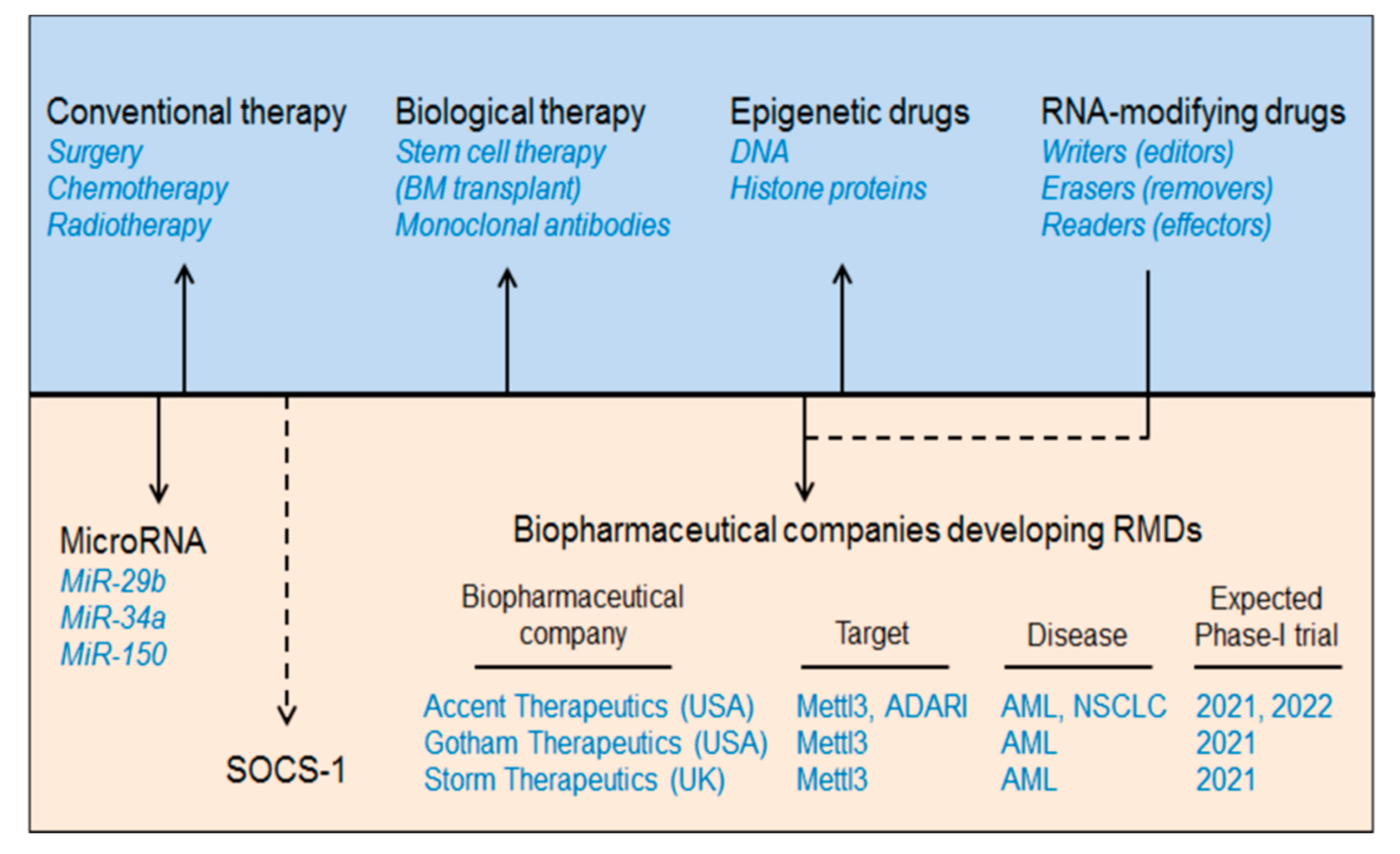
2. AML Therapeutics
3. Epitranscriptomics in AML Therapy
3.1. Editors (Writers)
3.1.1. Mettl3 in AML
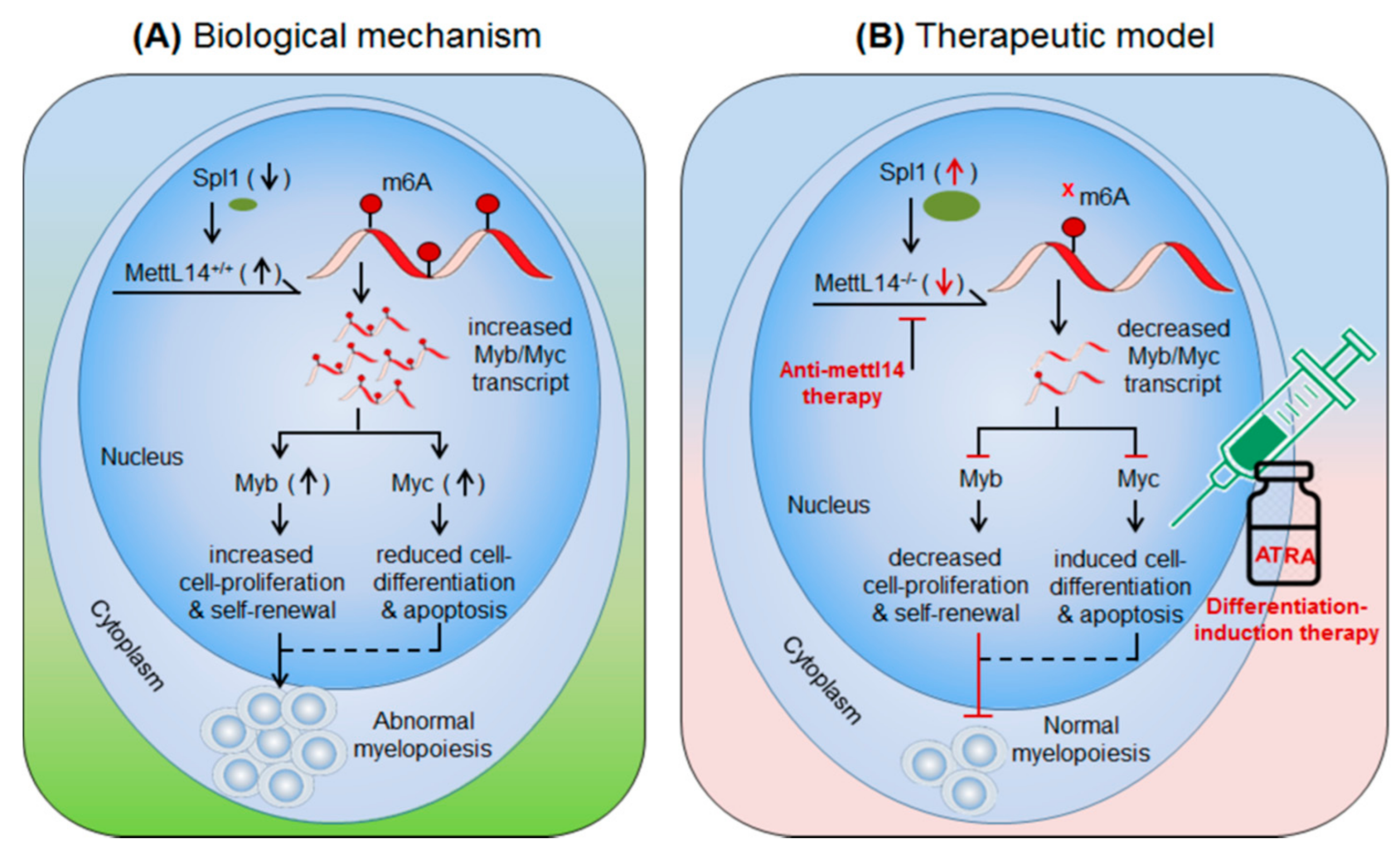
3.1.2. METTL14 in AML
3.2. Erasers (Removers)
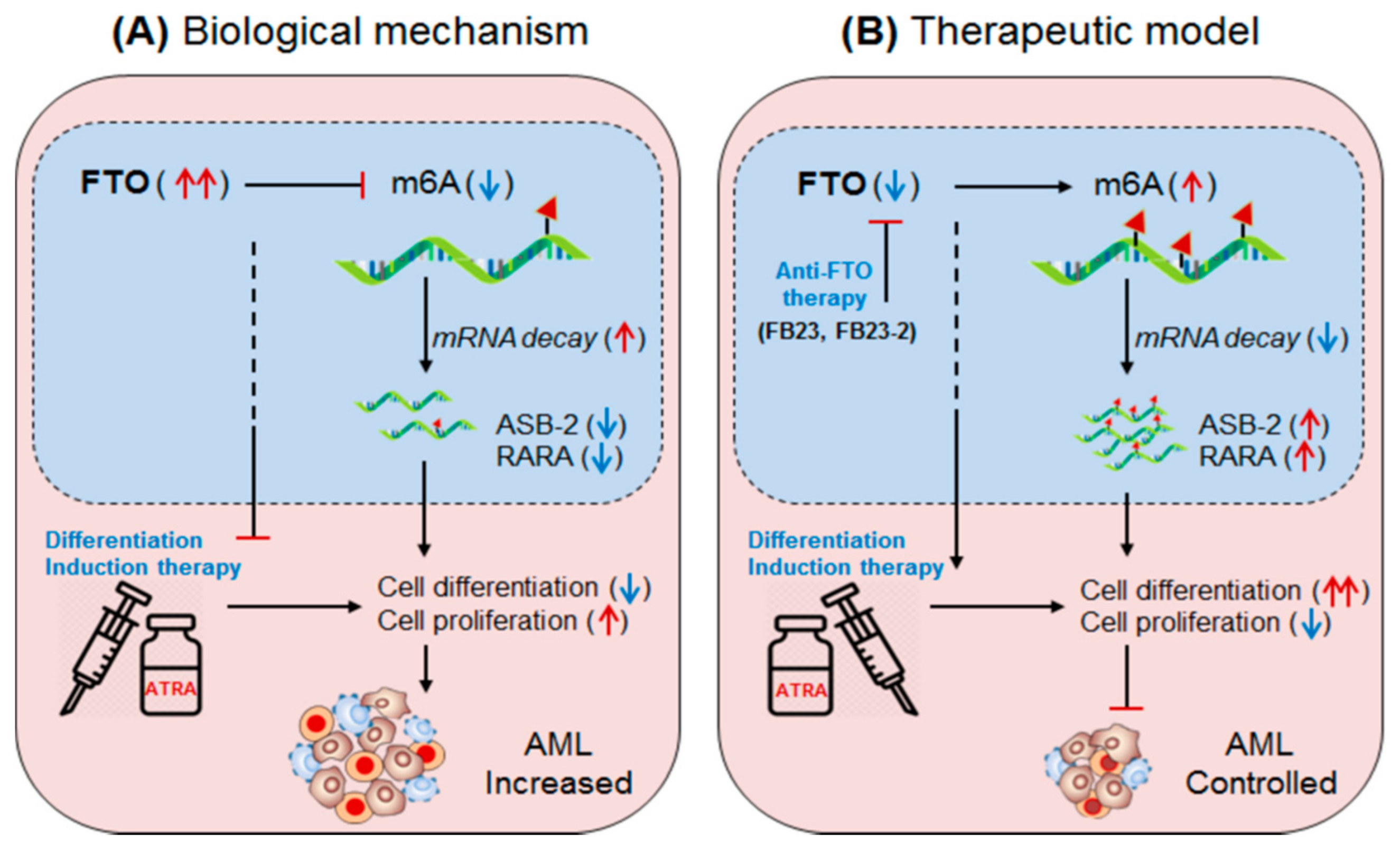
3.2.1. m6A-Demethylase FTO in AML
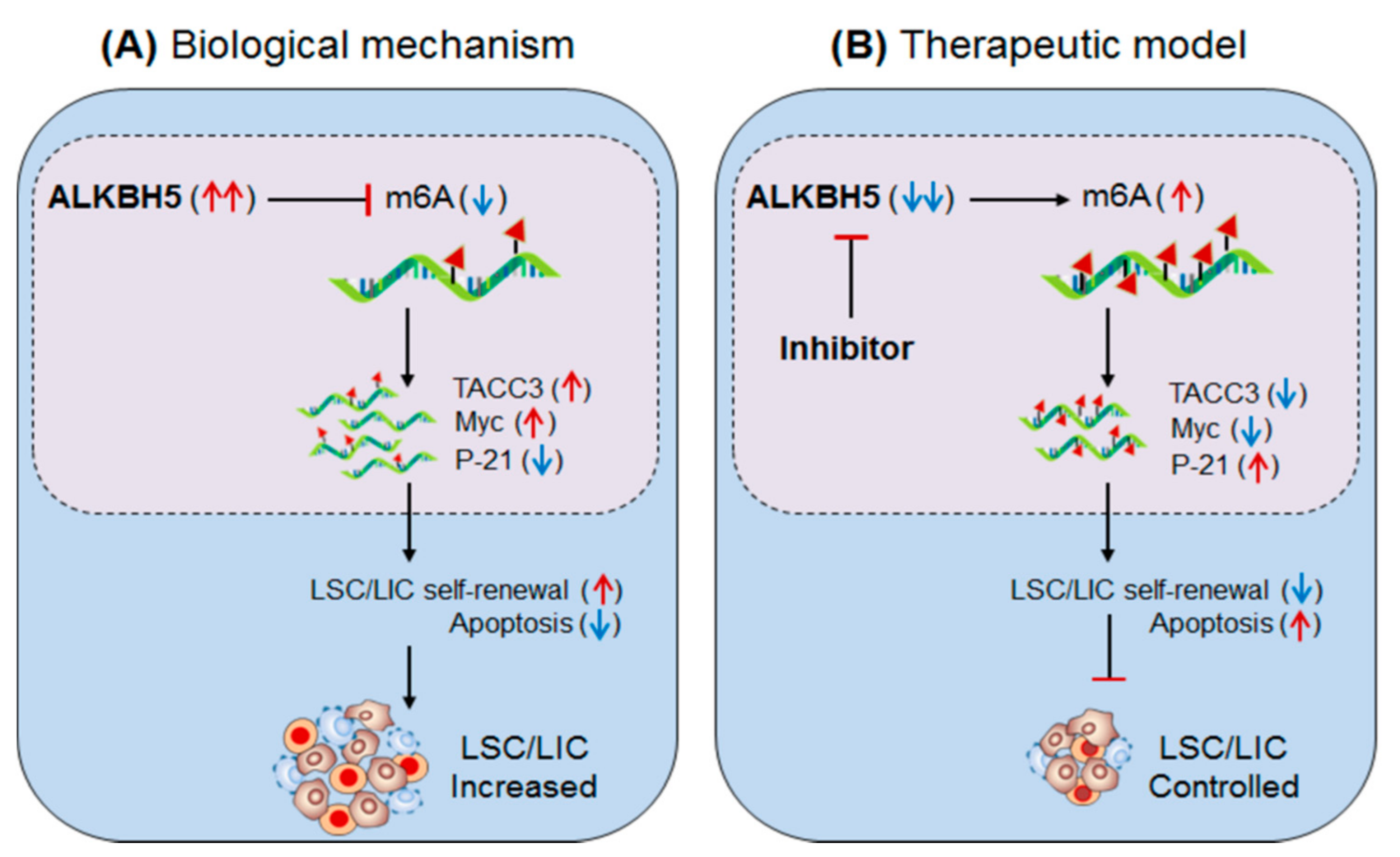
3.2.2. ALKBH5 (Eraser) in AML
3.3. Effectors (Readers)
3.3.1. YTHDF2 in AML
3.3.2. YTHDF2 in Stem Cell Expansion
4. Therapeutic Strategies
4.1. Mettl3 Inhibitor
4.2. FTO Inhibitor
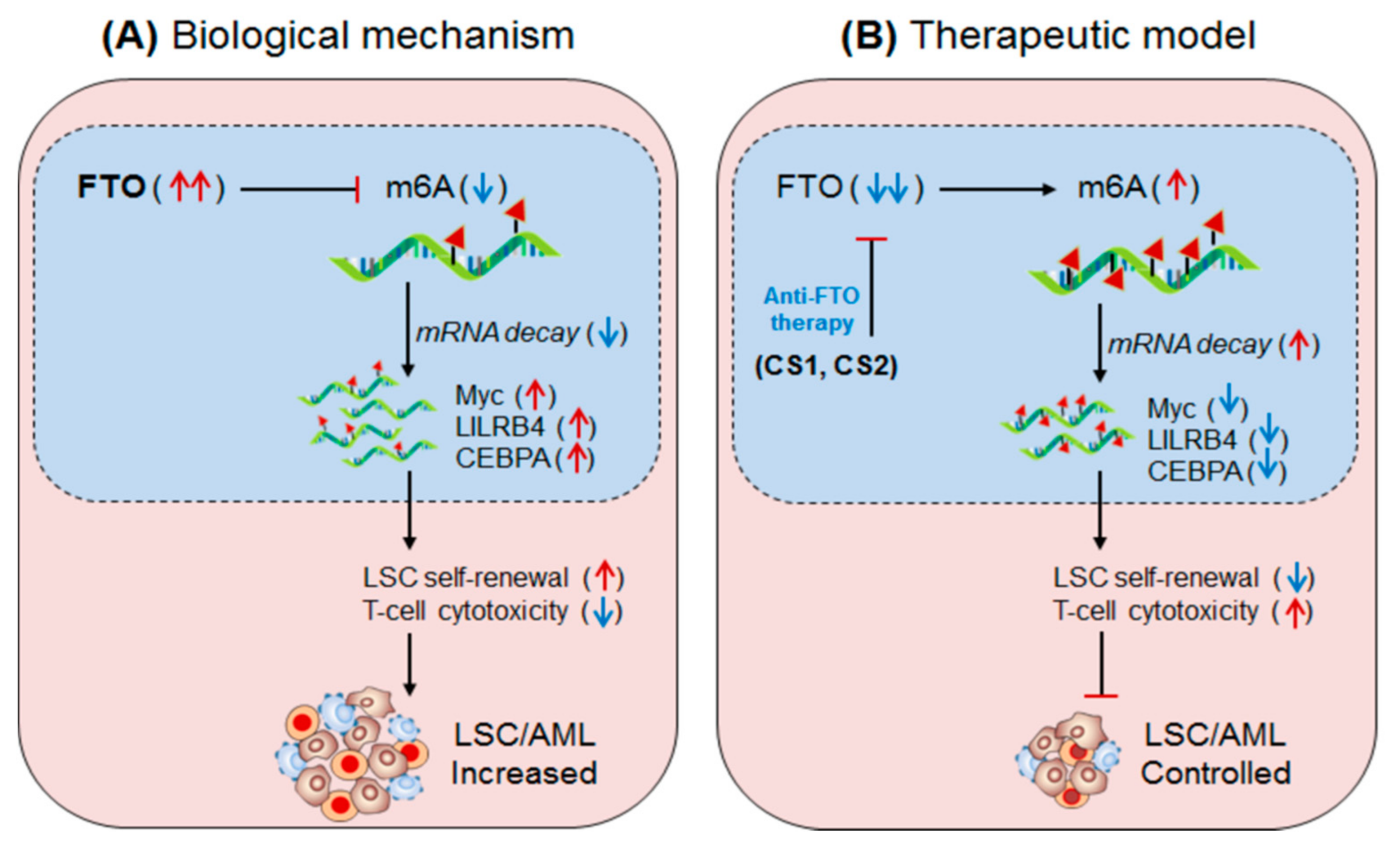
4.3. FTO in Anti-Tumour Immunity (AML)
5. MicroRNA in AML
5.1. miR-150 in AML
5.2. miR-34a in AML
5.3. miR-29b in AML
6. Suppressor of Cytokine Signalling (SOCS/CISH) in AML
7. Conclusions
8. Future Prospective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Desrosiers, R.; Friderici, K.; Rottman, F. Identification of Methylated Nucleosides in Messenger RNA from Novikoff Hepatoma Cells. Proc. Natl. Acad. Sci. USA 1974, 71, 3971–3975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, R.; Kelley, D. Existence of methylated messenger RNA in mouse L cells. Cell 1974, 1, 37–42. [Google Scholar] [CrossRef]
- Jia, G.; Yang, C.-G.; Yang, S.; Jian, X.; Yi, C.; Zhou, Z.; He, C. Oxidative demethylation of 3-methylthymine and 3-methyluracil in single-stranded DNA and RNA by mouse and human FTO. FEBS Lett. 2008, 582, 3313–3319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.-G.; et al. Erratum: Corrigendum: N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2012, 8, 1008. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Wei, J.; He, C. Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol. Cell 2019, 74, 640–650. [Google Scholar] [CrossRef]
- Cully, M. Chemical inhibitors make their RNA epigenetic mark. Nat. Rev. Drug Discov. 2019, 18, 892–894. [Google Scholar] [CrossRef]
- Van Der Werf, I.; Jamieson, C. The Yin and Yang of RNA Methylation: An Imbalance of Erasers Enhances Sensitivity to FTO Demethylase Small-Molecule Targeting in Leukemia Stem Cells. Cancer Cell 2019, 35, 540–541. [Google Scholar] [CrossRef] [Green Version]
- Kane, S.E.; Beemon, K. Precise localization of m6A in rous sarcoma virus RNA reveals clustering of methylation sites: Implications for rna processing. Mol. Cell Biol. 1985, 5, 2298–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, K.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [Green Version]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nat. Cell Biol. 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Pelcovits, A.; Niroula, R. Acute myeloid leukemia: A review. R. I. Med. J. 2020, 103, 38–40. [Google Scholar]
- Daver, N.; Wei, A.H.; Pollyea, D.A.; Fathi, A.T.; Vyas, P.; DiNardo, C.D. New directions for emerging therapies in acute myeloid leukemia: The next chapter. Blood Cancer J. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Leisch, M.; Jansko, B.; Zaborsky, N.; Greil, R.; Pleyer, L. Next generation sequencing in AML-on the way to becoming a new standard for treatment initiation and/or modulation? Cancers 2019, 11, 252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiNardo, C.D.; Cortes, J. Mutations in AML: Prognostic and therapeutic implications. Hematology 2016, 2016, 348–355. [Google Scholar] [CrossRef] [Green Version]
- Walasek, A. The new perspectives of targeted therapy in acute myeloid leukemia. Adv. Clin. Exp. Med. 2019, 28, 271–276. [Google Scholar] [CrossRef]
- Morsink, L.M.; Walter, R.B. Novel monoclonal antibody-based therapies for acute myeloid leukemia. Best Pract. Res. Clin. Haematol. 2019, 32, 116–126. [Google Scholar] [CrossRef]
- Yu, J.; Xie, T.; Wang, Z.; Wang, X.; Zeng, S.; Kang, Y.; Hou, T. DNA methyltransferases: Emerging targets for the discovery of in-hibitors as potent anticancer drugs. Drug Discov. Today 2019, 24, 2323–2331. [Google Scholar] [CrossRef]
