2. Alkaloids
Alkaloids are cyclic compounds containing one or more nitrogen atoms in their cycle or side chain that demonstrate weak alkali properties. Main natural sources of alkaloids belong to the plant kingdom, and mainly to higher plants. However, within the recent years the number of alkaloid molecules found in the marine sources is gradually growing [
36]. Those species include marine bacteria including cyanobacteria (or blue-green algae), actinomycetes, marine-derived fungi, marine algae, bryozoans, starfishes, holothurians, ascidians, and marine sponges [
37]. Bioactive alkaloids as well as officinal medicine alkaloids exert various pharmacological effects including antitumor properties [
38]. Alkaloids compounds demonstrating antiglioma activity are listed below with anticancer mechanisms given in details.
2.1. Imidazolone and Indole Alkaloids
A new alkaloid named zorrimidazolone isolated from the Mediterranean stolidobranch ascidian
Polyandrocarpa zorritensis was shown to exert cytotoxic effects against C6 cells resulting in 60% decreased viability when applied in concentration 250 μM for 48 h under in vitro conditions. IC
50 for zorrimidazolone providing inhibiting effects against C6 cells was within the micromolar range (
Table 1), which is generally considered as moderate cytotoxic activity [
39]. This new metabolite belongs to the 2-aminoimidazolone class of marine metabolites, which have been predominantly isolated from
Axinella and
Agelas marine sponges and very rarely found in the ascidians. The compounds from this class isolated from ascidians are the N,N-dimethylaminoimidazolone found in
Dendrodoa grossularia [
40] and the polyandrocarpamines A and B found in a Fijian
Polyandrocarpa sp. [
41]. Two monoindole alkaloids, 3-indolylglyoxylic acid and its methyl ester that were also found in
Polyandrocarpa zorritensis, demonstrated weaker cytotoxic activity toward the C6 cells, which was found selective and depending on the concentration. The structurally close compound, 4-hydroxy-3-methoxyphenylglyoxylic acid methyl ester, which does not belong to the alkaloid class, did not affect C6 cell viability. According to the author conclusion, zorrimidazolone may be of interest for the development of potential antiproliferative molecules against gliomas.
Indol alkaloids meridianins isolated from ascidian
Aplidium meridianum [
42] and variolin B found in Antarctic sponge
Kirkpatrickia varialosa [
43] became prototypes of the novel synthetic derivatives as well as hybrid molecules meriolins obtained from those compounds. The natural meridianins A, B, C, D, E, F, and G are brominated and/or hydroxylated 3-(2-aminopyrimidine)-indoles differing in the bromine and/or hydroxyl substitution (
Figure 1) [
44,
45]. Meridianins were shown to inhibit various protein kinases, such as CDKs, glycogen synthase kinase-3, cyclic nucleotide-dependent kinases, and casein kinase 1 [
46] playing important role in the cancer cell lifecycle. They also prevent cell proliferation and induce apoptosis due to their ability to penetrate cellular membrane and disturb activity of kinases responsible for cell division and death. Meridianin E was experimentally proved an effective protein kinase inhibitor with high selectivity regarding CDK1 and CDK5 [
46]. One of the meridianin C derivatives substituted at the C-5 position was found to be strong and selective inhibitor of pim kinases (including pim-1, pim-2, and pim-3 overexpressed in various cancer cell types) with IC
50 values within a nanomolar concentration range [
47].
Variolins (A, B, deoxyvariolin B, and N(3′)-methyl tetrahydrovariolin B) are natural marine alkaloids possessing an uncommon pyrido[3′,2′:4,5]pyrrolo[1,2-c]pyrimidine skeleton (
Figure 2) [
43]. Variolin B exerted anticancer activity on the P388 murine leukemia cell line with an IC
50 value 716 nM. Variolin A and N(3′)-methyl tetrahydrovariolin B displayed significantly weaker activity against these cancer cells. Variolin B had been shown to induce apoptosis exerting highly potent cytotoxic activity against various human cancer cell lines including the ones with overexpressed level of cell efflux pump p-glycoprotein (PGP). Variolin B and deoxyvariolin B were experimentally confirmed to exert strong cytotoxic activity against various cancer cell lines with IC
50 values varying from 50 to 100 nM. Both compounds were noted to inhibit the histone H1 phosphorylation mediated by cyclin E-CDK2, cyclin A-CDK2, cyclin B-CDK1, cyclin H-CDK7, and cyclin D-CDK4, with IC
50 values in the micromolar range [
48].
A new CDK inhibitory scaffold with antitumor activity has been composed by combining the common features of meridianins and variolins. Thus, the new class of synthetic 7-azaindole-containing analogues have been designed and the term ‘meriolin’ has been coined to describe this hybrid structure [
49,
50]. Meriolins exhibit better antiproliferative and proapoptotic properties in cell cultures than their ‘inspirational parent’ molecules. Meriolins was shown to exert significant inhibiting effects against CDKs and possess antiproliferative and pro-apoptotic activity in human cancer cell lines in vitro [
50]. In particular, phosphorylation at CDK1-, CDK4-, and CDK9-specific sites has been shown to be counteracted by meriolins in neuroblastoma SH-SY5Y [
49].
Nineteen different meriolin structures have been investigated in such human glioma cell lines as anaplastic astrocytoma SW1088 and glioblastoma U87. All mentioned meriolins efficiently decelerated tumor cell growth rate within IC
50 range from 1 nM to 1 μM. Meriolin types 3, 5, and 15 provoked proliferation rate inhibition in both glioma cell lines. IC
50 for SW1088 cells were 34, 32, and 46 nM, respectively, whereas these values for U87 were 76, 18.4, and 5.1 nM, respectively (
Table 2). Meriolin types 5 and 15 showed the strongest antiproliferative activity via induction of the cell cycle block and apoptosis in glioma cells [
51].
