
Therapeutic Potential of PARP Inhibitors in the Treatment of Gastrointestinal Cancers



Abstract
:1. Introduction
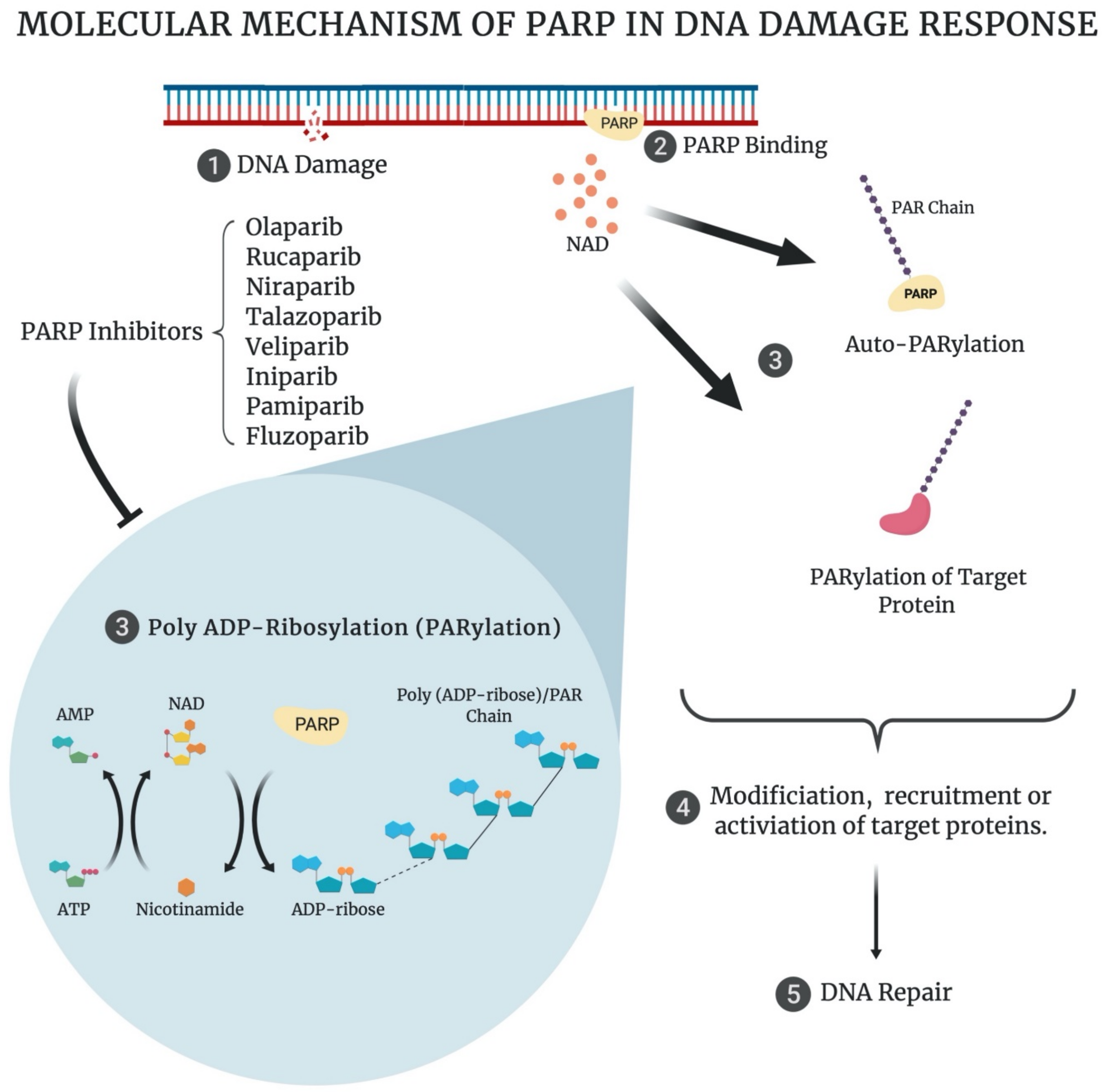
2. Poly (ADP-Ribose) Polymerase and the DNA Damage Response (DDR)
3. PARP Inhibitors and ‘Synthetic Lethality’
4. Current Clinical Applications of PARP Inhibitors in Cancer
5. The Role of the DDR PARP Enzymes in GI Cancer Development and Progression
5.1. Oesophageal Cancer
5.2. Gastric Cancer
5.3. Hepatic Cancer
5.4. Pancreatic Cancer
5.5. Colorectal Cancer
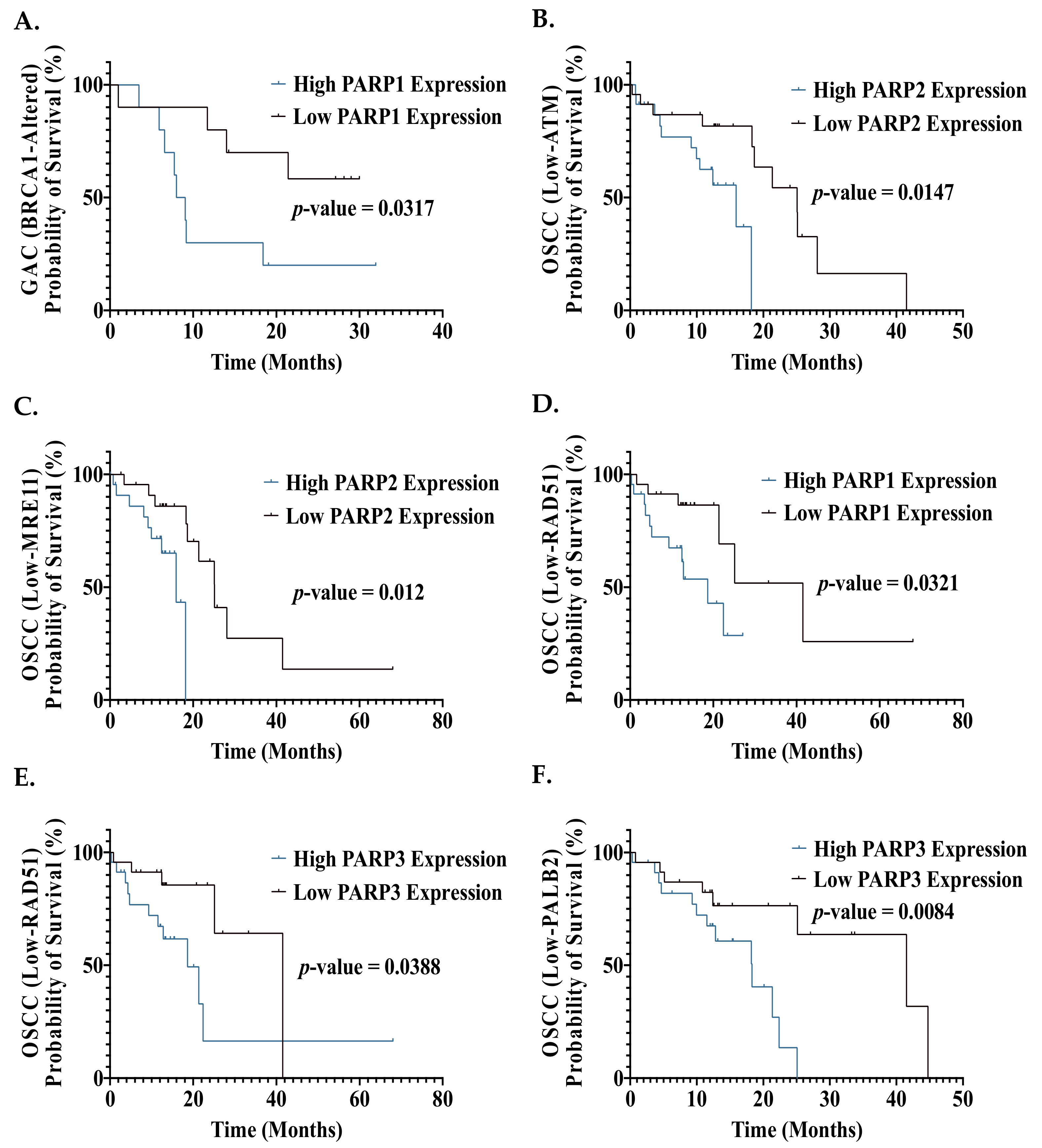
6. Homologous Recombination Status: A Predictive Biomarker of PARP Inhibitor Sensitivity in GI Cancers
7. The Role of DDR PARP Enzymes in the Response of GI Cancer to Radiation and Chemotherapy
7.1. Pre-Clinical Evidence
7.1.1. Radiotherapy
7.1.2. Chemotherapy
7.2. Clinical Trials Assessing PARPi and Cytotoxic Therapy Combination Treatment Approaches