- Yang, X.; Wong, M.P.M.; Ng, R.K. Aberrant DNA methylation in acute myeloid leukemia and its clinical implications. Int. J. Mol. Sci. 2019, 20, 4576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, X. DNA methyltransferase inhibitors in acute myeloid leukemia: Discovery, design and first therapeutic expe-riences. Expert Opin. Drug Discov. 2012, 7, 1039–1051. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Con-version of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by mll partner tet1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; VasanthaKumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.; Mitchell-Flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095.e20. [Google Scholar] [CrossRef] [Green Version]
- Golub, D.; Iyengar, N.; Dogra, S.; Wong, T.; Bready, D.; Tang, K.; Modrek, A.S.; Placantonakis, D.G. Mutant Isocitrate Dehydrogenase Inhibitors as Targeted Cancer Therapeutics. Front. Oncol. 2019, 9, 417. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Konopleva, M. Concurrent inhibition of IDH and methyltransferase maximizes therapeutic efficacy in IDH mutant acute myeloid leukemia. Haematologica 2021, 106, 324–326. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Koh, Y.; Hong, J.; Eom, H.-S.; Yoon, S.-S. Recent Clinical Update of Acute Myeloid Leukemia: Focus on Epigenetic Therapies. J. Korean Med. Sci. 2021, 36, e85. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Yang, H.; Bueso-Ramos, C.; Ferrajoli, A.; Cortes, J.; Wierda, W.G.; Faderl, S.; Koller, C.; Morris, G.; Rosner, G.; et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [saha]) in patients with advanced leukemias and myelodysplastic syndromes. Blood 2008, 111, 1060–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.-Y.; Yu, L. Epigenetic therapies in acute myeloid leukemia: The role of hypomethylating agents, histone deacetylase inhibitors and the combination of hypomethylating agents with histone deacetylase inhibitors. Chin. Med. J. 2020, 133, 699–715. [Google Scholar] [CrossRef] [PubMed]
- San Jose-Eneriz, E.; Gimenez-Camino, N.; Agirre, X.; Prosper, F. Hdac inhibitors in acute myeloid leukemia. Cancers 2019, 11, 1794. [Google Scholar] [CrossRef] [Green Version]
- Levis, M. Midostaurin approved for FLT3-mutated AML. Blood 2017, 129, 3403–3406. [Google Scholar] [CrossRef]
- Alfayez, M.; Kantarjian, H.; Kadia, T.; Ravandi-Kashani, F.; Daver, N. CPX-351 (vyxeos) in AML. Leuk. Lymphoma 2020, 61, 288–297. [Google Scholar] [CrossRef]
- Krauss, A.C.; Gao, X.; Li, L.; Manning, M.L.; Patel, P.; Fu, W.; Janoria, K.G.; Gieser, G.; Bateman, D.A.; Przepiorka, D.; et al. FDA Approval Summary: (Daunorubicin and Cytarabine) Liposome for Injection for the Treatment of Adults with High-Risk Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 2685–2690. [Google Scholar] [CrossRef] [PubMed]
- Dennis, M.; Russell, N.; Hills, R.K.; Hemmaway, C.; Panoskaltsis, N.; McMullin, M.-F.; Kjeldsen, L.; Dignum, H.; Thomas, I.F.; Clark, R.E.; et al. Vosaroxin and vosaroxin plus low-dose Ara-C (LDAC) vs low-dose Ara-C alone in older patients with acute myeloid leukemia. Blood 2015, 125, 2923–2932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, S. Gilteritinib: First Global Approval. Drugs 2019, 79, 331–339. [Google Scholar] [CrossRef]
- Zhao, J.; Song, Y.; Liu, D. Correction to: Gilteritinib: A novel FLT3 inhibitor for acute myeloid leukemia. Biomark. Res. 2019, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Guerra, V.A.; DiNardo, C.; Konopleva, M. Venetoclax-based therapies for acute myeloid leukemia. Best Pract. Res. Clin. Haematol. 2019, 32, 145–153. [Google Scholar] [CrossRef]
- Kirschbaum, M.H.; Foon, K.A.; Frankel, P.; Ruel, C.; Pulone, B.; Tuscano, J.M.; Newman, E.M. A phase 2 study of belinostat (pxd101) in patients with relapsed or refractory acute myeloid leukemia or patients over the age of 60 with newly diagnosed acute myeloid leukemia: A california cancer consortium study. Leuk. Lymphoma 2014, 55, 2301–2304. [Google Scholar]
- Quintas-Cardama, A.; Santos, F.P.; Garcia-Manero, G. Histone deacetylase inhibitors for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Leukemia 2011, 25, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Bug, G.; Burchert, A.; Wagner, E.-M.; Kröger, N.; Berg, T.; Güller, S.; Metzelder, S.K.; Wolf, A.; Hünecke, S.; Bader, P.; et al. Phase I/II study of the deacetylase inhibitor panobinostat after allogeneic stem cell transplantation in patients with high-risk MDS or AML (PANOBEST trial). Leukemia 2017, 31, 2523–2525. [Google Scholar] [CrossRef] [PubMed]