In the in vivo experiments, meriolin 15 inhibited proliferation of the glioma cells, induced apoptosis, and reduced the number of non-differentiated tumor cells in the U87 glioblastoma xenograft model in nude mice. The results suggested that meriolin 15 inhibits DK7/CDK9 consequently decreasing RNA polymerase II and its phosphorylation that results in downregulation of the survival factor Mcl-1, thereby allowing the activation of proapoptotic factors (Noxa, Bim, etc.) [
51]. The cytotoxic effect of meriolins have been also investigated in the rat primary proliferating astrocyte and neuron cultures. Meriolin type 5 and 15 demonstrated antiproliferative and pro-death activities. IC
50 of the meriolin 15 is 7.8 nM in astrocytes and 4.7 nM in neurons. Therefore, meriolins provide extremely potent antiglioma activity and at the same time exert strong toxic effect in healthy astrocytes. This requires further investigations focused on development of other derivative with less toxicity regarding normal healthy cells.
2.2. Fascaplysins
Fascaplysin is a red pigment and bis-indole alkaloid (12, 13-Dihydro-13-oxopyrido[1,2-a:3,4-b′] diindol-5-ium chloride) that was initially isolated from the Fijian sponge
Fascapfysinopsis sp. in 1988 and characterized as the novel 12H-pyrido[1,2-a:3,4-b′]diindole ring system (
Figure 3). It was considered a unique compound among the natural products [
52]. Later fascaplysin and some related compounds homofascaplysin A and 3-bromohomofascaplysin A were discovered and isolated from ascidian
Didemnum sp. [
53]. Then synthesis of fascaplysin and its derivatives was elaborated in a relatively short period, and this process is still ongoing [
54,
55].
Fascaplysin exerts various biological activities including selective kinase 4 (CDK-4) inhibition, DNA binding, and antiangiogenic effects [
56]. Fascaplysin was found to exert cytotoxic effects in a panel with at least 36 cancer cell lines with IC
50 in a range 0.6–4 μM [
54,
57,
58,
59]. It was also shown that fascaplysin and its derivatives are quite efficacious in in vivo mouse tumor models against human colon carcinoma HCT-116, human non-small-cell lung carcinoma NCI-H460 [
54], human malignant melanoma A375 [
58], and murine sarcoma S180 via induction of apoptosis and antiangiogenesis [
57].
The molecular mechanism of fascaplysin-induced apoptosis is directly linked to activation of the caspase-3, caspase-8, and caspase-9 pathways, cleavage of Bid, release of cytochrome C into cytosol and downregulation of the Bcl-2 level in cancer cells. Fascaplysin was also shown to block vascular endothelial growth factor (VEGF), inhibit proliferation, and induce apoptosis of human umbilical vein endothelial cells (HUVECs) [
56]. TNF and TNF receptor superfamily in HUVECs and hepatocarcinoma cells BeL-7402 cab be regulated by fascaplysin resulting in the tumor necrosis-related apoptosis-inducing ligand-(TRAIL)-induced apoptosis leading to activation of caspases 3 and 9 and Bid decrease [
60]. Fascaplysin was noted to induce high cytotoxicity against small-cell lung cancer (SCLC) cells resulting in the cell cycle arrest in G
1/G
0 at lower concentration of active compounds and in S-phase at the higher fascaplysin level. Its high cytotoxic activity against these cancer cells is due to multiple routes of action, affecting topoisomerase I, integrity of DNA and generation of reactive oxygen species (ROS) [
57].
Anticancer activity of fascaplysin under in vitro conditions results in reduced expression of CDK4, cyclin D1 and downregulation of the CDK4-specific Ser795 retinoblastoma protein (Rb) phosphorylation in HeLa cell line. Apoptosis induced by fascaplysin is related to activation of effector caspases, migration of cytochrome C into cytosol, and reduced Bcl-2 expression. Cytotoxic effects of fascaplysin were observed in chemosensitive promyelocytic HL-60 cancer cells as it activates both pro-apoptotic events like PARP-1 cleavage/caspase activation and triggered autophagy as shown by the increased expression of LC3-II, ATG7, and beclin [
61]. It should be emphasized that fascaplysin demonstrates significant anticancer activity in non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC) lines, which is not depending on the CDK4 pathway suggesting the direct effects on the DNA function and transcription of various proteins [
59]. Another mechanism of antitumor activity was found to be related in increase of the phosphorylation of Akt/PKB and adenosine monophosphate-activated protein kinase (AMPK) that play a key role in anti-apoptotic or pro-survival pathways in cancer [
58]. Fascaplysin in addition was shown to abolish phosphorylation of mTOR, 4EBP1, and p70S6K1 thus triggering the cap-dependent translation machinery and affecting expression of oncoproteins such as survivin, c-myc, cyclin D1, VEGF, and HIF-1α. Alkaloid derivative 7-chloro-fascaplysin similarly inhibits cell survival through interference with the PI3K/Akt/mTOR pathway, which in turn modulates HIF-1α, eNOS, and MMP-2/9 in the breast cancer cell line [
62]. Experimental treatment of the HCT116 (colorectal), A375 (malignant melanoma), and H1975 (lung) xenografted tumor tissues resulted in decreased tumor angiogenesis and increased cleaved-caspase-3. Consequently, survivin and HIF-1α are downregulated by suppressing 4EBP1-p70S6K1 axis-mediated de novo protein synthesis as was confirmed in the in vitro and in vivo experiments. In addition, fascaplysin inhibits vascular endothelial growth factor receptor 2 (VEGFR2) and tropomyosin-related kinase A (TRKA) via DFG-out non-competitive inhibition. These data suggest that fascaplysin inhibits TRKA and VEGFR2 and downregulates survivin and HIF-1α resulting in tumor growth inhibition [
58].