7.2.1. Oesophageal Cancer
7.2.2. Gastric Cancer
7.2.3. Hepatic and Biliary Cancer
7.2.4. Pancreatic Cancer
7.2.5. Colorectal Cancer
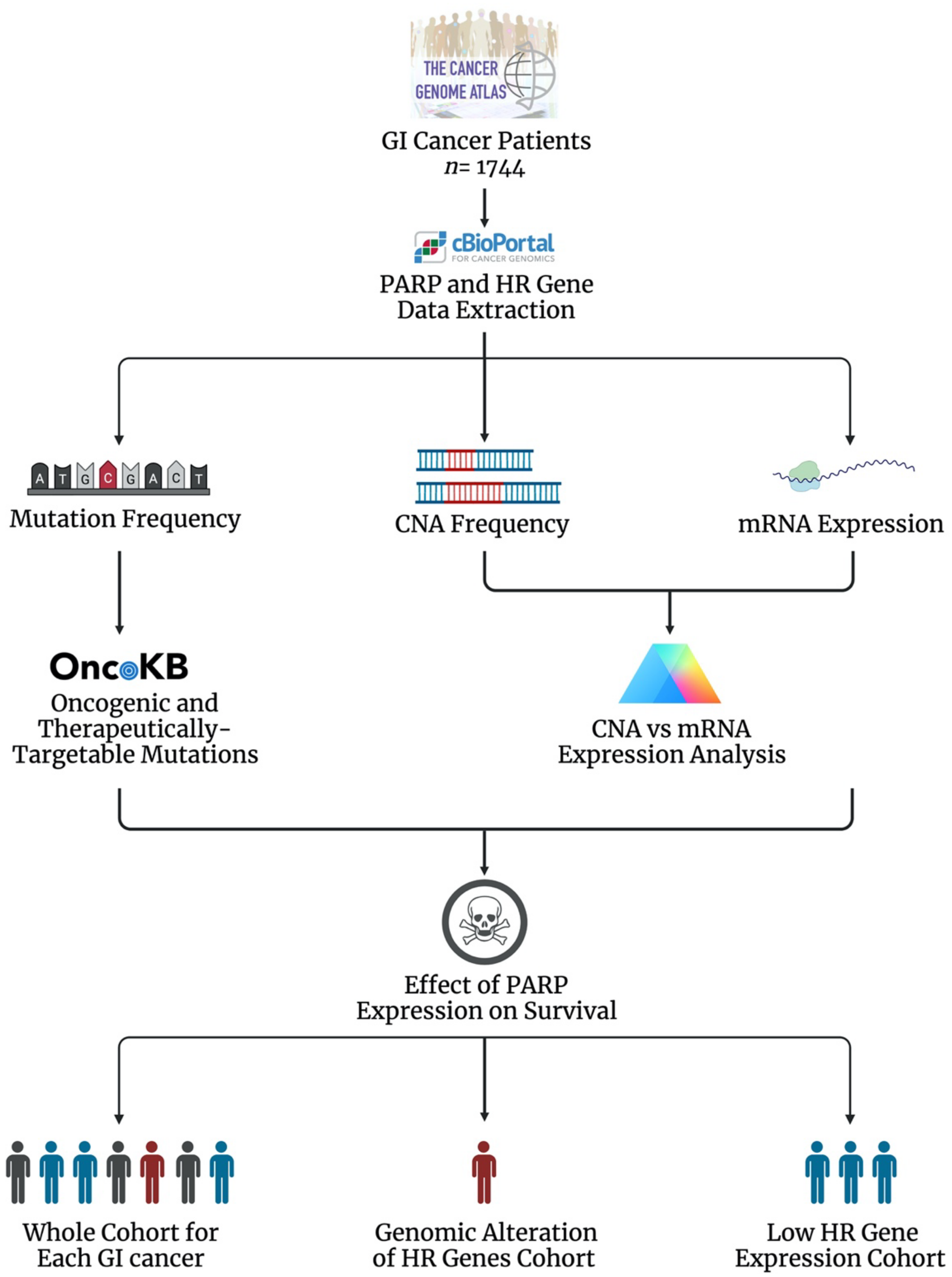
8. The Landscape of PARP and HR in Gastrointestinal Tumours
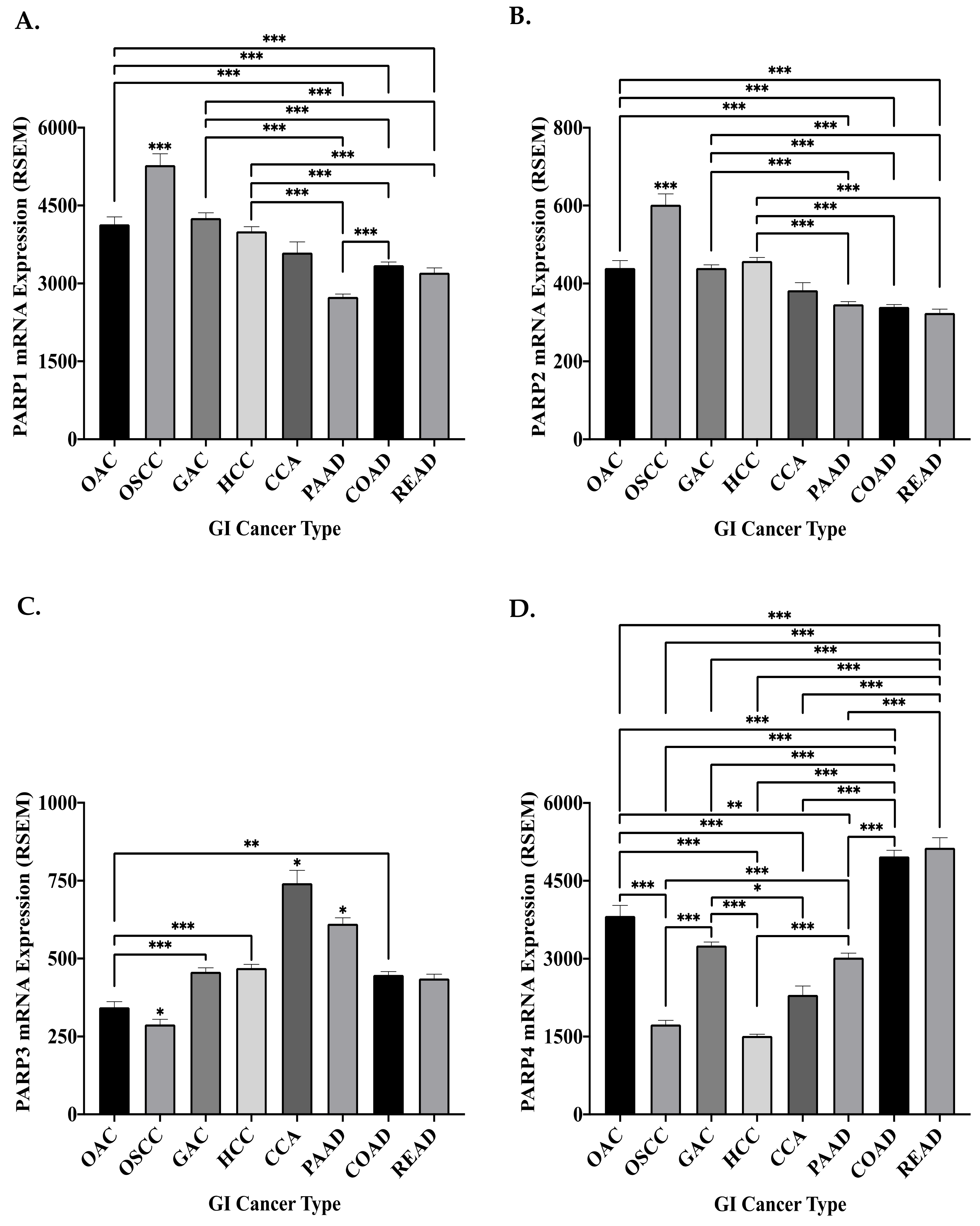
8.1. The Genomic and Transcriptomic Landscape of the DDR PARP Genes in GI Cancers
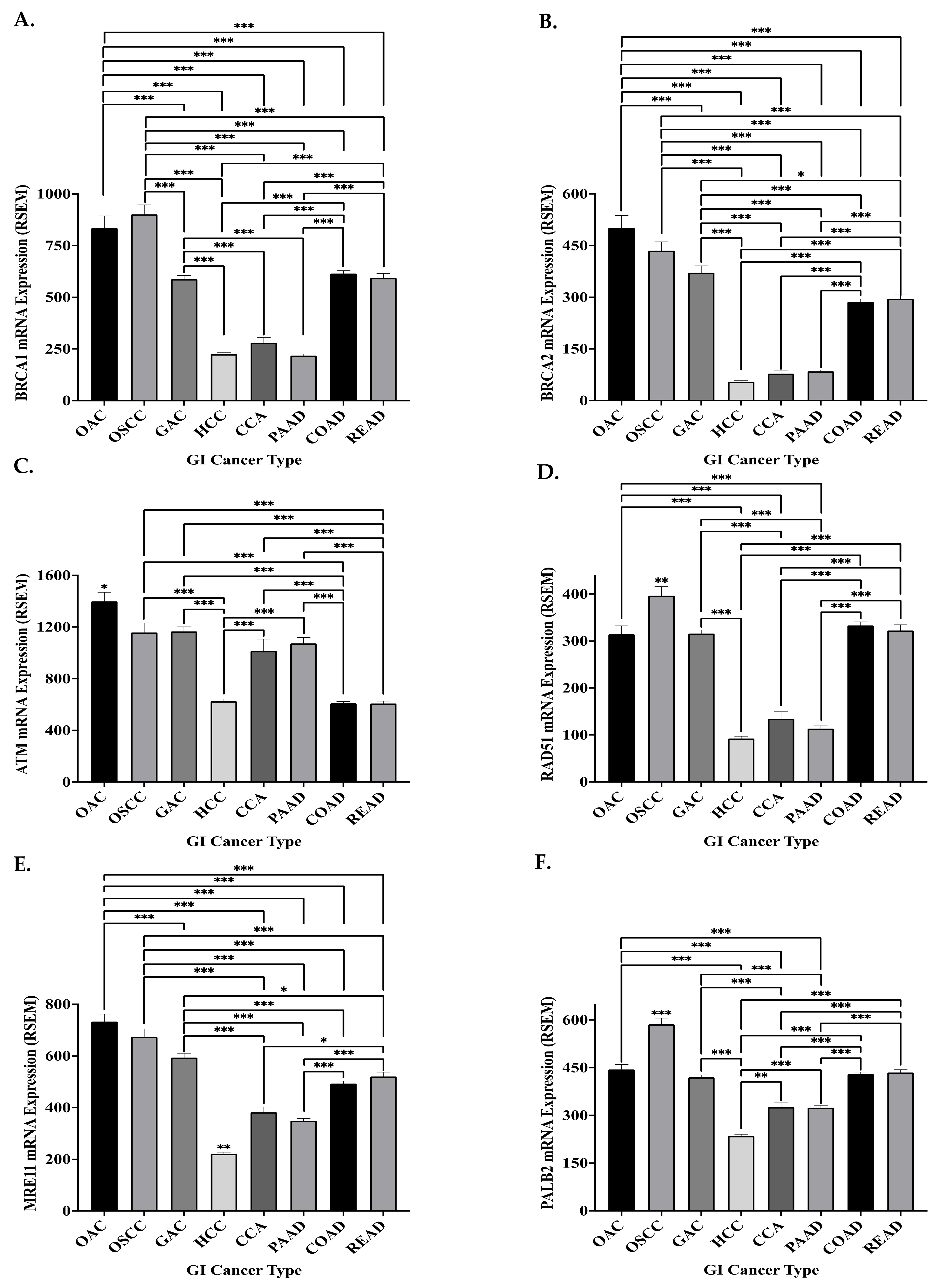
8.2. The Genomic and Transcriptomic Landscape of Key HR Genes in GI Cancers
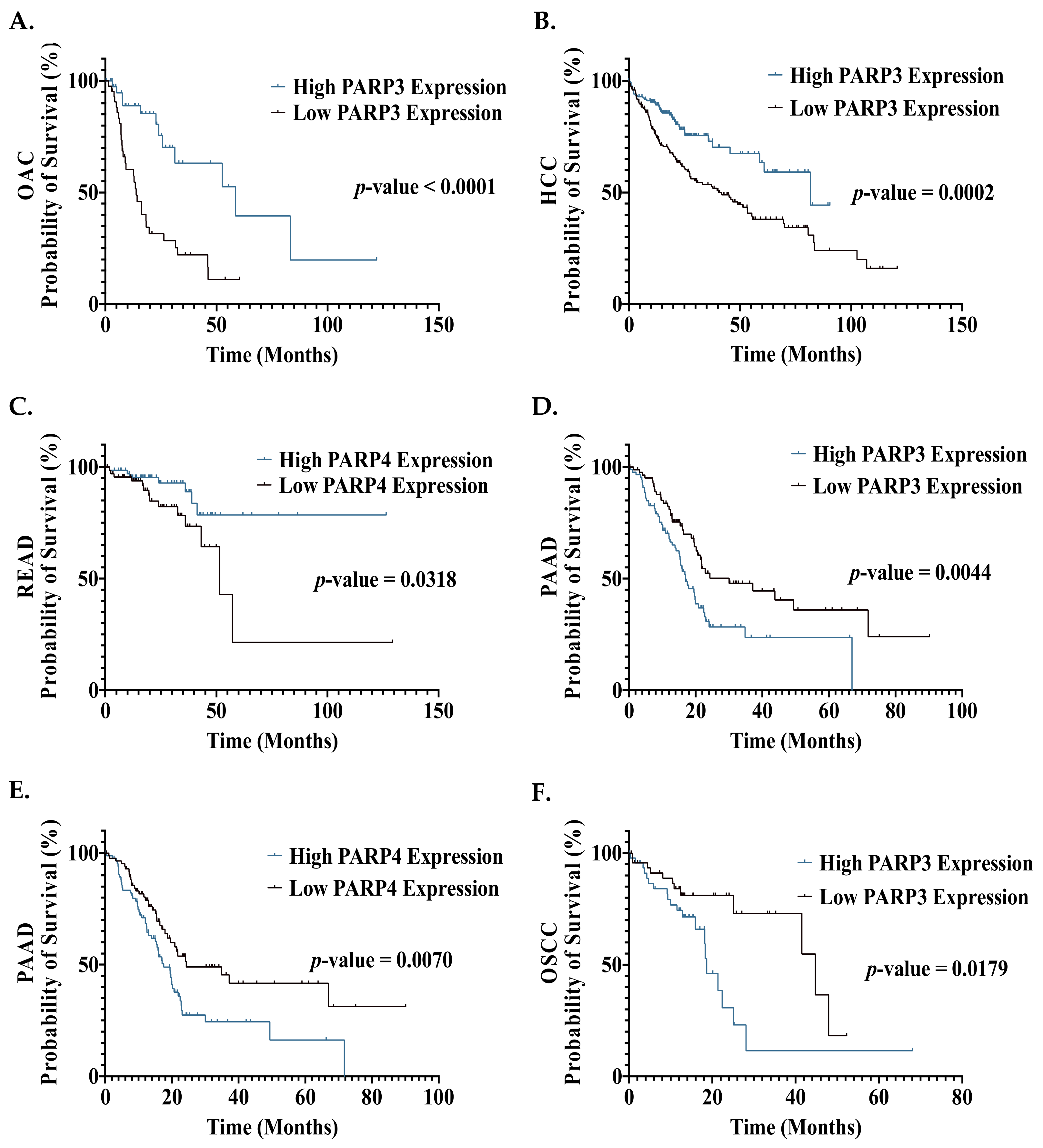
8.3. The Prognostic Effect of PARP mRNA Expression in GI Cancers
9. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References and Note
- The International Agency for Research on Cancer (IARC). Cancer Fact Sheets. Available online: http://gco.iarc.fr/today/fact-sheets-cancers (accessed on 15 January 2020).