- DeAngelo, D.J.; Walker, A.R.; Schlenk, R.F.; Sierra, J.; Medeiros, B.C.; Ocio, E.M.; Röllig, C.; Strickland, S.A.; Thol, F.; Valera, S.-Z.; et al. Safety and efficacy of oral panobinostat plus chemotherapy in patients aged 65 years or younger with high-risk acute myeloid leukemia. Leuk. Res. 2019, 85, 106197. [Google Scholar] [CrossRef]
- Wieduwilt, M.J.; Pawlowska, N.; Thomas, S.; Olin, R.; Logan, A.C.; Damon, L.E.; Martin, T.; Kang, M.; Sayre, P.H.; Boyer, W.; et al. Histone Deacetylase Inhibition with Panobinostat Combined with Intensive Induction Chemotherapy in Older Patients with Acute Myeloid Leukemia: Phase I Study Results. Clin. Cancer Res. 2019, 25, 4917–4923. [Google Scholar] [CrossRef]
- Kung Sutherland, M.S.; Walter, R.B.; Jeffrey, S.C.; Burke, P.J.; Yu, C.; Kostner, H.; Stone, I.; Ryan, M.C.; Sussman, D.; Lyon, R.P.; et al. Sgn-cd33a: A novel cd33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant aml. Blood 2013, 122, 1455–1463. [Google Scholar] [CrossRef]
- Vey, N.; Delaunay, J.; Martinelli, G.; Fiedler, W.; Raffoux, E.; Prebet, T.; Gomez-Roca, C.; Papayannidis, C.; Kebenko, M.; Paschka, P.; et al. Phase i clinical study of rg7356, an anti-cd44 humanized antibody, in patients with acute myeloid leu-kemia. Oncotarget 2016, 7, 32532–32542. [Google Scholar] [CrossRef] [Green Version]
- Vu, L.P.; Pickering, B.F.; Cheng, Y.; Zaccara, S.; Nguyen, D.; Minuesa, G.; Chou, T.; Chow, A.; Saletore, Y.; Mackay, M.; et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 2017, 23, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, I.; Tzelepis, K.; Pandolfini, L.; Namshik, H.; Millan-Zambrano, G.; Robson, S.C.; Aspris, D.; Migliori, V.; Bannister, A.J.; Hannes, P.; et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nat. Cell Biol. 2017, 552, 126–131. [Google Scholar] [CrossRef]
- Konstantinos, T.E.D.; Eliza, Y.; Justyna, R.; Demetrios, A.; Ana, F.D.; Richard, F.; Alan, H.; Dan, L.; Yaara, O.R.; Alexandra, S.; et al. Pharmacological inhibition of the rna m6a writer mettl3 as a novel ther-apeutic strategy for acute myeloid leukemia. Blood 2019, 134, 403. [Google Scholar]
- Schmiechen, A.C.; Batista, P.J. An epitranscriptomic vulnerability in myeloid malignancies. Nat. Med. 2017, 23, 1252–1254. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, O. RNA Methyltransferases as a New Therapeutic Target in Leukemia. Hematologist 2018, 15, 2. [Google Scholar] [CrossRef]
- Yankova, E.; Blackaby, W.; Albertella, M.; Rak, J.; De Braekeleer, E.; Tsagkogeorga, G.; Pilka, E.S.; Aspris, D.; Leggate, D.; Hendrick, A.G.; et al. Small molecule inhibition of mettl3 as a strategy against myeloid leukaemia. Nature 2021, 593, 597–601. [Google Scholar] [CrossRef]
- Weng, H.; Huang, H.; Wu, H.; Qin, X.; Zhao, B.S.; Dong, L.; Shi, H.; Skibbe, J.; Shen, C.; Hu, C.; et al. Mettl14 inhibits hema-topoietic stem/progenitor differentiation and promotes leukemogenesis via mrna m(6)a modification. Cell Stem Cell 2018, 22, 191–205.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guibal, F.C.; Moog-Lutz, C.; Smolewski, P.; Di Gioia, Y.; Darzynkiewicz, Z.; Lutz, P.G.; Cayre, Y.E. Asb-2 inhibits growth and pro-motes commitment in myeloid leukemia cells. J. Biol. Chem. 2002, 277, 218–224. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N 6-Methyladenosine RNA Demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Sheng, Y.; Zhu, A.C.; Robinson, S.; Jiang, X.; Dong, L.; Chen, H.; Su, R.; Yin, Z.; Li, W.; et al. RNA Demethylase ALKBH5 Selectively Promotes Tumorigenesis and Cancer Stem Cell Self-Renewal in Acute Myeloid Leukemia. Cell Stem Cell 2020, 27, 64–80.e9. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Qian, P.; Shao, W.; Shi, H.; He, X.C.; Gogol, M.; Yu, Z.; Wang, Y.; Qi, M.; Zhu, Y.; et al. Suppression of m6A reader Ythdf2 promotes hematopoietic stem cell expansion. Cell Res. 2018, 28, 904–917. [Google Scholar] [CrossRef]