In our laboratory, antitumor efficiency of fascaplysin and its synthetic derivatives such as 7-phenylfascaplysin, 3-chlorofascaplysin, 3-bromofascaplysin, and 10-bromofascaplysin in C6 glioma cells was compared under in vitro conditions. Fascaplysin was applied in concentration range from 0.5 to 2 µM and exerted significant dose- and time-dependent antiproliferative and cytotoxic effects with IC
50 about 1.0 µM (
Table 3). Inhibiting effects were noted to be associated with the dose-dependent increase of the glioma cell number being in apoptosis stage. Inhibiting effect induced by fascaplysin in that model was significantly higher than that of temozolomide [
63]. Cytotoxic influence of all fascaplysin derivatives investigated in our study was superior to the activity of unsubstituted fascaplysin. In particular, 3-bromofascaplysin and 7-phenylfascaplysin had shown the highest capacity to induce C6 glioma cell death [
64].
On the other hand, high cytotoxicity of fascaplysin regarding normal cells should be emphasized, because it may be associated with its planar structure contributing d-s DNA intercalation [
65]. Development of the regulating approaches for such unusual cytotoxicity requires further investigation of various fascaplysin derivatives with the better safety profile.
2.3. Carboline Alkaloids (Tricyclic Pyridoindoles)
A large group of natural, semisynthetic, and synthetic compounds is presented with tricyclic pyridoindoles, i.e., carbolines, which are classified as α-carbolines (pyrido[2,3-b]indoles), β-carbolines (pyrido[3,4-b]indoles, γ-carbolines (pyrido[4,3-b]indoles) and δ-carbolines (pyrido[3,2-b]indoles) depending on the position of the pyridine nitrogen relative to the indole. β-carbolines initially discovered in
Peganum harmala and later widely found in medicinal plants and natural herbal products are the well investigated compounds [
66]. Alkaloids composed of tricyclic moiety and structurally relayed to both indole and carbazole compounds belong to the group of α-carbolines. Among the marine species, the representatives of the Tunicata subtype, mostly ascidians, are the source of α-carboline alkaloids [
67]. Natural α-carbolines and their synthetic derivatives are of great interest as the lead compounds in the new drug development. α-Carboline related derivatives have been synthesized and experimentally demonstrated to show anticancer activities [
68].
Six α-carboline analogues were synthesized, designated as TJY-13, TJY-14, TJY-16, TJY-18, TJY-22, TJY-24, and then tested on the human glioma cell lines U87, U251, T98G and rat glioma C6. 48 h incubation of the glioma cells with those compounds resulted in inhibited proliferation as shown in
Table 4. α-Carboline analog TJY-16 (6-acetyl-9-(3,4,5-trimethoxybenzyl)-9H-pyrido[2,3-b]indole) was found to be the strongest and highly potent inhibitor of the glioma cell viability even when applied in the nanomole concentration [
69]. 50 nM concentration of TJY-16 induced cell circle arrest in the G
2/M phase in U87 and T98G glioma cells. Moreover, 24–48 treatment of the tumor cells with TJY-16 led to significantly greater portion of the sub-G
1 phase blocked cells. Microscopic images of the glioma cells treated with TJY-16 demonstrated apoptotic signs, such as nuclear shrinkage and DNA condensation, and increased level of the cleaved caspase-3. Caspase-8 activation and depolarization of the mitochondrial membrane potential (ΔΨm) indicated that both extrinsic and intrinsic apoptotic pathways were involved in TJY-16-induced apoptosis. Surprisingly, cell death was noted in three human glioma cell lines, but it was not observed in the C6 rat glioma cell culture.
TJY-16 administered intraperitoneally once per day for 10 days in a dose 24 mg/kg in the nude mice with xenograft tumor model of U87 glioma cells effectively inhibited tumor growth and induced caspase-3 activation. Anti-glioma effect of TJY-16 was significantly higher than that of temozolomide, which was administered orally once per day for 5 days in a dose 80 mg/kg [
69]. Based on these results, authors of the research study considered α-carboline derivative TJY-16 as a perspective agent for the therapy of malignant gliomas.
Tetrahydroisoquinoline alkaloids with antitumor activity isolated from marine species are commonly categorized into ecteinascidins and renieramycins. Ecteinascidin-743 (ET-743), which is also known as trabectedin, yondelis, and CID 108150 is the one of the most potent representatives of ecteinascidins, isolated from Carribean ascidian (subphylum
Tunicata)
Ecteinascidia turbinata [
70]. ET-743 was approved as a first line drug for the treatment of inoperable soft tissue sarcoma with high resistance to conventional chemotherapeutics. Mechanism of antitumor activity of the ET-743 is related to its capacity of making complexes with minor groove of DNA double helix and alkylate N2 guanin, thus preventing cell proliferation, DNA reparation, and activation of transcription. This cascade leads in apoptosis of the target cells [
71]. Three alkaloids namely ecteinascidin-770 (ET-770, a stabilized derivative of ET-743, isolated from ascidian
E. thurstoni), 2′-N-4″-pyridinecarbonyl derivative of ET-770 and renieramycin M, a major bis-1,2,3,4-tetrahydroisoquinolinequinone alkaloid from the marine sponge
Xestospongia sp. were tested in human glioblastoma cells U373MG. All tested compounds exerted strong anti-glioma influence at the nanomolar concentrations after a 72 h treatment (
Table 5) [
72].
Drug effects against U373MG cells exerted by each compound in IC50 concentration within 72 h resulted in induced PARP and CASP3 cleavage that reflect molecular markers of ongoing apoptosis. Investigation of the gene expression profile of the whole genome of the U373MG cells treated with alkaloids in IC50 concentrations within 24 h have shown that ecteinascidin-770 reduced expression of 426 genes and upregulated 45 genes, renieramycin M suppressed expression of 274 genes and increased expression of 9 genes, and 2′-N-4″-pyridinecarbonyl derivative of ET-770 downregulated 417 genes and upregulated 84 genes. Generally, upregulated genes significantly prevailed over the genes with reduced expression for each tested compound. It should be noted that a set of 196 downregulated genes and 6 upregulated genes has shown to be the same for all tested compounds suggesting the presence of joint pathway involved in induction of apoptosis.