- Allemani, C.; Matsuda, T.; Di Carlo, V.; Harewood, R.; Matz, M.; Nikšić, M.; Bonaventure, A.; Valkov, M.; Johnson, C.J.; Estève, J.; et al. Global surveillance of trends in cancer survival 2000–14 (CONCORD-3): Analysis of individual records for 37,513,025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet 2018, 391, 1023–1075. [Google Scholar] [CrossRef] [Green Version]
- Zare, A.; Mahmoodi, M.; Mohammad, K.; Zeraati, H.; Hosseini, M.; Holakouie Naieni, K. Factors Affecting the Survival of Patients with Gastric Cancer Undergone Surgery at Iran Cancer Institute: Univariate and Multivariate Analyses. Iran. J. Public Health 2014, 43, 800–808. [Google Scholar]
- Tustumi, F.; Kimura, C.M.S.; Takeda, F.R.; Uema, R.H.; Salum, R.A.A.; Ribeiro-Junior, U.; Cecconello, I. Prognostic factors and survival analysis in esophageal carcinoma. Arq. Bras. Cir. Dig. 2016, 29, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liu, H.; Chen, Y.; Zhu, C.; Fang, W.; Yu, Z.; Mao, W.; Xiang, J.; Han, Y.; Chen, Z.; et al. Neoadjuvant Chemoradiotherapy Followed by Surgery Versus Surgery Alone for Locally Advanced Squamous Cell Carcinoma of the Esophagus (NEOCRTEC5010): A Phase III Multicenter, Randomized, Open-Label Clinical Trial. J. Clin. Oncol. 2018, 36, 2796–2803. [Google Scholar] [CrossRef]
- Van Hagen, P.; Hulshof, M.C.C.M.; van Lanschot, J.J.B.; Steyerberg, E.W.; Henegouwen, M.I.v.B.; Wijnhoven, B.P.L.; Richel, D.J.; Nieuwenhuijzen, G.A.P.; Hospers, G.A.P.; Bonenkamp, J.J.; et al. Preoperative Chemoradiotherapy for Esophageal or Junctional Cancer. N. Engl. J. Med. 2012, 366, 2074–2084. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Shan, F.; Wang, Y.; Zhang, Y.; Zhang, L.; Li, S.; Jia, Y.; Xue, K.; Miao, R.; Li, Z.; et al. Correlation of pathological complete response with survival after neoadjuvant chemotherapy in gastric or gastroesophageal junction cancer treated with radical surgery: A meta-analysis. PLoS ONE 2018, 13, e0189294. [Google Scholar] [CrossRef] [PubMed]
- Zorcolo, L.; Rosman, A.S.; Restivo, A.; Pisano, M.; Nigri, G.R.; Fancellu, A.; Melis, M. Complete Pathologic Response after Combined Modality Treatment for Rectal Cancer and Long-Term Survival: A Meta-Analysis. Ann. Surg. Oncol. 2012, 19, 2822–2832. [Google Scholar] [CrossRef] [PubMed]
- Epidermoid anal cancer: Results from the UKCCCR randomised trial of radiotherapy alone versus radiotherapy, 5-fluorouracil, and mitomycin. Lancet 1996, 348, 1049–1054. [CrossRef]
- Christians, K.K.; Tsai, S.; Mahmoud, A.; Ritch, P.; Thomas, J.P.; Wiebe, L.; Kelly, T.; Erickson, B.; Wang, H.; Evans, D.B.; et al. Neoadjuvant FOLFIRINOX for borderline resectable pancreas cancer: A new treatment paradigm? Oncologist 2014, 19, 266–274. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.E.; Wo, J.Y.; Ryan, D.P.; Jiang, W.; Yeap, B.Y.; Drapek, L.C.; Blaszkowsky, L.S.; Kwak, E.L.; Allen, J.N.; Clark, J.W.; et al. Total Neoadjuvant Therapy With FOLFIRINOX Followed by Individualized Chemoradiotherapy for Borderline Resectable Pancreatic Adenocarcinoma: A Phase 2 Clinical Trial. JAMA Oncol. 2018, 4, 963–969. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Biau, J.; Chautard, E.; Verrelle, P.; Dutreix, M. Altering DNA Repair to Improve Radiation Therapy: Specific and Multiple Pathway Targeting. Front. Oncol. 2019, 9, 1009. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73 (Suppl. S1), e478s. [Google Scholar] [CrossRef] [PubMed]
- Maier, P.; Hartmann, L.; Wenz, F.; Herskind, C. Cellular Pathways in Response to Ionizing Radiation and Their Targetability for Tumor Radiosensitization. Int. J. Mol. Sci. 2016, 17, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torgovnick, A.; Schumacher, B. DNA repair mechanisms in cancer development and therapy. Front. Genet. 2015, 6, 157. [Google Scholar] [CrossRef] [Green Version]
- Blanpain, C.; Mohrin, M.; Sotiropoulou, P.A.; Passegué, E. DNA-Damage Response in Tissue-Specific and Cancer Stem Cells. Cell Stem Cell 2011, 8, 16–29. [Google Scholar] [CrossRef]
- Kiwerska, K.; Szyfter, K. DNA repair in cancer initiation, progression, and therapy-a double-edged sword. J. Appl. Genet. 2019, 60, 329–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza-Aghazadeh-Attari, M.; Darband, S.G.; Kaviani, M.; Mihanfar, A.; Aghazadeh Attari, J.; Yousefi, B.; Majidinia, M. DNA damage response and repair in colorectal cancer: Defects, regulation and therapeutic implications. DNA Repair 2018, 69, 34–52. [Google Scholar] [CrossRef]
- Rodrigues, A.S.; Gomes, B.C.; Martins, C.; Gromicho, M.; Oliveira, N.G.; Guerreiro, P.S.; Rueff, J. DNA Repair and Resistance to Cancer Therapy. In New Research Directions in DNA Repair; Chen, C., Ed.; InTechOpen: San Diego, CA, USA, 2013. [Google Scholar] [CrossRef]
- Vyas, S.; Matic, I.; Uchima, L.; Rood, J.; Zaja, R.; Hay, R.T.; Ahel, I.; Chang, P. Family-wide analysis of poly(ADP-ribose) polymerase activity. Nat. Commun. 2014, 5, 4426. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.O.; Khan, F.A.; Galindo-Campos, M.A.; Yélamos, J. Understanding specific functions of PARP-2: New lessons for cancer therapy. Am. J. Cancer Res. 2016, 6, 1842–1863. [Google Scholar]
- Sousa, F.G.; Matuo, R.; Soares, D.G.; Escargueil, A.E.; Henriques, J.A.P.; Larsen, A.K.; Saffi, J. PARPs and the DNA damage response. Carcinogenesis 2012, 33, 1433–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Eisemann, T.; Pascal, J.M. Poly(ADP-ribose) polymerase enzymes and the maintenance of genome integrity. Cell. Mol. Life Sci. 2020, 77, 19–33. [Google Scholar] [CrossRef]
- Boussios, S.; Karathanasi, A.; Cooke, D.; Neille, C.; Sadauskaite, A.; Moschetta, M.; Zakynthinakis-Kyriakou, N.; Pavlidis, N. PARP Inhibitors in Ovarian Cancer: The Route to “Ithaca”. Diagnostics 2019, 9, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, C.; Boehler, C.; Guirouilh Barbat, J.; Bonnet, M.-E.; Illuzzi, G.; Ronde, P.; Gauthier, L.R.; Magroun, N.; Rajendran, A.; Lopez, B.S.; et al. PARP3 affects the relative contribution of homologous recombination and nonhomologous end-joining pathways. Nucleic Acids Res. 2014, 42, 5616–5632. [Google Scholar] [CrossRef] [Green Version]
- Boehler, C.; Gauthier, L.R.; Mortusewicz, O.; Biard, D.S.; Saliou, J.-M.; Bresson, A.; Sanglier-Cianferani, S.; Smith, S.; Schreiber, V.; Boussin, F.; et al. Poly(ADP-ribose) polymerase 3 (PARP3), a newcomer in cellular response to DNA damage and mitotic progression. Proc. Natl. Acad. Sci. USA 2011, 108, 2783–2788. [Google Scholar] [CrossRef] [Green Version]
- Manke, I.A.; Lowery, D.M.; Nguyen, A.; Yaffe, M.B. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science 2003, 302, 636–639. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Kiyotani, K.; Yew, P.Y.; Kato, T.; Tamura, K.; Yap, K.L.; Nielsen, S.M.; Mester, J.L.; Eng, C.; Nakamura, Y.; et al. Germline PARP4 mutations in patients with primary thyroid and breast cancers. Endocr. Relat. Cancer 2016, 23, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Diagram created with BioRender.com.
- Wahlberg, E.; Karlberg, T.; Kouznetsova, E.; Markova, N.; Macchiarulo, A.; Thorsell, A.-G.; Pol, E.; Frostell, Å.; Ekblad, T.; Öncü, D.; et al. Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nat. Biotechnol. 2012, 30, 283–288. [Google Scholar] [CrossRef]
- Thorsell, A.-G.; Ekblad, T.; Karlberg, T.; Löw, M.; Pinto, A.F.; Trésaugues, L.; Moche, M.; Cohen, M.S.; Schüler, H. Structural Basis for Potency and Promiscuity in Poly(ADP-ribose) Polymerase (PARP) and Tankyrase Inhibitors. J. Med. Chem. 2017, 60, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.-y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Ashworth, A.; Lord, C.J.; Reis-Filho, J.S. Genetic Interactions in Cancer Progression and Treatment. Cell 2011, 145, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps317. [Google Scholar] [CrossRef]
- AstraZeneca Pharmaceuticals, LP. Lynparza (Olaparib); Food & Drug Administration: Silver Spring, MD, USA, 2021.
- GlaxoSmithKline. Zejula (Niraparib); Food & Drug Administration: Silver Spring, MD, USA, 2021.
- Clovis Oncology, Inc. Rubraca (Rucaparib); Food & Drug Administration: Silver Spring, MD, USA, 2020.
- Pfizer, Inc. Talzenna (Talazoparib); Food & Drug Administration: Silver Spring, MD, USA, 2020.