- Paris, J.; Morgan, M.; Campos, J.; Spencer, G.J.; Shmakova, A.; Ivanova, I.; Mapperley, C.; Lawson, H.; Wotherspoon, D.A.; Sepulveda, C.; et al. Targeting the RNA m6A Reader YTHDF2 Selectively Compromises Cancer Stem Cells in Acute Myeloid Leukemia. Cell Stem Cell 2019, 25, 137–148.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, K.D.; Vodyanik, M.; Slukvin, I.I. Hematopoietic differentiation and production of mature myeloid cells from human pluripotent stem cells. Nat. Protoc. 2011, 6, 296–313. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Wang, W.-T.; Yu, X.; Yang, L.; Chen, S.; Li, Y. Pathways related to PMA-differentiated THP1 human monocytic leukemia cells revealed by RNA-Seq. Sci. China Life Sci. 2015, 58, 1282–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chim, C.S.; Kwong, Y.L.; Liang, R.; Chu, Y.C.; Chan, C.H.; Chan, L.C.; Wong, K.F.; Chan, T.K. All-trans retinoic acid (atra) in the treatment of acute promyelocytic leukemia (apl). Hematol. Oncol. 1996, 14, 147–154. [Google Scholar] [CrossRef]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nat. Cell Biol. 2014, 505, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Gopal, V.; Hulette, B.; Li, Y.Q.; Kuvelkar, R.; Raza, A.; Larson, R.; Goldberg, J.; Tricot, G.; Bennett, J.; Preisler, H. C-myc and c-myb ex-pression in acute myelogenous leukemia. Leuk. Res. 1992, 16, 1003–1011. [Google Scholar] [CrossRef]
- Mundo, L.; Ambrosio, M.R.; Raimondi, F.; Del Porro, L.; Guazzo, R.; Mancini, V.; Granai, M.; Rocca, B.J.; Lopez, C.; Bens, S.; et al. Molecular switch from MYC to MYCN expression in MYC protein negative Burkitt lymphoma cases. Blood Cancer J. 2019, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Sakai, N.; Kubota, Y.; Shuin, T. Statistically significant expression of b-myb in clinically advanced human renal-cell carci-nomas. Int. J. Oncol. 1993, 2, 419–423. [Google Scholar]
- Carter, J.L.; Hege, K.; Yang, J.; Kalpage, H.A.; Su, Y.; Edwards, H.; Hüttemann, M.; Taub, J.W.; Ge, Y. Targeting multiple signaling pathways: The new approach to acute myeloid leukemia therapy. Signal. Transduct. Target. Ther. 2020, 5, 288. [Google Scholar] [CrossRef]
- Delgado, M.D.; León, J. Myc Roles in Hematopoiesis and Leukemia. Genes Cancer 2010, 1, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Glasow, A.; Prodromou, N.; Xu, K.; von Lindern, M.; Zelent, A. Retinoids and myelomonocytic growth factors cooperatively activate rara and induce human myeloid leukemia cell differentiation via map kinase pathways. Blood 2005, 105, 341–349. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Cui, X.; Rao, M.K.; Chen, Y.; Huang, Y. Exome-based analysis for RNA epigenome sequencing data. Bioinformatics 2013, 29, 1565–1567. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Mod-el-based analysis of chip-seq (macs). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Zhang, Y.; Ma, J.; Guo, F.; Cao, Q.; Zhang, Y.; Zhou, B.; Chai, J.; Zhao, W.; Zhao, R. The Demethylase Activity of FTO (Fat Mass and Obesity Associated Protein) Is Required for Preadipocyte Differentiation. PLoS ONE 2015, 10, e0133788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Wei, L.-H.; Wang, Y.; Xiao, Y.; Liu, J.; Zhang, W.; Yan, N.; Amu, G.; Tang, X.; Zhang, L.; et al. Structural insights into FTO’s catalytic mechanism for the demethylation of multiple RNA substrates. Proc. Natl. Acad. Sci. USA 2019, 116, 2919–2924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masetti, R.; Vendemini, F.; Zama, D.; Biagi, C.; Gasperini, P.; Pession, A. All-trans retinoic acid in the treatment of pediatric acute promyelocytic leukemia. Expert Rev. Anticancer Ther. 2012, 12, 1191–1204. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Hu, G.; Cai, X. The success and the challenge of all-trans retinoic acid in the treatment of cancer. Crit. Rev. Food Sci. Nutr. 2019, 59, S71–S80. [Google Scholar] [CrossRef]
- Rubio, V.; Calviño, E.; García-Pérez, A.; Herráez, A.; Diez, J.C. Human acute promyelocytic leukemia NB4 cells are sensitive to esculetin through induction of an apoptotic mechanism. Chem. Interact. 2014, 220, 129–139. [Google Scholar] [CrossRef]