Analysis of molecular network made possible identifying EGFR signal pathway in the U373MG cells as a supplementation to the axonal and cell adhesion as significant downregulating gene pathways. ErbB (EGFR) signal pathway was found to be composed of focal adhesion kinase (FAK)/PTK2, Akt3, and GSK3β acting as key molecules involved in cell migration and development of the nervous system. At the same time, a set of genes being upregulated by alkaloid have shown significant linkage via cell cycle with CDC25A working as a hub molecule. It was also shown that suppressed expression of Akt3 by RNA interference reduce expression of the Bad phosphorylated form resulting in induced caspase-dependent apoptosis in glioma cells [
73]. Akt3 is known to be required for anchorage-independent growth of glioma cells. Glycosynthase kinase 3β (GSK3β) involved in apoptotic pathways presents a serine/threonine kinase that regulates Wnt/β-catenin named Hedgehog in an integrated manner and receptor tyrosine kinase (RTK) signaling pathways that play a key role in such cellular functions as glycogen metabolism, cell differentiation, proliferation, and apoptosis. Downregulating effects of siRNA towards GSK3β activity inhibit cell migration and induce apoptosis of glioma cells via c-Myc activation and suppression of the nuclear factor-κB (NF-κB) activity [
74]. The cell cycle progression inhibiting and activating processes are known to be regulated by the complex checkpoint mechanism that includes cyclins A, B, D, and E along with cyclin dependent kinases (CDKs) and CDK inhibitors (CDKIs) of both the Cip/Kip and Ink4 families. The hypophosphorylated Rb protein interacts with the E2F family transcription factors E2F1, E2F2, and E2F3, and activates gene expression that is crucial for the cell cycle progression. Rb protein, hyperphosphorylated by cyclin D1-CDK4 and cyclin E1-CDK2 complexes, in turn releases E2Fs and represses the cell cycle gene expression [
75]. Thus, Rb/E2F pathway acts a molecular switch contributing to either progressing or arrest of the cell cycle. The U373MG molecular network in the glioma cells composed of the integral set of downregulated and upregulated genes generally affected by all tested tetrahydroisoquinoline alkaloids was found to have a significant relation with transcription regulating mechanisms via transcription factors Rb/E2F [
72]. These findings suggest those alkaloids work as the DNA-alkylating agents interfering cell division finally resulting in apoptosis of the target cells.
It is already known that the human glioblastoma cell lines MO59K and MO59J are basically characterized by the presence or absence of the DNA-dependent protein kinase (DNA-PK) catalytic subunit, respectively, which considered a part of the DNA double-strand-break repair pathway. These cells have been shown to have different responses to the treatment with ET-743. MO59J cells were much more sensitive to ET-743 treatment compared to the MO59K cells with 5-fold lower values of the ET-743 IC
50 (0.041 ± 0.004 vs. 0.2 ± 0.02 nM) (
p < 0.05) [
76]. These results confirm that tetrahydroisoquinoline alkaloids possess a unique mechanism of interaction with DNA.
2.4. Pyrrole Alkaloids
Alkaloids rigidins A, B, C, D, and E initially isolated from the tunicate
Eudistoma cf.
rigida [
77,
78] and later synthesized [
79] were the most notable ones among the group of pyrrole alkaloids but have been shown to have low activity regarding cultivated human cancer cells. Further investigations of synthetic compounds led to development of the promising approaches to modification of the 7-deazaxanthine skeleton that typical of rigidins converting into corresponding 7-deazahypoxanthines (
Figure 4).
Novel marine rigidin analogues C2-aryl- and C2-alkyl-7-deazahypoxanthines obtained via synthesis techniques were proposed for construction of the pyrrolo[2,3-d]pyrimidine ring system and applied in submicromolar and nanomolar quantities exerting strong anti-proliferating activity against different cell lines including multidrug resistant tumors such as glioblastoma, melanoma, and non-small cell lung cancer. As the only difference between 7-deazaxantine rigidin scaffold and 7-deazahypoxanthines skeleton structure is the lack of carbonyl group at C2 in the latter structure, such modifications in this position are supposed to be critical for their activity. It was demonstrated that one of the C2-methyl-7-deazahypoxanthines, namely 6-benzoyl-2-methyl-5-phenyl-1H-pyrrolo[2,3-d]pyrimidin-4(7H)-one, exerted the greatest antiproliferative activity against glioblastoma cell line U-87 (GI
50 = 0.077 ± 0.002 μM), human cervical adenocarcinoma HeLa (GI
50 = 0.029 ± 0.001 μM), breast adenocarcinoma MCF-7 (0.035 ± 0.003 μM), and lung carcinoma cell line A549 (0.25 ± 0.01 μM). Other synthetic C2-methyl-7-deazahypoxanthines also exerted substantial antiproliferative effects against U-87 cell line with a concentration range from 0.90 ± 0.16 μM to 9.23 ± 2.13 μM being inferior to the original C7-phenyl analogue but still preserving submicromolar potency, except only one derivative demonstrating micromolar potency (
Table 6). The drop of the activity of this analogue could be also explained by its polar character impairing cell membrane permeability.
Synthetic 7-deazahypoxanthines were shown to be capable of disrupting microtubule cytoskeleton organization in the tumor cells via binding to the colchicine site of β-tubulin [
79]. One of the 7-deazahypoxanthines at concentrations between 1 and 2 μM induced significant alterations of the mitotic microtubule organization when cultivating on the HeLa cell line. The interphase microtubules were less affected at these concentration range suggesting that this compound was primarily affecting dynamic microtubules. It exerted slight influence on the stable interphase or spindle microtubules but at the same time induced a marked mitotic spindle shift that may be related to the astral microtubule defects. Astral microtubules as the most dynamic microtubule population at mitotic stage are likely to be the most sensitive targets for tubulin-targeting drugs such as 7-deazahypoxanthines [
79].