- EMA. Lynparza. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/lynparza (accessed on 23 June 2021).
- EMA. Zejula. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/zejula (accessed on 23 June 2021).
- EMA. Rubraca. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/rubraca (accessed on 23 June 2021).
- EMA. Talzenna. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/talzenna (accessed on 23 June 2021).
- Vilar, E.; Bartnik, C.M.; Stenzel, S.L.; Raskin, L.; Ahn, J.; Moreno, V.; Mukherjee, B.; Iniesta, M.D.; Morgan, M.A.; Rennert, G.; et al. MRE11 deficiency increases sensitivity to poly(ADP-ribose) polymerase inhibition in microsatellite unstable colorectal cancers. Cancer Res. 2011, 71, 2632–2642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, L.; Qin, Q.; Lu, J.; Liu, J.; Zhu, H.; Yang, X.; Zhang, C.; Xu, L.; Liu, Z.; Cai, J.; et al. Novel poly (ADP-ribose) polymerase inhibitor, AZD2281, enhances radiosensitivity of both normoxic and hypoxic esophageal squamous cancer cells. Dis. Esophagus 2016, 29, 215–223. [Google Scholar] [CrossRef]
- Boussios, S.; Karihtala, P.; Moschetta, M.; Karathanasi, A.; Sadauskaite, A.; Rassy, E.; Pavlidis, N. Combined Strategies with Poly (ADP-Ribose) Polymerase (PARP) Inhibitors for the Treatment of Ovarian Cancer: A Literature Review. Diagnostics 2019, 9, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonçalves, A.; Bertucci, A.; Bertucci, F. PARP Inhibitors in the Treatment of Early Breast Cancer: The Step Beyond? Cancers 2020, 12, 1378. [Google Scholar] [CrossRef]
- Kurnit, K.C.; Avila, M.; Hinchcliff, E.M.; Coleman, R.L.; Westin, S.N. PARP inhibition in the ovarian cancer patient: Current approvals and future directions. Pharmacol. Ther. 2020, 213, 107588. [Google Scholar] [CrossRef]
- Yamamoto, M.; Yamasaki, M.; Tsukao, Y.; Tanaka, K.; Miyazaki, Y.; Makino, T.; Takahashi, T.; Kurokawa, Y.; Nakajima, K.; Takiguchi, S.; et al. Poly (ADP-ribose) polymerase-1 inhibition decreases proliferation through G2/M arrest in esophageal squamous cell carcinoma. Oncol. Lett. 2017, 14, 1581–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Nasuno, T.; Mimaki, S.; Okamoto, M.; Esumi, H.; Tsuchihara, K. Effect of a poly(ADP-ribose) polymerase-1 inhibitor against esophageal squamous cell carcinoma cell lines. Cancer Sci. 2014, 105, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Liu, X.; Sun, J.; Yuan, Q.; Li, J. CircRAD23B facilitates proliferation and invasion of esophageal cancer cells by sponging miR-5095. Biochem. Biophys. Res. Commun. 2019, 516, 357–364. [Google Scholar] [CrossRef]
- Wu, H.; Li, Y.; Hou, Q.; Zhou, R.; Li, Z.; Wu, S.; Yu, J.; Jiang, M. Single-cell intratumoral stemness analysis reveals the involvement of cell cycle and DNA damage repair in two different types of esophageal cancer. Oncol. Rep. 2019, 41, 3201–3208. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, J.; Diaz, E.; Reya, T. Stem cells in cancer initiation and progression. J. Cell Biol. 2019, 219, e201911053. [Google Scholar] [CrossRef] [PubMed]
- De Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhang, Y.; Zhao, Y.; Gao, D.; Xing, J.; Liu, H. High PARP-1 expression is associated with tumor invasion and poor prognosis in gastric cancer. Oncol. Lett. 2016, 12, 3825–3835. [Google Scholar] [CrossRef]
- Afzal, H.; Yousaf, S.; Rahman, F.; Ahmed, M.W.; Akram, Z.; Akhtar Kayani, M.; Mahjabeen, I. PARP1: A potential biomarker for gastric cancer. Pathol. Res. Pract. 2019, 215, 152472. [Google Scholar] [CrossRef]
- Nossa, C.W.; Jain, P.; Tamilselvam, B.; Gupta, V.R.; Chen, L.-F.; Schreiber, V.; Desnoyers, S.; Blanke, S.R. Activation of the abundant nuclear factor poly(ADP-ribose) polymerase-1 by Helicobacter pylori. Proc. Natl. Acad. Sci. USA 2009, 106, 19998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.-P.; Hou, M.-C.; Lan, K.-H.; Li, C.-P.; Chao, Y.; Lin, H.-C.; Lee, S.-D. Helicobacter pylori-induced chronic inflammation causes telomere shortening of gastric mucosa by promoting PARP-1-mediated non-homologous end joining of DNA. Arch. Biochem. Biophys. 2016, 606, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-H.; Jang, K.Y.; Kim, M.J.; Yoon, S.; Jo, Y.; Kwon, S.M.; Kim, K.M.; Kwon, K.S.; Kim, C.Y.; Woo, H.G. Tumor suppressive effect of PARP1 and FOXO3A in gastric cancers and its clinical implications. Oncotarget 2015, 6, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, S.; Nomura, F.; Tomonaga, T.; Sunaga, M.; Noda, M.; Ebara, M.; Saisho, H. Expression of poly(ADP-ribose) polymerase in human hepatocellular carcinoma and analysis of biopsy specimens obtained under sonographic guidance. Oncol. Rep. 2004, 12, 821–825. [Google Scholar] [CrossRef]
- Qi, H.; Lu, Y.; Lv, J.; Wu, H.; Lu, J.; Zhang, C.; Zhang, S.; Bao, Q.; Zhang, X.; Xie, C.; et al. The long noncoding RNA lncPARP1 contributes to progression of hepatocellular carcinoma through up-regulation of PARP1. Biosci. Rep. 2018, 38, BSR20180703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, F.; Yaguchi, M.; Togawa, A.; Miyazaki, M.; Isobe, K.; Miyake, M.; Noda, M.; Nakai, T. Enhancement of poly-adenosine diphosphate-ribosylation in human hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2000, 15, 529–535. [Google Scholar] [CrossRef]
- Lin, L.; Zhang, Y.-D.; Chen, Z.-y.; Chen, Y.; Ren, C.-P. The clinicopathological significance of miR-149 and PARP-2 in hepatocellular carcinoma and their roles in chemo/radiotherapy. Tumor Biol. 2016, 37, 12339–12346. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liu, Z.; Wang, J.; Xie, H.; Li, J.; Cao, J.; Zhou, L.; Zheng, S. Global proteomic profiling in multistep hepatocarcinogenesis and identification of PARP1 as a novel molecular marker in hepatocellular carcinoma. Oncotarget 2016, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Quiles-Perez, R.; Muñoz-Gámez, J.A.; Ruiz-Extremera, Á.; O’Valle, F.; Sanjuán-Nuñez, L.; Martín-Álvarez, A.B.; Martín-Oliva, D.; Caballero, T.; Muñoz de Rueda, P.; León, J.; et al. Inhibition of poly adenosine diphosphate-ribose polymerase decreases hepatocellular carcinoma growth by modulation of tumor-related gene expression. Hepatology 2010, 51, 255–266. [Google Scholar] [CrossRef]
- Mao, X.; Du, S.; Yang, Z.; Zhang, L.; Peng, X.; Jiang, N.; Zhou, H. Inhibitors of PARP-1 exert inhibitory effects on the biological characteristics of hepatocellular carcinoma cells in vitro. Mol. Med. Rep. 2017, 16, 208–214. [Google Scholar] [CrossRef] [Green Version]
- Radnai, B.; Antus, C.; Racz, B.; Engelmann, P.; Priber, J.K.; Tucsek, Z.; Veres, B.; Turi, Z.; Lorand, T.; Sumegi, B.; et al. Protective effect of the poly(ADP-ribose) polymerase inhibitor PJ34 on mitochondrial depolarization-mediated cell death in hepatocellular carcinoma cells involves attenuation of c-Jun N-terminal kinase-2 and protein kinase B/Akt activation. Mol. Cancer 2012, 11, 34. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, C.; Tian, Y.; Zhang, F.; Xu, W.; Li, X.; Shu, Z.; Wang, Y.; Huang, K.; Huang, D. Inhibition of Poly(ADP-Ribose) Polymerase-1 Protects Chronic Alcoholic Liver Injury. Am. J. Pathol. 2016, 186, 3117–3130. [Google Scholar] [CrossRef] [Green Version]
- Gottschalk, A.J.; Timinszky, G.; Kong, S.E.; Jin, J.; Cai, Y.; Swanson, S.K.; Washburn, M.P.; Florens, L.; Ladurner, A.G.; Conaway, J.W.; et al. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc. Natl. Acad. Sci. USA 2009, 106, 13770–13774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, N.-F.; Hu, L.; Fung, J.M.; Xie, D.; Zheng, B.-J.; Chen, L.; Tang, D.-J.; Fu, L.; Wu, Z.; Chen, M.; et al. Isolation and characterization of a novel oncogene, amplified in liver cancer 1, within a commonly amplified region at 1q21 in hepatocellular carcinoma. Hepatology 2008, 47, 503–510. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhang, W.; Wang, Z.; Liu, S. Long non-coding RNA PTTG3P functions as an oncogene by sponging miR-383 and up-regulating CCND1 and PARP2 in hepatocellular carcinoma. BMC Cancer 2019, 19, 731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, D.; Huang, H.; Wang, J.; Zhao, Y.; Hu, X.; He, F.; Yu, L.; Wu, J. NF90 regulates PARP1 mRNA stability in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2017, 488, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Longnecker, D.S. Pathology of Exocrine Pancreatic Neoplasms. In UpToDate Clinical Summaries, 35th ed.; Goldberg, R.M., Ed.; Wolters Kluwer: Philadelphia, PA, USA, 2020. [Google Scholar]
- Martinez-Bosch, N.; Iglesias, M.; Munne-Collado, J.; Martinez-Caceres, C.; Moreno, M.; Guerra, C.; Yelamos, J.; Navarro, P. Parp-1 genetic ablation in Ela-myc mice unveils novel roles for Parp-1 in pancreatic cancer. J. Pathol. 2014, 234, 214–227. [Google Scholar] [CrossRef]
- Klauschen, F.; von Winterfeld, M.; Stenzinger, A.; Sinn, B.V.; Budczies, J.; Kamphues, C.; Bahra, M.; Wittschieber, D.; Weichert, W.; Striefler, J.; et al. High nuclear poly-(ADP-ribose)-polymerase expression is prognostic of improved survival in pancreatic cancer. Histopathology 2012, 61, 409–416. [Google Scholar] [CrossRef]
- Xu, F.; Sun, Y.; Yang, S.Z.; Zhou, T.; Jhala, N.; McDonald, J.; Chen, Y. Cytoplasmic PARP-1 promotes pancreatic cancer tumorigenesis and resistance. Int. J. Cancer 2019, 145, 474–483. [Google Scholar] [CrossRef]