- Kohroki, J.; Fujita, S.; Itoh, N.; Yamada, Y.; Imai, H.; Yumoto, N.; Nakanishi, T.; Tanaka, K. ATRA-regulatedAsb-2gene induced in differentiation of HL-60 leukemia cells. FEBS Lett. 2001, 505, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.; Imamura, T.; Yano, M.; Yoshida, H.; Fujiki, A.; Hirashima, Y.; Hosoi, H. Sensitivity of mll-rearranged aml cells to all-trans retinoic acid is associated with the level of h3k4me2 in the raralpha promoter region. Blood Cancer J. 2014, 4, e205. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.-Y.; Chen, Z. Acute promyelocytic leukemia: From highly fatal to highly curable. Blood 2008, 111, 2505–2515. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Su, R.; Sheng, Y.; Dong, L.; Dong, Z.; Xu, H.; Ni, T.; Zhang, Z.; Zhang, T.; Li, C.; et al. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell 2019, 35, 677–691.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Kennedy, S.; Hajian, T.; Gibson, E.; Seitova, A.; Xu, C.; Arrowsmith, C.H.; Vedadi, M. A radioactivity-based assay for screen-ing human m6a-rna methyltransferase, mettl3-mettl14 complex, and demethylase alkbh5. J. Biomol. Screen 2016, 21, 290–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moroz-Omori, E.V.; Huang, D.; Bedi, R.K.; Cheriyamkunnel, S.J.; Bochenkova, E.; Dolbois, A.; Rzeczkowski, M.D.; Wiedmer, L.; Śledź, P.; Caflisch, P. Mettl3 inhibitors for epitranscriptomic modulation of cellular processes. bio-Rxiv 2020. [Google Scholar] [CrossRef]
- Kuipers, K.J.J.; Hellweg, S.; Verones, F. Potential consequences of regional species loss for global species richness: A quanti-tative approach for estimating global extinction probabilities. Environ. Sci. Technol. 2019, 53, 4728–4738. [Google Scholar] [CrossRef]
- Su, R.; Dong, L.; Li, C.; Nachtergaele, S.; Wunderlich, M.; Qing, Y.; Deng, X.; Wang, Y.; Weng, X.; Hu, C.; et al. R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m6A/MYC/CEBPA Signaling. Cell 2018, 172, 90–105.e23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, S.N.; Armstrong, S.A. It’s Not What You Say but How You Say It: Targeting RNA Methylation in AML. Mol. Cell 2020, 78, 996–998. [Google Scholar] [CrossRef]
- Su, R.; Dong, L.; Li, Y.; Gao, M.; Han, L.; Wunderlich, M.; Deng, X.; Li, H.; Huang, Y.; Gao, L.; et al. Targeting fto suppresses can-cer stem cell maintenance and immune evasion. Cancer Cell 2020, 38, 79–96.e11. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Shi, H.; Ye, P.; Li, L.; Qu, Q.; Sun, G.; Sun, G.; Lu, Z.; Huang, Y.; Yang, C.-G.; et al. m 6 A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 2017, 18, 2622–2634. [Google Scholar] [CrossRef]
- Huang, Y.; Yan, J.; Li, Q.; Li, J.; Gong, S.; Zhou, H.; Gan, J.; Jiang, H.; Jia, G.-F.; Luo, C.; et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015, 43, 373–384. [Google Scholar] [CrossRef]
- Singh, B.; Kinne, H.E.; Milligan, R.D.; Washburn, L.J.; Olsen, M.; Lucci, A. Important Role of FTO in the Survival of Rare Panresistant Triple-Negative Inflammatory Breast Cancer Cells Facing a Severe Metabolic Challenge. PLoS ONE 2016, 11, e0159072. [Google Scholar] [CrossRef]
- Zheng, G.; Cox, T.; Tribbey, L.; Wang, G.Z.; Iacoban, P.; Booher, M.E.; Gabriel, G.J.; Zhou, L.; Bae, N.; Rowles, J.; et al. Synthesis of a FTO Inhibitor with Anticonvulsant Activity. ACS Chem. Neurosci. 2014, 5, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T.; Huntly, B.J. Targeting Epigenetic Readers in Cancer. N. Engl. J. Med. 2012, 367, 647–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.-I.; Robson, S.C.; Chung, C.-W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nat. Cell Biol. 2011, 478, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Fong, C.Y.; Gilan, O.; Lam, E.Y.; Rubin, A.F.; Ftouni, S.; Tyler, D.; Stanley, K.; Sinha, D.; Yeh, P.; Morison, J.; et al. Bet in-hibitor resistance emerges from leukaemia stem cells. Nature 2015, 525, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Biel, M.; Kretsovali, A.; Karatzali, E.; Papamatheakis, J.; Giannis, A. Design, Synthesis, and Biological Evaluation of a Small-Molecule Inhibitor of the Histone Acetyltransferase Gcn5. Angew. Chem. Int. Ed. 2004, 43, 3974–3976. [Google Scholar] [CrossRef] [PubMed]
- Tzelepis, K.; Koike-Yusa, H.; De Braekeleer, E.; Li, Y.; Metzakopian, E.; Dovey, O.M.; Mupo, A.; Grinkevich, V.; Li, M.; Mazan, M.; et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep. 2016, 17, 1193–1205. [Google Scholar] [CrossRef] [Green Version]
- Krivtsov, A.V.; Twomey, D.; Feng, Z.; Stubbs, M.C.; Wang, Y.; Faber, J.; Levine, J.E.; Wang, J.; Hahn, W.C.; Gilliland, D.G.; et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL–AF9. Nat. Cell Biol. 2006, 442, 818–822. [Google Scholar] [CrossRef]