2.5. Pyrrospirone Alkaloids
Novel pyrrospirone alkaloids C, D, E, F, G, H, and J as well as penicipyrroether A were isolated from marine fungus
Penicillium sp. (ZZ380 strains) generally found in the wild sea crab
Pachygrapsus crassipes. These alkaloids along with other chemically related compounds make a family of fungal secondary metabolites, which are also called hirsutellones and contain a unique 13-membered ether ring composed of specific structural units such as decahydrofluorene, para-cyclophane, and pyrrolidinone. That fungal metabolite family were isolated from fungi belongings to the genera Cylindrocarpon, Embellisia, Hirsutella, Lewia, Neonectria, Penicillium, and Trichoderma and are divided into four chemical groups: hirsutellones, pyrrospirones, pyrrocidines, and GKK1032s [
80]. Pyrrospirones C and D have the same molecular formula (
Figure 5). From the structural point of view, both of them are composed of two carbonyls, six aromatic carbons, two olefin carbons, four quaternary carbons, two oxymethines, one methoxyl, six methines, five methylenes, and five methyls as well as they have closed spiro ring system but different configurations at C-17. In a similar fashion, pyrrospirones E and F have the same molecular formula. The
13C NMR data of pyrrospirones E and F show close similarities with those of pyrrospirones C and D, respectively, except the different chemical shifts for C-19 to C-21 due to the absence of a methoxy at C-19 in pyrrospirones E and F. This indicates pyrrospirones E and F are the analogues of pyrrospirones C and D without methoxyl at C-19. Similar to pyrrospirones C and D, the structural difference between pyrrospirones E and F is related to different C-17 configurations. Molecular formula of pyrrospirones G is two protons less than that of pyrrospirones E. Structural difference between pyrrospirones G and E presented with oxymethine at C-17 in E is replaced by a carbonyl group in G compound. That is 16 mass units less than that of pyrrospirone E. Pyrrospirone H is considered a pyrrospirone E analogue with a methylene attached at C-17. Pyrrospirone I is two protons less than that of pyrrospirone H. The pyrrospirone I structure is considered as analogue of pyrrospirone H with a double bond at C-16 and C-17 [
80]. Despite structural parts of rings A–C, F, and G for penicipyrroether A and pyrrospirones C–I are the same, the structures of penicipyrroether A and pyrrospirones have specific differences. First of all, D ring contains five-membered ether ring for penicipyrroether A and cyclohexanone for pyrrospirones. Then, penicipyrroether A does not have spiro junction for rings D and E. NMR spectroscopic data analysis and HRESIMS data (high resolution electrospray ionization mass spectroscopy) demonstrated that compound pyrrospirone J is an analogue of penicipyrroether A with the same structural part of rings A, B, F, and G but possesses an epoxy moiety at C-10 and C-11 and a different five-membered ether ring D fused with ring E through a spiro carbon of C-15. In addition, the dehydro-pyrrolidinone moiety (ring E) in penicipyrroether A is replaced by a pyrrolidinone moiety in pyrrospirone J [
81].
Inhibiting activity regarding proliferation of the glioma cells U87MG, U251, SHG44, and C6 of the pyrrospirone alkaloids isolated from the ZZ380 strain were investigated. The results showed that pyrrospirone G exerts relatively strong activity towards aforementioned cell lines with IC
50 values approximately between 1.06 and 8.52 μM. Pyrrospirones C, D, E, F, H, I, and J have demonstrated moderate anti-glioma activity with IC
50 values generally between 7.44 and 26.64 μM (
Table 7) [
80]. Penicipyrroether A exerted strong inhibiting influence on the cell proliferation in glioma lines U87MG and U251 with the IC
50 values 1.64 and 5.50 μM, relatively [
81]. Doxorubicin used as a positive control have suppressed glioma cell proliferation with IC
50 approximately 1.20 and 8.03 μM, respectively. These results suggest that antiglioma activity of penicipyrroether A is equivalent or slightly stronger than doxorubicin potency. Furthermore, cytotoxicity of penicipyrroether A and doxorubicin against normal human astrocytes (HA, cat. no. 1800, ScienCell, Carlsbad, CA, USA), expressed in CC
50 was shown to be 23.28 ± 1.05 μM and 8.57 ± 0.16 μM, respectively. Selectivity index (CC
50/IC
50) of penicipyrroether A towards U87MG and U251 cells was found to be 14.2 and 4.2, respectively, which is substantially higher than that of doxorubicin i.e., 7.1 and 1.1, respectively [
81].
Penicipyrrodiether A containing phenol A derivative fused to the pyrrolidinone core via the five membered ether ring added have shown the moderate antiglioma activity manifesting in reduced proliferation of the glioma U87-MG, U251, SHG-44, and C6 cells with IC
50 values between 11.32 and 29.10 μM. Doxorubicin used as positive control was found to exert its activity with IC
50 values 0.70 to 9.61 μM [
80].
Therefore, among the pyrrospirone alkaloids that have been studied, at least two compounds namely pyrrospirone G and penicipyrroether A are worth attention as promising antiglioblastoma agents.
2.6. Pyrrocidine Alkaloid
Novel pyrrocidine alkaloid trichobamide A isolated from the ascidian-derived fungus
Trichobotrys effuse 4729 had shown significant inhibition of the cell proliferation in two glioma lines such as U251 and SNB19. This novel alkaloid is different due to the presence of unprecedented tetrahydro-5H-furo[2,3-b]pyrrol-5-one moiety (
Figure 6). Structurally, trichobamide A represents the first example of tetrahydro-5H-furo[2,3-b]pyrrol-5-one ring system and biosynthetically is related to a family of pyrrocidine alkaloids bearing a macrocyclic ether and succinimide-derived moieties.