- Vyas, S.; Chesarone-Cataldo, M.; Todorova, T.; Huang, Y.-H.; Chang, P. A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nat. Commun. 2013, 4, 2240. [Google Scholar] [CrossRef] [Green Version]
- Nosho, K.; Yamamoto, H.; Mikami, M.; Taniguchi, H.; Takahashi, T.; Adachi, Y.; Imamura, A.; Imai, K.; Shinomura, Y. Overexpression of poly(ADP-ribose) polymerase-1 (PARP-1) in the early stage of colorectal carcinogenesis. Eur. J. Cancer 2006, 42, 2374–2381. [Google Scholar] [CrossRef]
- Dziaman, T.; Ludwiczak, H.; Ciesla, J.M.; Banaszkiewicz, Z.; Winczura, A.; Chmielarczyk, M.; Wisniewska, E.; Marszalek, A.; Tudek, B.; Olinski, R. PARP-1 Expression is Increased in Colon Adenoma and Carcinoma and Correlates with OGG1. PLoS ONE 2014, 9, e115558. [Google Scholar] [CrossRef] [PubMed]
- Dörsam, B.; Seiwert, N.; Foersch, S.; Stroh, S.; Nagel, G.; Begaliew, D.; Diehl, E.; Kraus, A.; McKeague, M.; Minneker, V.; et al. PARP-1 protects against colorectal tumor induction, but promotes inflammation-driven colorectal tumor progression. Proc. Natl. Acad. Sci. USA 2018, 115, E4061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, L.; Threadgill, M.D.; Wang, Y.; Li, M. Effect of Poly (ADP-ribose) Polymerase-1 Inhibition on the Proliferation of Murine Colon Carcinoma CT26 Cells. Pathol. Oncol. Res. 2008, 15, 323. [Google Scholar] [CrossRef]
- Li, M.; Threadgill, M.D.; Wang, Y.; Cai, L.; Lin, X. Poly(ADP-Ribose) Polymerase Inhibition Down-Regulates Expression of Metastasis-Related Genes in CT26 Colon Carcinoma Cells. Pathobiology 2009, 76, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Hiroshi, Y.; Takashi, T.; Masatoshi, H.; Hideaki, K.; Shigekazu, H.; Terumitsu, S.; Hiroyuki, Y.; Toru, Y.; Tohru, N.; Yutaka, T. Elevated Expression of Poly(ADP-Ribose) Polymerase-1 is Associated with Liver Metastasis in Colorectal Cancer. Acta Med. Nagasaki 2002, 47, 111–115. [Google Scholar]
- Abdelrahman, A.E.; Ibrahim, D.A.; El-Azony, A.; Alnagar, A.A.; Ibrahim, A. ERCC1, PARP-1, and AQP1 as predictive biomarkers in colon cancer patients receiving adjuvant chemotherapy. Cancer Biomark 2020, 27, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Young, E.L.; Thompson, B.A.; Neklason, D.W.; Firpo, M.A.; Werner, T.; Bell, R.; Berger, J.; Fraser, A.; Gammon, A.; Koptiuch, C.; et al. Pancreatic cancer as a sentinel for hereditary cancer predisposition. BMC Cancer 2018, 18, 697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shindo, K.; Yu, J.; Suenaga, M.; Fesharakizadeh, S.; Cho, C.; Macgregor-Das, A.; Siddiqui, A.; Witmer, P.D.; Tamura, K.; Song, T.J.; et al. Deleterious Germline Mutations in Patients With Apparently Sporadic Pancreatic Adenocarcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 3382–3390. [Google Scholar] [CrossRef]
- Lowery, M.A.; Wong, W.; Jordan, E.J.; Lee, J.W.; Kemel, Y.; Vijai, J.; Mandelker, D.; Zehir, A.; Capanu, M.; Salo-Mullen, E.; et al. Prospective Evaluation of Germline Alterations in Patients with Exocrine Pancreatic Neoplasms. J. Natl. Cancer Inst. 2018, 110, 1067–1074. [Google Scholar] [CrossRef] [Green Version]
- Schneider, R.; Slater, E.P.; Sina, M.; Habbe, N.; Fendrich, V.; Matthäi, E.; Langer, P.; Bartsch, D.K. German national case collection for familial pancreatic cancer (FaPaCa): Ten years experience. Fam. Cancer 2011, 10, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Varadhachary, G.R.; Sela, T.; Fogelman, D.R.; Halperin, N.; Shroff, R.T.; Halparin, S.; Xiao, L.; Aderka, D.; Maitra, A.; et al. Phase II study of olaparib for BRCAness phenotype in pancreatic cancer. J. Clin. Oncol. 2018, 36 (Suppl. S4), 297. [Google Scholar] [CrossRef]
- Binder, K.A.R.; Mick, R.; Hara, M.; Teitelbaum, U.; Karasic, T.; Schneider, C.; Dwyer, P.J.; Carpenter, E.; Pantel, A.; Makvandi, M.; et al. Abstract CT234: A Phase II, single arm study of maintenance rucaparib in patients with platinum-sensitive advanced pancreatic cancer and a pathogenic germline or somatic mutation in BRCA1, BRCA2 or PALB2. Cancer Res. 2019, 79 (Suppl. S13), CT234. [Google Scholar] [CrossRef]
- Kasi, A.; Chalise, P.; Williamson, S.K.; Baranda, J.C.; Sun, W.; Al-Rajabi, R.M.d.T.; Saeed, A.; Kumer, S.; Schmitt, T.; Foster, C.; et al. Niraparib in metastatic pancreatic cancer after previous chemotherapy (NIRA-PANC): A phase 2 trial. J. Clin. Oncol. 2019, 37 (Suppl. S15), TPS4168. [Google Scholar] [CrossRef]
- Sakogawa, K.; Aoki, Y.; Misumi, K.; Hamai, Y.; Emi, M.; Hihara, J.; Shi, L.; Kono, K.; Horikoshi, Y.; Sun, J.; et al. Involvement of homologous recombination in the synergism between cisplatin and poly (ADP-ribose) polymerase inhibition. Cancer Sci. 2013, 104, 1593–1599. [Google Scholar] [CrossRef] [Green Version]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. γH2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef]
- Mamdani, H.; Mehta, R.; Fountzilas, C.; Radovich, M.; Perkins, S.; Jalal, S.I. A phase II study evaluating safety and efficacy of niraparib in patients with previously treated homologous recombination (HR) defective or loss of heterozygosity (LOH) high-metastatic esophageal/GEJ/proximal gastric adenocarcinoma: A Big Ten Cancer Research Consortium study. J. Clin. Oncol. 2020, 38 (Suppl. S4), TPS472. [Google Scholar] [CrossRef]
- Min, A.; Im, S.A.; Yoon, Y.K.; Song, S.H.; Nam, H.J.; Hur, H.S.; Kim, H.P.; Lee, K.H.; Han, S.W.; Oh, D.Y.; et al. RAD51C-deficient cancer cells are highly sensitive to the PARP inhibitor olaparib. Mol. Cancer 2013, 12, 865–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, E.; Williamson, C.T.; Ye, R.; Elegbede, A.; Peterson, L.; Lees-Miller, S.P.; Bebb, D.G. Low ATM protein expression and depletion of p53 correlates with olaparib sensitivity in gastric cancer cell lines. Cell Cycle 2014, 13, 2129–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, D.H. A Phase II Study of Olaparib in Patients with Advanced Biliary Tract Cancer with Aberrant DNA Repair Gene Mutations. Available online: https://clinicaltrials.gov/ct2/show/NCT04042831?term=olaparib&recrs=abdfm&cond=Biliary+Cancer&draw=2&rank=1 (accessed on 3 March 2021).
- Wang, C.; Jette, N.; Moussienko, D.; Bebb, D.G.; Lees-Miller, S.P. ATM-Deficient Colorectal Cancer Cells Are Sensitive to the PARP Inhibitor Olaparib. Transl. Oncol. 2017, 10, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef]
- Leichman, L.; Groshen, S.; O’Neil, B.H.; Messersmith, W.; Berlin, J.; Chan, E.; Leichman, C.G.; Cohen, S.J.; Cohen, D.; Lenz, H.-J.; et al. Phase II Study of Olaparib (AZD-2281) After Standard Systemic Therapies for Disseminated Colorectal Cancer. Oncologist 2016, 21, 172–177. [Google Scholar] [CrossRef] [Green Version]
- Giannini, G.; Ristori, E.; Cerignoli, F.; Rinaldi, C.; Zani, M.; Viel, A.; Ottini, L.; Crescenzi, M.; Martinotti, S.; Bignami, M.; et al. Human MRE11 is inactivated in mismatch repair-deficient cancers. EMBO Rep. 2002, 3, 248–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.; Kamjoo, M.; Thomas, H.D.; Kyle, S.; Pavlovska, I.; Babur, M.; Telfer, B.A.; Curtin, N.J.; Williams, K.J. The clinically active PARP inhibitor AG014699 ameliorates cardiotoxicity but does not enhance the efficacy of doxorubicin, despite improving tumor perfusion and radiation response in mice. Mol. Cancer Ther. 2011, 10, 2320–2329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastak, K.; Bhutra, S.; Parry, R.; Ford, J.M. Poly (ADP-ribose) polymerase inhibitor, an effective radiosensitizer in lung and pancreatic cancers. Oncotarget 2017, 8, 26344. [Google Scholar] [CrossRef] [Green Version]
- Lohse, I.; Kumareswaran, R.; Cao, P.; Pitcher, B.; Gallinger, S.; Bristow, R.G.; Hedley, D.W. Effects of Combined Treatment with Ionizing Radiation and the PARP Inhibitor Olaparib in BRCA Mutant and Wild Type Patient-Derived Pancreatic Cancer Xenografts. PLoS ONE 2016, 11, e0167272. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Huang, X.; Shuang, Z.; Lin, G.; Wang, J.; Duan, F.; Chen, J.; Li, S. PARP inhibitor olaparib sensitizes cholangiocarcinoma cells to radiation. Cancer Med. 2018, 7, 1285–1296. [Google Scholar] [CrossRef]
- Shelton, J.W.; Waxweiler, T.V.; Landry, J.; Gao, H.; Xu, Y.; Wang, L.; El-Rayes, B.; Shu, H.K. In vitro and in vivo enhancement of chemoradiation using the oral PARP inhibitor ABT-888 in colorectal cancer cells. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 469–476. [Google Scholar] [CrossRef]
- Hegan, D.C.; Lu, Y.; Stachelek, G.C.; Crosby, M.E.; Bindra, R.S.; Glazer, P.M. Inhibition of poly(ADP-ribose) polymerase down-regulates BRCA1 and RAD51 in a pathway mediated by E2F4 and p130. Proc. Natl. Acad. Sci. USA 2010, 107, 2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirai, T.; Saito, S.; Fujimori, H.; Matsushita, K.; Nishio, T.; Okayasu, R.; Masutani, M. Radiosensitization by PARP inhibition to proton beam irradiation in cancer cells. Biochem. Biophys. Res. Commun. 2016, 478, 234–240. [Google Scholar] [CrossRef]
- Hirai, T.; Shirai, H.; Fujimori, H.; Okayasu, R.; Sasai, K.; Masutani, M. Radiosensitization effect of poly(ADP-ribose) polymerase inhibition in cells exposed to low and high liner energy transfer radiation. Cancer Sci. 2012, 103, 1045–1050. [Google Scholar] [CrossRef]