- Song, H.; Liu, C.; Shen, N.; Yi, P.; Dong, F.; Li, X.; Zhang, N.; Huang, T. Overexpression of TACC3 in Breast Cancer Associates With Poor Prognosis. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.-S.; Wang, H.-B.; Zhou, Z.-G.; Zhang, Y.-J.; Zhong, Q.; Xu, L.; Huang, Y.-H.; Yeung, S.-C.; Chen, M.-S.; Zeng, M.-S. TACC3 promotes stemness and is a potential therapeutic target in hepatocellular carcinoma. Oncotarget 2015, 6, 24163–24177. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, E.C.; Jain, L.; Vickers, M.H.; Olins, A.L.; Olins, D.E.; Perry, J.K.; O’Sullivan, J.M. TNF-α Differentially Regulates Cell Cycle Genes in Promyelocytic and Granulocytic HL-60/S4 Cells. Genes Genomes Genet. 2019, 9, 2775–2786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raetz, E.A.; Teachey, D.T. T-cell acute lymphoblastic leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 580–588. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.H.; Bertulfo, F.C.; Sanda, T. Leukemia-Initiating Cells in T-Cell Acute Lymphoblastic Leukemia. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mapperley, C.; Van De Lagemaat, L.N.; Lawson, H.; Tavosanis, A.; Paris, J.; Campos, J.; Wotherspoon, D.; Durko, J.; Sarapuu, A.; Choe, J.; et al. The mRNA m6A reader YTHDF2 suppresses proinflammatory pathways and sustains hematopoietic stem cell function. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef]
- Qing, Y.; Dong, L.; Gao, L.; Li, C.; Li, Y.; Han, L.; Prince, E.; Tan, B.; Deng, X.; Wetzel, C.; et al. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/m6A/PFKP/LDHB axis. Mol. Cell 2021, 81, 922–939.e9. [Google Scholar] [CrossRef]
- Bhate, A.; Sun, T.; Li, J.B. ADAR1: A New Target for Immuno-oncology Therapy. Mol. Cell 2019, 73, 866–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Q.; Zhang, Q.; Shi, Y.; Shi, Q.; Jiang, Y.; Gu, Y.; Li, Z.; Li, X.; Zhao, K.; Wang, C.; et al. Tet2 promotes pathogen infec-tion-induced myelopoiesis through mrna oxidation. Nature 2018, 554, 123–127. [Google Scholar] [CrossRef]
- Fang, Z.H.; Wang, S.L.; Zhao, J.T.; Lin, Z.J.; Chen, L.Y.; Su, R.; Xie, S.T.; Carter, B.Z.; Xu, B. miR-150 exerts antileukemia activity in vitro and in vivo through regulating genes in multiple pathways. Cell Death Dis. 2016, 7, e2371. [Google Scholar] [CrossRef]
- Kumar, S.; Ashraf, M.U.; Kumar, A.; Bae, Y.-S. Therapeutic Potential of microRNA Against Th2-associated Immune Disorders. Curr. Top. Med. Chem. 2021, 21, 753–766. [Google Scholar] [CrossRef]
- Cao, Y.; Liu, Y.; Shang, L.; Wei, W.; Shen, Y.; Gu, Q.; Xie, X.; Dong, W.; Lin, Y.; Yue, Y.; et al. Decitabine and all-trans retinoic acid synergistically exhibit cytotoxicity against elderly AML patients via miR-34a/MYCN axis. Biomed. Pharmacother. 2020, 125, 109878. [Google Scholar] [CrossRef] [PubMed]
- León, J.; Ferrandiz, N.; Acosta, J.C.; Delgado, M.D. Inhibition of cell differentiation: A critical mechanism for MYC-mediated carcinogenesis? Cell Cycle 2009, 8, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Melnik, S.; Werth, N.; Boeuf, S.; Hahn, E.-M.; Gotterbarm, T.; Anton, M.; Richter, W. Impact of c-MYC expression on proliferation, differentiation, and risk of neoplastic transformation of human mesenchymal stromal cells. Stem Cell Res. Ther. 2019, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Ma, H.; Liu, Q.; Xiao, Y.; Pan, S.; Zhou, H.; Jia, L. Mir-29b/sp1/fut4 axis modulates the malignancy of leukemia stem cells by regulating fucosylation via wnt/beta-catenin pathway in acute myeloid leukemia. J. Exp. Clin. Cancer Res. 2019, 38, 200. [Google Scholar] [CrossRef] [Green Version]
- Palmer, D.C.; Guittard, G.C.; Franco, Z.; Crompton, J.G.; Eil, R.L.; Patel, S.J.; Ji, Y.; Van Panhuys, N.; Klebanoff, C.A.; Sukumar, M.; et al. Cish actively silences TCR signaling in CD8+ T cells to maintain tumor tolerance. J. Exp. Med. 2015, 212, 2095–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delconte, R.B.; Kolesnik, T.B.; Dagley, L.F.; Rautela, J.; Shi, W.; Putz, E.M.; Stannard, K.; Zhang, J.-G.; Teh, C.; Firth, M.; et al. CIS is a potent checkpoint in NK cell–mediated tumor immunity. Nat. Immunol. 2016, 17, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Putz, E.M.; Guillerey, C.; Kos, K.; Stannard, K.; Miles, K.; Delconte, R.B.; Takeda, K.; Nicholson, S.E.; Huntington, N.D.; Smyth, M.J. Tar-geting cytokine signaling checkpoint cis activates nk cells to protect from tumor initiation and metastasis. Oncoimmunology 2017, 6, e1267892. [Google Scholar] [CrossRef] [Green Version]