Within the concentration range from 1 to 10 µM trichobamide A exerted concentration- and time-dependent inhibiting effects against glioma cells. It was found out that the mechanism of antiproliferative activity of trichobamide A is directly related to increased relative expression of proteins P53, Bax, caspase 3, and caspase 9 as well as reduced expression of Bcl-2 in glioma cells. Trichobamide A was discussed to induce apoptosis in glioma cells through a pathway upregulated by P53/Bax/Bcl-2 [
82].
2.7. Alkaloid Pseurotin A
An alkaloid pseurotin A initially isolated from the cultures of some fungus species such as
Pseudeurotium ovalis,
Aspergillus spp. and
Hericium erinaceum, was later isolated from a culture broth of marine bacterium
Bacillus sp. (the strain FS8D), marine barnacle
Lepas anatifera. The pseurotin A was shown to be highly active in reducing proliferation of four different glioma cells with IC
50 values of 0.51–29.3 μM [
83] (
Table 8). Doxorubicin used as positive control in the same cell lines had exerted antiglioma activity with IC
50 0.5–9.6 μM.
Mechanisms of antitumor activity were investigated in the U87-MG cell line. Pseurotin A was shown to reduce the expression levels of PKM2 and especially LDH5, which is a key enzyme responsible for accelerating conversion of pyruvate to lactate, the final product of tumor glycolysis. In addition, pseurotin A upregulates expression of pyruvate dehydrogenase beta (PDHB), adenosine triphosphate synthase beta (ATPB), and cytochrome C (Cyto-C) [
83], which are main enzyme components in the processes of tricarboxylic acid (TCA) cycle and oxidative phosphorylation in normal cells.
Based in the results obtained through the study, authors have proposed that pseurotin A exerts anti-glioma effects via inhibition of the accelerated rate of glycolysis int the glioma cells through the downregulation of PKM2 and LDH5 expression. Also, it alters tumor metabolic pathway to the process of oxidative phosphorylation by enhancing TCA cycle and oxidative phosphorylation activities through the upregulation of PDHB, ATPB, and Cyto-C.
2.8. Polycyclic Diamine Alkaloids
Polycyclic diamine alkaloids represent the specific class of alkaloid compounds that are mostly composed ammonia, propenal, and long-chain dialdehydes as the universal building blocks. These alkaloids are typically found in the marine sponges belonging to the order
Haplosclerida with a major part of species represented with four families of that order (
Callyspongiidae,
Chalinidae,
Niphatidae, and
Petrosiidae). Two recently described alkaloids namely papuamine and haliclonadiamine as well as two newly discovered compounds called neopetrocyclamine A and neopetrocyclamine B were isolated from the Indonesian sponge
Neopetrosia cf
exigua and then screened for their efficacy in vitro against glioblastoma cell line SF-295. A symmetrical diamine compound papuamine was originally discovered from a marine sponge belonging to the genus
Haliclona from Papua-New Guinea [
84] and from Palau [
85]. In the Palau specimens, papuamine was just a minor component in its mixture with haliclonadiamine, which is an unsymmetrical diastereoisomer of papuamine.
Neither neopetrocyclamine A nor neopetrocyclamine B had shown significant cytotoxicity at 20 μM. Nevertheless, both alkaloids papuamine and haliclonadiamine demonstrated strong inhibiting influence against glioblastoma cells (
Table 9). As it is shown in the table, papuamine is more potent than haliclonadiamine against glioblastoma SF-295 cells because its GI
50 value is almost 8-fold lower [
86]. Authors suggested that the stereogenic center at C-6 of the papuamine play important role in the anti-glioma activity.
Papuamine was recently shown to possess antimetastatic activity against MDA-MB-231 breast cancer cells and reduce tumor cell vitality via mitochondria damage and JNK activation [
87]. However, specific molecular targets and mechanisms of the anti-glioma effects exerted by these compounds are to be studied yet.
2.9. Polycyclic Granulatimide Alkaloids
Alkaloids granulatimide and isogranulatimide isolated from ascidian
Didemnum granulatum were shown to exert inhibiting activity against checkpoint kinase 1 (Chk1) [
88].
The result of the molecular docking study suggested that amino group at the para-indolic position in the granulatimide framework could be directed towards the binding site opening. After that, 17 amido and amino analogues of granulatimide and isogranulatimide were tested in the oligodendroglioma Hs683 and glioblastoma U373 cells and the results demonstrated that two derivatives with open structure exerted antiglioma activity exceeding that of the granulatimide. Their anticancer effects were noted at micromolar concentrations (
Table 10) [
89]. It is worth noting that the closure of the open structure into the corresponding final product usually resulted in the lowered in vitro tumor growth inhibition. Nevertheless, no relevant inhibition of Chk1 was detectable with these structures [
89]. That may suggest the presence of other pathways playing important role in the inhibition of the glioma cell proliferation.
8. Antraquinones
One novel antraquinone and two already known ones were extracted from actinomycete
Streptomyces sp. ZZ406 isolated from the sea anemone
Haliplanella lineata. A new anthraquinone was elucidated as 1-hydroxymethyl-8-hydroxy-anthraquinone-3-carboxylic acid. Two antraquinones were previously described as 1,8-dihydroxy-3-methyl-anthraquinone (also known as chrysophanol or chrysophanic acid), and 3,8-dihydroxy-1-methyl-anthraquinone-2-carboxylic acid. It has been found that new anthraquinone had a promising activity against different glioma cells, which is comparable with antiglioma effects exerted by doxorubicin (
Table 30). At the same time, cytotoxicity of the novel compounds toward normal astrocytes was extremely low with CC
50 values greater than 100 μM whereas that of doxorubicin is 8.7 ± 1.2 μM. Therefore, selectivity index (CC
50/IC
50) of doxorubicin is 0.9 (for U251 cells), 3.5 (for SHG44), and 4.6 (for U87MG), whereas those values for the novel antraquinone were—>17.5, >12.3, and >21.3, respectively. Presented parameters of antiproliferative activity and selectivity index indicate the presence of substantial advantages of the new antraquinone, at least, in comparison to doxorubicin. Already known antraquinones isolated from
Streptomyces sp. ZZ406 also demonstrated high antiglioma efficacy and activity of chrysophanol was significantly greater than that of doxorubicin [
143].