- Calabrese, C.R.; Almassy, R.; Barton, S.; Batey, M.A.; Calvert, A.H.; Canan-Koch, S.; Durkacz, B.W.; Hostomsky, Z.; Kumpf, R.A.; Kyle, S.; et al. Anticancer Chemosensitization and Radiosensitization by the Novel Poly(ADP-ribose) Polymerase-1 Inhibitor AG14361. J. Natl. Cancer Inst. 2004, 96, 56–67. [Google Scholar] [CrossRef]
- Donawho, C.K.; Luo, Y.; Luo, Y.; Penning, T.D.; Bauch, J.L.; Bouska, J.J.; Bontcheva-Diaz, V.D.; Cox, B.F.; DeWeese, T.L.; Dillehay, L.E.; et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin. Cancer Res. 2007, 13, 2728–2737. [Google Scholar] [CrossRef] [Green Version]
- Grimes, D.R.; Partridge, M. A mechanistic investigation of the oxygen fixation hypothesis and oxygen enhancement ratio. Biomed. Phys. Eng. Express 2015, 1, 045209. [Google Scholar] [CrossRef]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enríquez, S.; Llop-Guevara, A.; Ibrahim, Y.H.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.M.; et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.K.; Coackley, C.; Krause, M.; Jalali, F.; Chan, N.; Bristow, R.G. A novel poly(ADP-ribose) polymerase inhibitor, ABT-888, radiosensitizes malignant human cell lines under hypoxia. Radiother. Oncol. 2008, 88, 258–268. [Google Scholar] [CrossRef]
- Dungey, F.A.; Löser, D.A.; Chalmers, A.J. Replication-Dependent Radiosensitization of Human Glioma Cells by Inhibition of Poly(ADP-Ribose) Polymerase: Mechanisms and Therapeutic Potential. Int. J. Radiat. Oncol. Biol. Phys. 2008, 72, 1188–1197. [Google Scholar] [CrossRef] [PubMed]
- Lynam-Lennon, N.; Reynolds, J.V.; Marignol, L.; Sheils, O.M.; Pidgeon, G.P.; Maher, S.G. MicroRNA-31 modulates tumour sensitivity to radiation in oesophageal adenocarcinoma. J. Mol. Med. 2012, 90, 1449–1458. [Google Scholar] [CrossRef]
- Zhang, J.; Stevens, M.F.; Bradshaw, T.D. Temozolomide: Mechanisms of action, repair and resistance. Curr. Mol. Pharm. 2012, 5, 102–114. [Google Scholar] [CrossRef]
- Reynolds, P.; Cooper, S.; Lomax, M.; O’Neill, P. Disruption of PARP1 function inhibits base excision repair of a sub-set of DNA lesions. Nucleic Acids Res. 2015, 43, 4028–4038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, K.; Minegaki, T.; Tanahashi, M.; Yamamoto, A.; Moriyama, Y.; Wada, A.; Matsumoto, A.; Ota, K.; Tanaka, M.; Masuda, U.; et al. Synergistic Effects of Olaparib and DNA-damaging Agents in Oesophageal Squamous Cell Carcinoma Cell Lines. Anticancer Res. 2019, 39, 1813–1820. [Google Scholar] [CrossRef]
- Liu, X.; Shi, Y.; Guan, R.; Donawho, C.; Luo, Y.; Palma, J.; Zhu, G.-d.; Johnson, E.F.; Rodriguez, L.E.; Ghoreishi-Haack, N.; et al. Potentiation of Temozolomide Cytotoxicity by Poly(ADP)Ribose Polymerase Inhibitor ABT-888 Requires a Conversion of Single-Stranded DNA Damages to Double-Stranded DNA Breaks. Mol. Cancer Res. 2008, 6, 1621. [Google Scholar] [CrossRef] [Green Version]
- Delaney, C.A.; Wang, L.-Z.; Kyle, S.; White, A.W.; Calvert, A.H.; Curtin, N.J.; Durkacz, B.W.; Hostomsky, Z.; Newell, D.R. Potentiation of Temozolomide and Topotecan Growth Inhibition and Cytotoxicity by Novel Poly(adenosine Diphosphoribose) Polymerase Inhibitors in a Panel of Human Tumor Cell Lines. Clin. Cancer Res. 2000, 6, 2860. [Google Scholar]
- Muñoz-Gámez, J.A.; López Viota, J.; Barrientos, A.; Carazo, Á.; Sanjuán-Nuñez, L.; Quiles-Perez, R.; Muñoz-de-Rueda, P.; Delgado, Á.; Ruiz-Extremera, Á.; Salmerón, J. Synergistic cytotoxicity of the poly (ADP-ribose) polymerase inhibitor ABT-888 and temozolomide in dual-drug targeted magnetic nanoparticles. Liver Int. 2015, 35, 1430–1441. [Google Scholar] [CrossRef]
- Rabik, C.A.; Dolan, M.E. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat. Rev. 2007, 33, 9–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamble, D.B.; Mu, D.; Reardon, J.T.; Sancar, A.; Lippard, S.J. Repair of cisplatin—DNA adducts by the mammalian excision nuclease. Biochemistry 1996, 35, 10004–10013. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.H.; Xiong, M.; Chen, X.P.; Xiao, Z.Y.; Zhao, Y.F.; Huang, Z.Y. PJ34, an inhibitor of PARP-1, suppresses cell growth and enhances the suppressive effects of cisplatin in liver cancer cells. Oncol. Rep. 2008, 20, 567–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streppel, M.M.; Pai, S.; Campbell, N.R.; Hu, C.; Yabuuchi, S.; Canto, M.I.; Wang, J.S.; Montgomery, E.A.; Maitra, A. MicroRNA 223 is upregulated in the multistep progression of Barrett’s esophagus and modulates sensitivity to chemotherapy by targeting PARP1. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 4067–4078. [Google Scholar] [CrossRef] [Green Version]
- McPherson, L.A.; Shen, Y.; Ford, J.M. Poly (ADP-ribose) polymerase inhibitor LT-626: Sensitivity correlates with MRE11 mutations and synergizes with platinums and irinotecan in colorectal cancer cells. Cancer Lett. 2014, 343, 217–223. [Google Scholar] [CrossRef]
- Pommier, Y.; Barcelo, J.M.; Rao, V.A.; Sordet, O.; Jobson, A.G.; Thibaut, L.; Miao, Z.-H.; Seiler, J.A.; Zhang, H.; Marchand, C.; et al. Repair of topoisomerase I-mediated DNA damage. Prog. Nucleic Acid Res. Mol. Biol. 2006, 81, 179–229. [Google Scholar] [CrossRef] [Green Version]
- Das, B.B.; Huang, S.-Y.N.; Murai, J.; Rehman, I.; Amé, J.-C.; Sengupta, S.; Das, S.K.; Majumdar, P.; Zhang, H.; Biard, D.; et al. PARP1-TDP1 coupling for the repair of topoisomerase I-induced DNA damage. Nucleic Acids Res. 2014, 42, 4435–4449. [Google Scholar] [CrossRef]
- Pfizer, Inc. Camptosar (Irinotecan); Food & Drug Administration: Silver Spring, MD, USA, 2020.
- Tahara, M.; Inoue, T.; Sato, F.; Miyakura, Y.; Horie, H.; Yasuda, Y.; Fujii, H.; Kotake, K.; Sugano, K. The use of Olaparib (AZD2281) potentiates SN-38 cytotoxicity in colon cancer cells by indirect inhibition of Rad51-mediated repair of DNA double-strand breaks. Mol. Cancer 2014, 13, 1170–1180. [Google Scholar] [CrossRef] [Green Version]
- Davidson, D.; Wang, Y.; Aloyz, R.; Panasci, L. The PARP inhibitor ABT-888 synergizes irinotecan treatment of colon cancer cell lines. Investig. New Drugs 2013, 31, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Tentori, L.; Leonetti, C.; Muzi, A.; Dorio, A.S.; Porru, M.; Dolci, S.; Campolo, F.; Vernole, P.; Lacal, P.M.; Praz, F.; et al. Influence of MLH1 on colon cancer sensitivity to poly(ADP-ribose) polymerase inhibitor combined with irinotecan. Int. J. Oncol. 2013, 43, 210–218. [Google Scholar] [CrossRef]
- Miknyoczki, S.; Chang, H.; Grobelny, J.; Pritchard, S.; Worrell, C.; McGann, N.; Ator, M.; Husten, J.; Deibold, J.; Hudkins, R.; et al. The selective poly(ADP-ribose) polymerase-1(2) inhibitor, CEP-8983, increases the sensitivity of chemoresistant tumor cells to temozolomide and irinotecan but does not potentiate myelotoxicity. Mol. Cancer Ther. 2007, 6, 2290. [Google Scholar] [CrossRef] [Green Version]
- Subhash, V.V.; Tan, S.H.; Yeo, M.S.; Yan, F.L.; Peethala, P.C.; Liem, N.; Krishnan, V.; Yong, W.-P. ATM expression predicts Veliparib and Irinotecan sensitivity in gastric cancer by mediating P53 independent regulation of cell cycle and apoptosis. Mol. Cancer Ther. 2016, 15, 3087–3096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genther Williams, S.M.; Kuznicki, A.M.; Andrade, P.; Dolinski, B.M.; Elbi, C.; O’Hagan, R.C.; Toniatti, C. Treatment with the PARP inhibitor, niraparib, sensitizes colorectal cancer cell lines to irinotecan regardless of MSI/MSS status. Cancer Cell Int. 2015, 15, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augustine, T.; Maitra, R.; Zhang, J.; Nayak, J.; Goel, S. Sensitization of colorectal cancer to irinotecan therapy by PARP inhibitor rucaparib. Investig. New Drugs 2019, 37, 948–960. [Google Scholar] [CrossRef] [PubMed]
- Miknyoczki, S.J.; Jones-Bolin, S.; Pritchard, S.; Hunter, K.; Zhao, H.; Wan, W.; Ator, M.; Bihovsky, R.; Hudkins, R.; Chatterjee, S.; et al. Chemopotentiation of Temozolomide, Irinotecan, and Cisplatin Activity by CEP-6800, a Poly(ADP-Ribose) Polymerase Inhibitor. Mol. Cancer Ther. 2003, 2, 371. [Google Scholar]
- George, J.W.; Bessho, M.; Bessho, T. Inactivation of XPF Sensitizes Cancer Cells to Gemcitabine. J. Nucleic Acids 2019, 2019, 6357609. [Google Scholar] [CrossRef] [Green Version]
- Jacob, D.A.; Bahra, M.; Langrehr, J.M.; Boas-Knoop, S.; Stefaniak, R.; Davis, J.; Schumacher, G.; Lippert, S.; Neumann, U.P. Combination therapy of poly (ADP-ribose) polymerase inhibitor 3-aminobenzamide and gemcitabine shows strong antitumor activity in pancreatic cancer cells. J. Gastroenterol. Hepatol. 2007, 22, 738–748. [Google Scholar] [CrossRef]
- Wyatt, M.D.; Wilson, D.M., III. Participation of DNA repair in the response to 5-fluorouracil. Cell. Mol. Life Sci. 2009, 66, 788–799. [Google Scholar] [CrossRef] [Green Version]
- Vitiello, P.P.; Cardone, C.; Ciardiello, D.; Belli, V.; Matrone, N.; Borrelli, C.; Poliero, L.; De Falco, V.; Giunta, E.F.; Vitale, P.; et al. 18P—Combination treatment with the PARP inhibitor niraparib and chemotherapeutics in a preclinical model of KRAS/BRAF mutated colorectal cancer cell lines across the four consensus molecular subtypes. Ann. Oncol. 2018, 29 (Suppl. S8), VIII5. [Google Scholar] [CrossRef]