- Li, H.B.; Tong, J.; Zhu, S.; Batista, P.J.; Duffy, E.E.; Zhao, J.; Bailis, W.; Cao, G.; Kroehling, L.; Chen, Y.; et al. M(6)a mrna methyl-ation controls t cell homeostasis by targeting the il-7/stat5/socs pathways. Nature 2017, 548, 338–342. [Google Scholar] [CrossRef] [Green Version]
- Sprent, J.; Surh, C.D. Writer’s block: Preventing m 6 A mRNA methylation promotes T cell naivety. Immunol. Cell Biol. 2017, 95, 855–856. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Barr, T.; Zhang, J.; Chen, Z.; Wang, L.S.; Sun, J.C.; Sun, J.C.; Chen, J.; Caligiuri, M.A.; Yu, J. The rna m6a reader ythdf2 controls nk cell anti-tumor and anti-viral immunity. bioRxiv 2021. [Google Scholar] [CrossRef]
- Zhu, H.; Blum, R.H.; Bernareggi, D.; Ask, E.H.; Wu, Z.; Hoel, H.J.; Meng, Z.; Wu, C.; Guan, K.L.; Malmberg, K.J.; et al. Metabolic reprograming via deletion of cish in human ipsc-derived nk cells promotes in vivo persistence and enhances anti-tumor activity. Cell Stem Cell 2020, 27, 224–237.e226. [Google Scholar] [CrossRef]
- Kumar, S.; Jeong, Y.; Ashraf, M.U.; Bae, Y.-S. Dendritic Cell-Mediated Th2 Immunity and Immune Disorders. Int. J. Mol. Sci. 2019, 20, 2159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miah, M.A.; Bae, Y.S. Regulation of dc development and dc-mediated t-cell immunity via cish. Oncoimmunology 2013, 2, e23404. [Google Scholar] [CrossRef] [PubMed]
- Anguille, S.; Van Tendeloo, V.; Berneman, Z. Dendritic cell-based therapeutic vaccination for acute myeloid leukemia. Bull. Cancer 2012, 99, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.A.; Lu, J.W.; Lin, T.Y.; Tsai, C.H.; Chou, W.C.; Lin, C.C.; Kuo, Y.Y.; Liu, C.Y.; Tseng, M.H.; Chiang, Y.C.; et al. Clini-co-biological significance of suppressor of cytokine signaling 1 expression in acute myeloid leukemia. Blood Cancer J. 2017, 7, e588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musa, J.; Aynaud, M.-M.; Mirabeau, O.; Delattre, O.; Grünewald, T.G. MYBL2 (B-Myb): A central regulator of cell proliferation, cell survival and differentiation involved in tumorigenesis. Cell Death Dis. 2017, 8, e2895. [Google Scholar] [CrossRef]
- Wang, J.Y.; Chen, L.J.; Qiang, P. The potential role of n6-methyladenosine (m6a) demethylase fat mass and obesity-associated gene (fto) in human cancers. Onco Targets Ther. 2020, 13, 12845–12856. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, M.; Kapoor, U.; Jantsch, M.F. Understanding RNA modifications: The promises and technological bottlenecks of the ‘epitranscriptome’. Open Biol. 2017, 7. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conventional Drugs | ||||
|---|---|---|---|---|
| Brand/Other Name | Drug | Drug Type | Clinical Trial | Ref. |
| Rydapt (Novartis) | Midostaurin | Multikinase FLT3 inhibitor | FDA approved | [29] |
| Vyxeos (Jazz Pharma) | CPX-351 | cytarabine and daunorubicin combination (5:1 molar ratio) | FDA approved | [30,31] |
| Formerly SNS-595 (Sunesis Pharma) | Vosaroxin | Topoisomerase II inhibitor: anticancer quinolone derivative (AQD) | FDA approved | [32] |
| ASP2215, Xospata (Astellas Pharma) | Gilteritinib | Dual inhibitor of FLT3/AXL | FDA approved | [33,34] |
| Venclexta, Venclyxto (AbbVie, Genentech) | Venetoclax | Bcl2-inhibitor | Phase III | NCT02993523 NCT03069352 [35] |
| Epigenetic Drugs | ||||
| Vidaza | Azacitidine | DNMT inhibitor (Hypomethylating agent) | FDA approved | [19] |
| Dacogen | Decitabine | DNMT inhibitor (Hypomethylating agent) | FDA approved | [19] |
| Tibsovo (AG-120) | Ivosidenib | IDH1 inhibitor | FDA approved | [23,24] |
| Idhifa (AG-221) | Enasidenib | IDH2 inhibitor | FDA approved | |
| Beleodaq (PXD101) | Belinostat | Pan-HDAC inhibitor | Phase II | [36] |
| Zolin | Vorinostat | Pan-HDAC inhibitor | Phase I/II | [37] |
| – | Panobinostat | Pan-HDAC inhibitor | Phase I/II | [38,39,40] |
| Istodax | Romidepsin | Selective HDAC inhibitor | Preclinical | [28] |