New antraquinone was applied in concentration 30.0 μM and significantly reduced HK2, PFKFB3, PKM2, and LDH5 expression level in the U87MG cells. Based on the results indicating strong activity against glioma cells with extremely high selectivity index and unique antiglioma mechanism the authors of that work assume that the new antraquinone possesses a good potential of antiglioma agent [
143].
Marine strain of
Streptomyces sp. 182SMLY isolated from a sediment sample collected from the East China Sea was found to be a source of two new polycyclic antraquinones, which were elucidated as N-acetyl-N-demethylmayamycin and streptoanthraquinone A. Cytotoxic and antibacterial agent mayamycin was previously isolated from
Streptomyces sp. strain HB202, a symbiotic bacterium of the marine sponge
Halichondria panicea [
144]. Both anthraquinones remarkably suppressed the proliferation of four different glioma cell lines with IC
50 values within a range from 0.5 to 7.3 μM (
Table 31). Doxorubicin as a positive control exerted its activity with IC
50 0.9–9.0 μM. IC
50 values towards the normal human astrocytes were about 25 ± 1.3 μM for N-acetyl-N-demethylmayamycin and greater than 100 μM for streptoanthraquinone A. IC
50 ratios between normal astrocytes and glioma cells were 6.4–50 for N-acetyl-N-demethylmayamycin and greater than 14–31 for streptoanthraquinone A. Those values indicate high selectivity index of antraquinones. Also, N-acetyl-N-demethylmayamycin (0.7 μM) and streptoanthraquinone A (3.3 μM) significantly induced apoptosis in the glioma U251 cells: the total number of apoptotic cells (early and late apoptotic cells) were increased by 38.76% and 36.67%, respectively, after 36 h of treatment when compared to control (3.58%) [
145].
The two anthraquinone derivatives 1′-deoxyrhodoptilometrin and (S)-(−)-rhodoptilometrin, isolated from the sea lily
Colobometra perspinosa, exerted moderate anticancer activity in vitro against human glioblastoma cell line SF-268 [
146]. Both compounds from the sea lilies
Comanthus sp. also demonstrated toxic effects against C6 glioma cells (
Table 32). The higher cytotoxic activity of 1′-deoxyrhodoptilometrin indicates the relevance of the hydroxyl group at C-1′ position for anti-glioma activity of the quinone structure. 1′-Deoxyrhodoptilometrin, but the (S)-(−)-rhodoptilometrin induced a detected apoptotic cell death via caspase 3/7 activity increase: significant higher enzyme activity was found in C6 glioma cells in 24 h of agitation with concentrations 25 μM and higher. 1′-Deoxyrhodoptilometrin applied in concentration 25 μM also induced necrotic cell death in glioma cells in 24 h agitation period that was detected due to increased LDH activity in the supernatant. (S)-(−)-rhodoptilometrin caused a slight increase in the LDH activity only at the highest concentration used (50 μM). Both compounds did not cause oxidative stress as no increased accumulation of the reactive oxygen species (up to 50 μM) were noted in the C6 cells. They also did not activate the Nrf2/ARE signaling pathway. In addition, both these compounds did not affect transcription factor NF-κB in the H4IIE cell used as a model system that was stimulated by the cytokine TNF-α. Effects of those compounds on the protein kinases involved in different signal transduction pathways associated with cell proliferation (Aurora-A, Aurora-B, CDK2, CDK4, EGF-R, ERBB2, and others), survival (Akt1), angiogenesis (VEGF-R2, VEGF-R3), and metastasis (FAK, MET, SRC) were analyzed. The results demonstrated both antraqionones are highly potent inhibitors of various kinases, in particular, the ones involved into the cell proliferation and angiogenesis [
147]. The protein kinases Aurora-A and Aurora-B were inhibited by both compounds with IC
50 values of 3.0 and 1.81 μM for 1′-deoxyrhodoptilometrin and 4.14 and 4.14 μM for (S)-(−)-rhodoptilometrin, respectively. Aurora kinases are known as potential anticancer drug targets, since they are involved in the control of chromosome assembly and segregation during mitosis.
1′-Deoxyrhodoptilometrin and (S)-(−)-rhodoptilometrin inhibited the VEGF with IC50 values of 1.87 and 18.76 μM. That result demonstrates high activity of 1′-deoxyrhodoptilometrin inhibiting mentioned protein kinase 10 times more effectively than (S)-(−)-rhodoptilometrin. Other important protein kinases that were inhibited by 1′-deoxyrhodoptilometrin and (S)-(−)-rhodoptilometrin were cyclin-dependent kinases (regulation of cell cycle), SAK kinase (mitotic regulator), insulin-like growth factor receptor, FAK (cellular adhesion and spreading processes), and EGFR (cell proliferation).
The EGFR family of the tyrosine kinases receptors consists of distinct receptors. EGFR was inhibited by those compounds with IC
50 values 4.0 µM (1′-deoxyrhodoptilometrin) and 12.4 µM ((S)-(−)-rhodoptilometrin), ERBB-2 was inhibited with IC
50 values 6.7 µM (1′-deoxyrhodoptilometrin) and 12.1 µM ((S)-(−)-rhodoptilometrin). ERBB-4 was inhibited only by 1′-deoxyrhodoptilometrin with an IC
50 value 9.4 µM [
147]. It is already known that a constitutive activation of the MAPK signaling pathway occurs in many tumors. It was discovered that 1′-deoxyrhodoptilometrin substantially decreased ERK MAP kinase (p44/p42) phosphorylation in the glioma C6 cells despite the whole amount of ERK protein remained unchanged. Therefore, protein kinase inhibition may be one of the main mechanisms contributing antraquinone’s anti-glioma activity.