- Abal, M.; Andreu, J.M.; Barasoain, I. Taxanes: Microtubule and centrosome targets, and cell cycle dependent mechanisms of action. Curr. Cancer Drug Targets 2003, 3, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Mikuła-Pietrasik, J.; Witucka, A.; Pakuła, M.; Uruski, P.; Begier-Krasińska, B.; Niklas, A.; Tykarski, A.; Książek, K. Comprehensive review on how platinum- and taxane-based chemotherapy of ovarian cancer affects biology of normal cells. Cell. Mol. Life Sci. 2019, 76, 681–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Yang, C.; Xie, C.; Jiang, J.; Gao, M.; Fu, L.; Li, Y.; Bao, X.; Fu, H.; Lou, L. Pharmacologic characterization of fluzoparib, a novel poly(ADP-ribose) polymerase inhibitor undergoing clinical trials. Cancer Sci. 2019, 110, 1064–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appleman, L.J.; Beumer, J.H.; Jiang, Y.; Lin, Y.; Ding, F.; Puhalla, S.; Swartz, L.; Owonikoko, T.K.; Donald Harvey, R.; Stoller, R.; et al. Phase 1 study of veliparib (ABT-888), a poly (ADP-ribose) polymerase inhibitor, with carboplatin and paclitaxel in advanced solid malignancies. Cancer Chemother. Pharmacol. 2019, 84, 1289–1301. [Google Scholar] [CrossRef]
- Martin-Richard, M.; Díaz Beveridge, R.; Arrazubi, V.; Alsina, M.; Galan Guzmán, M.; Custodio, A.B.; Gómez, C.; Muñoz, F.L.; Pazo, R.; Rivera, F. SEOM Clinical Guideline for the diagnosis and treatment of esophageal cancer. Clin. Transl. Oncol. 2016, 18, 1179–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.-M.; Liu, R.; Ba, Y.; Jiang, D.; Wang, M.; Zheng, Y.; Wei, J.; Bai, Y.-X.; Lin, L.; Xiong, J.; et al. Phase I study of fluzoparib, a PARP1 Inhibitor in combination with apatinib and paclitaxel in patients (pts) with advanced gastric and gastroesophageal junction (GEJ) adenocarcinoma. J. Clin. Oncol. 2019, 37 (Suppl. S15), 4060. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.S. Phase I Study of Irinotecan Liposome (Nal-IRI), Fluorouracil, Leucovorin and Rucaparib in the Treatment of Select Gastrointestinal Metastatic Malignancies Followed by a Phase Ib of First and Second Line Treatment of Both Unselected and Selected (for BRCA 1/2 and PALB2 Mutations) Patients With Metastatic Adenocarcinoma of the Pancreas Then Followed by a Phase II Study of First Line Treatment of Selected Patients With Metastatic Adenocarcinoma of the Pancreas With Genomic Markers (Signature) of Homologous Recombination Deficiency (HRD). In Liposomal Irinotecan, Fluorouracil, Leucovorin Calcium, and Rucaparib in Treating Patients With Metastatic Pancreatic, Colorectal, Gastroesophageal, or Biliary Cancer; Mayo Clinic: Arizona, FL, USA, 2017. [Google Scholar]
- Martin-Richard, M.; Custodio, A.; García-Girón, C.; Grávalos, C.; Gomez, C.; Jimenez-Fonseca, P.; Manzano, J.L.; Pericay, C.; Rivera, F.; Carrato, A. Seom guidelines for the treatment of gastric cancer 2015. Clin. Transl. Oncol. 2015, 17, 996–1004. [Google Scholar] [CrossRef] [Green Version]
- Bang, Y.-J.; Im, S.-A.; Lee, K.-W.; Cho, J.Y.; Song, E.-K.; Lee, K.H.; Kim, Y.H.; Park, J.O.; Chun, H.G.; Zang, D.Y.; et al. Randomized, Double-Blind Phase II Trial With Prospective Classification by ATM Protein Level to Evaluate the Efficacy and Tolerability of Olaparib Plus Paclitaxel in Patients With Recurrent or Metastatic Gastric Cancer. J. Clin. Oncol. 2015, 33, 3858–3865. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Xu, R.-H.; Chin, K.; Lee, K.-W.; Park, S.H.; Rha, S.Y.; Shen, L.; Qin, S.; Xu, N.; Im, S.-A.; et al. Olaparib in combination with paclitaxel in patients with advanced gastric cancer who have progressed following first-line therapy (GOLD): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1637–1651. [Google Scholar] [CrossRef]
- Berlin, J.; Ramanathan, R.K.; Strickler, J.H.; Subramaniam, D.S.; Marshall, J.; Kang, Y.-K.; Hetman, R.; Dudley, M.W.; Zeng, J.; Nickner, C.; et al. A phase 1 dose-escalation study of veliparib with bimonthly FOLFIRI in patients with advanced solid tumours. Br. J. Cancer 2018, 118, 938–946. [Google Scholar] [CrossRef]
- Nuthalapati, S.; Munasinghe, W.; Giranda, V.; Xiong, H. Clinical Pharmacokinetics and Mass Balance of Veliparib in Combination with Temozolomide in Subjects with Nonhematologic Malignancies. Clin. Pharmacokinet. 2018, 57, 51–58. [Google Scholar] [CrossRef]
- Gabrielson, A.; Tesfaye, A.A.; Marshall, J.L.; Pishvaian, M.J.; Smaglo, B.; Jha, R.; Dorsch-Vogel, K.; Wang, H.; He, A.R. Phase II study of temozolomide and veliparib combination therapy for sorafenib-refractory advanced hepatocellular carcinoma. Cancer Chemother. Pharmacol. 2015, 76, 1073–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Grazie, M.; Biagini, M.R.; Tarocchi, M.; Polvani, S.; Galli, A. Chemotherapy for hepatocellular carcinoma: The present and the future. World J. Hepatol. 2017, 9, 907–920. [Google Scholar] [CrossRef]
- Lampiasi, N.; Umezawa, K.; Montalto, G.; Cervello, M. Poly (ADP-ribose) polymerase inhibition synergizes with the NF-κB inhibitor DHMEQ to kill hepatocellular carcinoma cells. Biochim. Biophys. Acta BBA Mol. Cell Res. 2014, 1843, 2662–2673. [Google Scholar] [CrossRef] [Green Version]
- Liang, B.-Y.; Xiong, M.; Ji, G.-B.; Zhang, E.-L.; Zhang, Z.-Y.; Dong, K.-S.; Chen, X.-P.; Huang, Z.-Y. Synergistic suppressive effect of PARP-1 inhibitor PJ34 and HDAC inhibitor SAHA on proliferation of liver cancer cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2015, 35, 535–540. [Google Scholar] [CrossRef]
- Luo, Q.; Li, Y.; Deng, J.; Zhang, Z. PARP-1 inhibitor sensitizes arsenic trioxide in hepatocellular carcinoma cells via abrogation of G2/M checkpoint and suppression of DNA damage repair. Chem. Biol. Interact. 2015, 226, 12–22. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. Pancreatic Patients; National Comprehensive Cancer Network: Plymouth Meeting, PA, USA, 2019. [Google Scholar]
- Bendell, J.; O’Reilly, E.M.; Middleton, M.R.; Chau, I.; Hochster, H.; Fielding, A.; Burke, W.; Burris, H., III. Phase I study of olaparib plus gemcitabine in patients with advanced solid tumours and comparison with gemcitabine alone in patients with locally advanced/metastatic pancreatic cancer. Ann. Oncol. 2015, 26, 804–811. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Lowery, M.A.; Yu, K.H.; Capanu, M.; Stadler, Z.K.; Epstein, A.S.; Golan, T.; Segal, A.; Segal, M.; Salo-Mullen, E.E.; et al. Randomized phase II study of gemcitabine (G), cisplatin (C) with or without veliparib (V) (arms A, B) and a phase II single-arm study of single-agent veliparib (arm C) in patients with BRCA or PALB2-mutated pancreas adenocarcinoma (PC). J. Clin. Oncol. 2013, 31 (Suppl. S15), TPS4144. [Google Scholar] [CrossRef]
- Tuli, R.; Shiao, S.L.; Nissen, N.; Tighiouart, M.; Kim, S.; Osipov, A.; Bryant, M.; Ristow, L.; Placencio-Hickok, V.; Hoffman, D.; et al. A phase 1 study of veliparib, a PARP-1/2 inhibitor, with gemcitabine and radiotherapy in locally advanced pancreatic cancer. EBioMedicine 2019, 40, 375–381. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Myzak, M.C.; Johnson, B.A., III; De Jesus-Acosta, A.; Le, D.T.; Jaffee, E.M.; Azad, N.S.; Donehower, R.C.; Zheng, L.; Oberstein, P.E.; et al. Olaparib in combination with irinotecan, cisplatin, and mitomycin C in patients with advanced pancreatic cancer. Oncotarget 2017, 8, 44073–44081. [Google Scholar] [CrossRef]
- Chiorean, E.G.; Guthrie, K.A.; Philip, P.A.; Swisher, E.M.; Jalikis, F.; Pishvaian, M.J.; Berlin, J.; Noel, M.S.; Suga, J.M.; Garrido-Laguna, I.; et al. Randomized phase II study of second-line modified FOLFIRI with PARP inhibitor ABT-888 (Veliparib) (NSC-737664) versus FOLFIRI in metastatic pancreatic cancer (mPC): SWOG S1513. J. Clin. Oncol. 2019, 37 (Suppl. S15), 4014. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Wang, H.; He, A.R.; Hwang, J.J.; Smaglo, B.G.; Kim, S.S.; Weinberg, B.A.; Weiner, L.M.; Marshall, J.L.; Brody, J.R. A Phase I/II Study of Veliparib (ABT-888) in Combination with 5-Fluorouracil and Oxaliplatin in Patients with Metastatic Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 5092–5101. [Google Scholar] [CrossRef] [PubMed]
- Berman, R.S.; Lee, V.; Ryan, D.P. Colon Cancer Treatment (PDQ®)—Health Professional Version. Available online: https://www.cancer.gov/types/colorectal/hp/colon-treatment-pdq#link/_265 (accessed on 1 April 2021).
- Gorbunova, V.; Beck, J.T.; Hofheinz, R.-D.; Garcia-Alfonso, P.; Nechaeva, M.; Cubillo Gracian, A.; Mangel, L.; Elez Fernandez, E.; Deming, D.A.; Ramanathan, R.K.; et al. A phase 2 randomised study of veliparib plus FOLFIRI±bevacizumab versus placebo plus FOLFIRI±bevacizumab in metastatic colorectal cancer. Br. J. Cancer 2019, 120, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Pishvaian, M.J.; Slack, R.S.; Jiang, W.; He, A.R.; Hwang, J.J.; Hankin, A.; Dorsch-Vogel, K.; Kukadiya, D.; Weiner, L.M.; Marshall, J.L.; et al. A phase 2 study of the PARP inhibitor veliparib plus temozolomide in patients with heavily pretreated metastatic colorectal cancer. Cancer 2018, 124, 2337–2346. [Google Scholar] [CrossRef]
- Cecchini, M. Temozolomide and Olaparib for O6-Methylguanine DNA Methyltransferase Promoter Hypermethylated Colorectal Cancer; Yale University: New Haven, CT, USA, 2019. [Google Scholar]
- Berman, R.S.; Lee, V.; Ryan, D.P. Rectal Cancer Treatment (PDQ®)—Health Professional Version. Available online: https://www.cancer.gov/types/colorectal/hp/rectal-treatment-pdq#_706_toc (accessed on 22 April 2021).