| SGI-110 | Guadecitabine | Dinucleotide of decitabine and deoxyguanosine | Phase III | NCT02348489 |
| Monoclonal Antibodies (mAbs) | ||||
| GO (Mylotarg, Wyeth Pharma) | Gemtuzumab Ozogamicin | CD33-targeted | Phase 2 | NCT03374332 NCT00372593 |
| SGN-CD33A (Seattle Genetics) | Vadastuximab talirine | CD33-targeted | Phase I | [41] |
| Darzalex Faspro (Janssen Biotech) | Daratumumab | CD38-targeted | Phase II | NCT03067571 |
| RG7356 | CD44-targeted | Phase I | [42] | |
| Iomab-B | Apamistamab | CD45-targeted | Phase III, SIERRA | NCT02665065 |
| RNA Modifiers | Disease Condition | Target | Mechanism of Action | Therapeutic Strategies | Ref. |
|---|---|---|---|---|---|
| Writers Mettl3 | upregulated in AML | MYB/MYC, Bcl2, pTEN, Spl1 (PU.1), PI3K-AKT pathway | Inhibiting cell-differentiation and apoptosis and promoting cell proliferation (self-renewal) capacity. | Selective Mettl-3/14 inhibitor or Targeted therapy | [43,46] |
| Mettl-14 | [46,47,48,49] | ||||
| Erasers FTO | upregulated in APL and LSC/LICs | Myc, CEBPA | Regulated by FTO related with Leukaemia | [50] | |
| ASB2, RARA, MYC, LILRB4, CEBPA | By inhibiting ASB2 and RARA gene targets as well as ATRA-induced cell-differentiation and apoptosis | Targeted silencing or specific FTO/ALKBH5 inhibitor | [51] | ||
| ALKBH5 | TACC3, Myc, P21 | By impairing self-renewal capacities | [52] | ||
| Readers YTHDF2 | upregulated in AML | Tal1 | YTHDF2 inhibit the expression of essential Tal1 gene. | Targeted silencing/ therapy | [53] |
| YTHDF2 | upregulated | TNFα | YTHDF2 inhibit the expression of TNFα required for cell necrosis and apoptosis. | [54] |
| Name | Therapeutic Application | Ref. | ||
|---|---|---|---|---|
| Disease | Epigenetic Inhibitor | Function | ||
| Writers Mettl3, Mettl14 Inhibitors | AML | STM2457 (STC-15) | By selective inhibition of Mettl-3/14 (proposed human clinical trial in 2022 by Storm Therapeutics) | [45,47,48] |
| Metabolic disease | SAH (S-Adenosyl-homo cysteine) | Selective inhibition of Mettl-3/14 by targeting endogenous metabolite | [76] | |
| Cancer | UZH1/UZH1a | Promotes apoptosis and enhance T-cell anti-tumour activity | [45,77] | |
| Erasers FTO Inhibitors | Upregulated in AML | FB23 | Selective FTO inhibitor works by retaining m6A-demethylase activity in LSCs | [75,78] |
| FB23-2 | [7,75] | |||
| R-2-hydroxyglutarate (R-2HG) | Selective FTO inhibitor targeting A/MYC/CEBPα signalling and increase anti-tumour immunity | [79] | ||
| CS1 and CS2 | Selective FTO inhibitor targeting LILRB4 immune checkpoint gene | [80,81] | ||
| Glioblastoma Stem Cells | Non-steroidal anti-inflammatory drug, (MA2) an ethyl ester form of meclofenamic acid | Selective FTO inhibitor targeting ADAM19 gene by m6A hyper-methylation | [82,83] | |
| Triple-negative inflammatory breast cancer | MO-I-500 | Selective inhibitor of FTO targeting IRX3 gene in SUM149-MA cells | [84] | |
| CNS (epilepsy) | BBB-penetrating small molecule inhibitor of FTO | First FTO inhibitor with anticonvulsant activity by targeting various microRNAs | [85] | |
| Readers YTHDF2 Inhibitors | AML | BET inhibitor (OTX015) | Bromodomain inhibition | [86,87,88] |
| MB-3 inhibitor of KAT2A | MB-3 inhibits the expression of KAT2A gene | [89,90] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, S.; Nagpal, R.; Kumar, A.; Ashraf, M.U.; Bae, Y.-S. Immunotherapeutic Potential of m6A-Modifiers and MicroRNAs in Controlling Acute Myeloid Leukaemia. Biomedicines 2021, 9, 690. https://doi.org/10.3390/biomedicines9060690
Kumar S, Nagpal R, Kumar A, Ashraf MU, Bae Y-S. Immunotherapeutic Potential of m6A-Modifiers and MicroRNAs in Controlling Acute Myeloid Leukaemia. Biomedicines. 2021; 9(6):690. https://doi.org/10.3390/biomedicines9060690
Chicago/Turabian StyleKumar, Sunil, Ravinder Nagpal, Amit Kumar, Muhammad Umer Ashraf, and Yong-Soo Bae. 2021. "Immunotherapeutic Potential of m6A-Modifiers and MicroRNAs in Controlling Acute Myeloid Leukaemia" Biomedicines 9, no. 6: 690. https://doi.org/10.3390/biomedicines9060690
APA StyleKumar, S., Nagpal, R., Kumar, A., Ashraf, M. U., & Bae, Y. -S. (2021). Immunotherapeutic Potential of m6A-Modifiers and MicroRNAs in Controlling Acute Myeloid Leukaemia. Biomedicines, 9(6), 690. https://doi.org/10.3390/biomedicines9060690