- Czito, B.G.; Deming, D.A.; Jameson, G.S.; Mulcahy, M.F.; Vaghefi, H.; Dudley, M.W.; Holen, K.D.; DeLuca, A.; Mittapalli, R.K.; Munasinghe, W.; et al. Safety and tolerability of veliparib combined with capecitabine plus radiotherapy in patients with locally advanced rectal cancer: A phase 1b study. Lancet Gastroenterol. Hepatol. 2017, 2, 418–426. [Google Scholar] [CrossRef]
- George, T.J.; Yothers, G.; Hong, T.S.; Russell, M.M.; You, Y.N.; Parker, W.; Jacobs, S.A.; Lucas, P.C.; Gollub, M.J.; Hall, W.A.; et al. NRG-GI002: A phase II clinical trial platform using total neoadjuvant therapy (TNT) in locally advanced rectal cancer (LARC)—First experimental arm (EA) initial results. J. Clin. Oncol. 2019, 37 (Suppl. S15), 3505. [Google Scholar] [CrossRef]
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of radiotherapy in cancer treatment. Cancer 2005, 104, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175. [CrossRef] [Green Version]
- Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [CrossRef] [Green Version]
- Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e1323. [CrossRef] [Green Version]
- Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [CrossRef] [PubMed] [Green Version]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Rumiato, E.; Pasello, G.; Montagna, M.; Scaini, M.C.; De Salvo, G.L.; Parenti, A.; Cagol, M.; Ruol, A.; Ancona, E.; Amadori, A.; et al. DNA copy number profile discriminates between esophageal adenocarcinoma and squamous cell carcinoma and represents an independent prognostic parameter in esophageal adenocarcinoma. Cancer Lett. 2011, 310, 84–93. [Google Scholar] [CrossRef]
- Bonde, P.; Gao, D.; Chen, L.; Duncan, M.; Miyashita, T.; Montgomery, E.; Harmon, J.W.; Wei, C. Selective decrease in the DNA base excision repair pathway in squamous cell cancer of the esophagus. J. Thorac. Cardiovasc. Surg. 2007, 133, 74–81.e73. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Li, S.; Tang, X.; Wang, Y.; Guo, W.; Cao, G.; Chen, K.; Zhang, M.; Guan, M.; Yang, D. Copy Number Amplification of DNA Damage Repair Pathways Potentiates Therapeutic Resistance in Cancer. Theranostics 2020, 10, 3939–3951. [Google Scholar] [CrossRef]
- Dewalt, R.I.; Kesler, K.A.; Hammoud, Z.T.; Baldridge, L.; Hattab, E.M.; Jalal, S.I. Gastroesophageal junction adenocarcinoma displays abnormalities in homologous recombination and nucleotide excision repair. Lung Cancer 2014, 5, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Park, P.H.; Yamamoto, T.M.; Li, H.; Alcivar, A.L.; Xia, B.; Wang, Y.; Bernhardy, A.J.; Turner, K.M.; Kossenkov, A.V.; Watson, Z.L.; et al. Amplification of the mutation-carrying BRCA2 allele promotes RAD51 loading and PARP inhibitor resistance in the absence of reversion mutations. Mol. Cancer Ther. 2019, 19, 602–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waks, A.G.; Cohen, O.; Kochupurakkal, B.; Kim, D.; Dunn, C.E.; Buendia Buendia, J.; Wander, S.; Helvie, K.; Lloyd, M.R.; Marini, L.; et al. Reversion and non-reversion mechanisms of resistance to PARP inhibitor or platinum chemotherapy in BRCA1/2-mutant metastatic breast cancer. Ann. Oncol. 2020, 31, 590–598. [Google Scholar] [CrossRef]
- Golan, T.; Oh, D.Y.; Reni, M.; Macarulla, T.M.; Tortora, G.; Hall, M.J.; Reinacher-Schick, A.C.; Borg, C.; Hochhauser, D.; Walter, T.; et al. POLO: A randomized phase III trial of olaparib maintenance monotherapy in patients (pts) with metastatic pancreatic cancer (mPC) who have a germline BRCAI/2 mutation (gBRCAm). J. Clin. Oncol. 2016, 34, TPS4152. [Google Scholar] [CrossRef]
- Yan, X.; Wu, T.; Tang, M.; Chen, D.; Huang, M.; Zhou, S.; Zhang, H.; Yang, X.; Li, G. Methylation of the ataxia telangiectasia mutated gene (ATM) promoter as a radiotherapy outcome biomarker in patients with hepatocellular carcinoma. Medicine 2020, 99, e18823. [Google Scholar] [CrossRef]
- Luo, J.; Si, Z.-Z.; Li, T.; Li, J.-Q.; Zhang, Z.-Q.; Chen, G.-S.; Qi, H.-Z.; Yao, H.-L. MicroRNA-146a-5p enhances radiosensitivity in hepatocellular carcinoma through replication protein A3-induced activation of the DNA repair pathway. Am. J. Physiol. Cell Physiol. 2018, 316, C299–C311. [Google Scholar] [CrossRef] [PubMed]
- Beger, C.; Ramadani, M.; Meyer, S.; Leder, G.; Krüger, M.; Welte, K.; Gansauge, F.; Beger, H.G. Down-regulation of BRCA1 in chronic pancreatitis and sporadic pancreatic adenocarcinoma. Clin. Cancer Res. 2004, 10, 3780–3787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maacke, H.; Jost, K.; Opitz, S.; Miska, S.; Yuan, Y.; Hasselbach, L.; Lüttges, J.; Kalthoff, H.; Stürzbecher, H.W. DNA repair and recombination factor Rad51 is over-expressed in human pancreatic adenocarcinoma. Oncogene 2000, 19, 2791–2795. [Google Scholar] [CrossRef]
- Bhardwaj, V.; Horvat, A.; Korolkova, O.; Washington, M.K.; El-Rifai, W.; Dikalov, S.I.; Zaika, A.I. Prevention of DNA damage in Barrett’s esophageal cells exposed to acidic bile salts. Carcinogenesis 2016, 37, 1161–1169. [Google Scholar] [CrossRef] [Green Version]
- Helleday, T. Homologous recombination in cancer development, treatment and development of drug resistance. Carcinogenesis 2010, 31, 955–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | PARP Inhibitor | Combination Therapy | Cancer Type | Trial Number |
|---|---|---|---|---|
| I | Talazoparib | Trifluridine/Tipiracil | Gastroesophageal Adenocarcinoma, Colorectal Cancer | NCT04511039 |
| I | Fluzoparib | Paclitaxel + Apatinib | Gastroesophageal Adenocarcinoma | NCT03026881 |
| I | Fluzoparib | FOLFIRINOX | Resectable Pancreatic Cancer | NCT04425876 |
| I/II | Fluzoparib | FOLFIRINOX | Advanced Pancreatic Cancer | NCT04228601 |
| I/II | Olaparib | Oxaliplatin + Tegafur/Gimeracil Oteracil Potassium | Gastric Cancer | NCT04410887 |
| I/II | Veliparib | FOLFOX | Pancreatic Cancer | NCT01489865 |
| I/II | Rucaparib | Irinotecan, Leucovorin, Fluorouracil | Gastrointestinal Malignancies | NCT03337087 |
| II | Olaparib | Temozolomide | MGMT-Hypermethylated Colorectal Cancer | NCT04166435 |
| II | Olaparib | Paclitaxel + Pembrolizumab | Gastric Cancer | NCT04209686 |
| II | Olaparib | Paclitaxel + Durvalumab | Gastric Cancer | NCT03579784 |
| II | Olaparib | Paclitaxel | Gastric Cancer | NCT01063517 |
| II | Veliparib | Gemcitabine + Cisplatin | BRCA/PALB2-mutated Pancreatic Cancer | NCT01585805 |
| II | Veliparib | FOLFIRI | Pancreatic Cancer | NCT02890355 |
| PARP Gene Alteration | Genomic Alteration Frequency (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| OAC | OSCC | GAC | HCC | CCA | PAAD | COAD | READ | Total | |
| n = 87 | n = 95 | n = 440 | n = 369 | n = 36 | n = 184 | n = 378 | n = 155 | n = 1744 | |
| Mutation | |||||||||
| PARP1 | 1.1 | 1.1 | 2.7 | 0.5 | - | 1.1 | 2.1 | 0.6 | 1.5 |
| PARP2 | - | - | 1.8 | 0.3 | - | 0.5 | 0.8 | 0.6 | 0.8 |
| PARP3 | 2.3 | - | 1.6 | - | - | - | 1.9 | - | 0.9 |
| PARP4 | 2.3 | 2.1 | 4.3 | 0.8 | - | - | 3.2 | 1.3 | 2.3 |
| CNA | |||||||||
| PARP1 | 1.1 | 2.1 | 2.0 | 5.1 | 5.6 | 2.2 | 0.5 | 0.6 | 2.3 |
| PARP2 | 1.1 | 3.2 | 1.6 | 0.8 | - | 0.5 | - | 0.6 | 0.9 |
| PARP3 | 3.4 | 2.1 | 0.9 | 0.3 | - | - | 0.3 | 0.6 | 0.7 |
| PARP4 | 4.6 | 1.1 | 0.9 | 0.8 | - | 0.5 | 4.5 | 4.5 | 2.1 |
| HR Gene Alteration | Genomic Alteration Frequency (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| OAC | OSCC | GAC | HCC | CCA | PAAD | COAD | READ | Total | |
| n = 87 | n = 95 | n = 440 | n = 369 | n = 36 | n = 184 | n = 378 | n = 155 | n = 1744 | |
| Mutation | |||||||||
| BRCA1 | 1.1 | 1.1 | 3.6 | 1.4 | - | 1.1 | 2.9 | 1.9 | 2.2 |
| BRCA2 | 3.4 | 4.2 | 8.9 | 2.2 | 2.8 | 1.1 | 5.8 | 5.2 | 5.0 |
| ATM | 6.9 | 3.2 | 10.5 | 3.5 | 2.8 | 4.3 | 9.8 | 9.0 | 7.3 |
| RAD51 | - | - | 0.5 | 0.5 | - | - | - | - | 0.2 |
| MRE11 | - | - | 1.6 | - | - | 0.5 | 1.1 | 1.9 | 0.9 |
| PALB2 | - | - | 2.5 | 0.3 | - | 0.5 | 1.9 | 1.3 | 1.3 |
| CNA | |||||||||
| BRCA1 | 3.4 | - | 2.3 | 1.6 | - | 2.2 | 0.3 | 0.6 | 1.4 |
| BRCA2 | 6.9 | 1.1 | 3.2 | 0.8 | - | - | 2.6 | 3.9 | 2.3 |
| ATM | 2.3 | - | 2.3 | 0.8 | - | - | 0.5 | 0.6 | 1.0 |
| RAD51 | 1.1 | 1.1 | 0.2 | 0.3 | - | 0.5 | 1.9 | 1.3 | 0.8 |
| MRE11 | 1.1 | 2.1 | 1.6 | 0.5 | - | - | 0.3 | - | 0.7 |
| PALB2 | - | - | 0.5 | - | - | - | 1.1 | - | 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhusaini, A.; Cannon, A.; Maher, S.G.; Reynolds, J.V.; Lynam-Lennon, N. Therapeutic Potential of PARP Inhibitors in the Treatment of Gastrointestinal Cancers. Biomedicines 2021, 9, 1024. https://doi.org/10.3390/biomedicines9081024
Alhusaini A, Cannon A, Maher SG, Reynolds JV, Lynam-Lennon N. Therapeutic Potential of PARP Inhibitors in the Treatment of Gastrointestinal Cancers. Biomedicines. 2021; 9(8):1024. https://doi.org/10.3390/biomedicines9081024
Chicago/Turabian StyleAlhusaini, Abdullah, Aoife Cannon, Stephen G. Maher, John V. Reynolds, and Niamh Lynam-Lennon. 2021. "Therapeutic Potential of PARP Inhibitors in the Treatment of Gastrointestinal Cancers" Biomedicines 9, no. 8: 1024. https://doi.org/10.3390/biomedicines9081024
APA StyleAlhusaini, A., Cannon, A., Maher, S. G., Reynolds, J. V., & Lynam-Lennon, N. (2021). Therapeutic Potential of PARP Inhibitors in the Treatment of Gastrointestinal Cancers. Biomedicines, 9(8), 1024. https://doi.org/10.3390/biomedicines9081024