9. Marine Nucleosides: Trachycladines
Naturally occurring modified nucleosides and its synthetic analogues exerting a wide spectrum of biological activity including anticancer effects [
148] are considered as a key lead compounds for the drug discovery and development. Initially marine modified nucleosides trachycladines A and B were isolated from the sponges
Trachycladus laevispirulifer [
149] and later from another sponge of the genus
Theonella. They both contain carbohydrate 2-C-methyl-D-5-deoxyribofuranose part, that was never found in natural sources before, and they differentiate by the nucleic bases (2-chloroadenine for trachycladines A and hypoxanthine for trachycladines B) attached to the carbohydrate part. Trachycladine A represents a carbohydrate-modified analogue of 2-chloro-2′-adenosine, whereas trachycladine B is a carbohydrate-modified analogue of inosine (
Figure 16). The first studies revealed a potent in vitro cytotoxic activity of trachycladine A against human colon cancer, leukemia, and breast cancer cell lines with IC
50 values ranging from 0.3 to 3.0 μM [
149]. Trachycladine A exerted significant cytostatic effects on the glioblastoma T98 cell line when applied in concentrations 50 and 100 µM, and against U87 cell in concentrations 10, 50, and 100 µM. U87 cell line was found to be more sensitive to trachycladines. 50 μM concentration of trachycladine A reduced viability of those cells by 70%, and viability of the T98 cells by 50% [
150].
Two trachycladine analogues, diacetate of the 2,6-dichloropurine derivative (
Figure 17,
3) and N-cyclopropyl trachycladine A (
Figure 17,
4) exerted greater activity against glioblastoma cells.
Comparison of the antiglioma properties of two natural trachycladines showed that trachycladine A definitely is more active compound. Cytotoxicity analysis of several synthetic analogues showed that nucleosides containing chlorine atom at C2 of purine ring are generally more active. On the other hand, N-cyclopentyl analogue of trachycladine A (
Figure 17,
5) demonstrated much lower activity than the corresponding N-cyclopropyl analogue and trachycladine A itself. Diacetate structure is following the same C2 chlorination pattern but it lacks the C-6 amino group, it also proved to be highly cytotoxic, probably due to the better cellular uptake because of its higher lipophilic nature. Additional experiments helped reveal the mode of action of active trachycladines relying more on mitotic catastrophes rather than DNA damage. Their activity as autophagic flux blockers was also postulated [
150].
10. Glycosphingolipids and Sphingosins
Glycosphingolipids (GSLs) represent a group of biomolecules containing two basic structural units: hydrophobic ceramide moiety and hydrophilic oligosaccharide chain. The ceramide part is made up of a sphingosine moiety composed a long amino alcohol chain with 18–20 carbon atoms and a long fatty acid chain [
151].
The cancer-associated GSLs have been considered as the tumor markers, and used as diagnostic markers and targets of cancer treatment [
152,
153]. On the other hand, bioactive GSLs exert some pharmacological effects—e.g., antimalarial activity—as well as immunomodulating and antitumor activities [
154,
155].
Various cerebrosides namely glycosylceramides were isolated from marine sponges (Porifera), ascidians (Chordata), octocorals and sea anemones (Cnidaria), and starfishes and sea cucumbers (Echinodermata) [
151]. Thus, these marine animals are considered a source of bioactive GSLs.
A bioactive glycolipid fraction obtained from the lipid extract of starfish
Narcissia canariensis was selected for its ability to significantly inhibit KB (human oral epidermoid carcinoma) cells proliferation. The fraction contained three homologous glycosphingolipids with β-glucopyranoside as a sugar head, 9-methyl-branched 4,8,10-triunsaturated long-chain aminoalcohol as sphingoid base and amide-linked 2-hydroxy fatty acid chains. Their majority (63%) had an amide-linked 2-hydroxydocosanoic acid chain and was identified as the ophidiacerebroside C (firstly isolated from the starfish
Ophidiaster ophidiamus). The minor components differed by the presence one methylene group were corresponding to ophidiacerebroside B and ophidiacerebroside D. It was found that the final glycolipid fraction demonstrated moderate cytotoxic activity on astrocytoma cells obtained after tumor resection of patients with glioblastoma multiforme-primary culture after 24 h of treatment (
Table 33) [
154].
Three glycosphingolipids isolated from the marine sponge
Axinyssa djiferi were named axidjiferosides A, B, and C. They contained an unsaturated long-chain amino alcohol as a sphingoid base. The sugar linked to the ceramide was identified as galactopyranose. The sphingoid base was identified as 2-amino-1,3,4-trihydroxy-octadecene. Generally, axidjiferosides were identified as three homologous β-galactopyranosylceramides composed of 2-amino-(6E)-octadec-6-en-1,3,4-triol and the major one, axidjiferoside A, containing 2-hydroxytetracosanoic acid [
155]. These glycosphingolipids in a form of total fraction exerted low cytotoxicity against astrocytoma cells obtained from the tumor resection in patients with glioblastoma multiforme (
Table 34).
Sphingosins
Two modified long-chain sphingoid C18 bases: (2R,3R,6R,7Z)-2aminooctadec-7-ene-1,3,6-triol and (2R,3R,6R)-2-aminooctadec-1,3,6-triol, named halisphingosine A and halisphingosine B, respectively, were isolated from ethyl acetate fraction of methanol extract of the marine sponge
Haliclona tubifera. Ethyl acetate fraction in contrast to the aqueous and hexane fractions exerted cytotoxic effect in the U87 glioma and SH-SY5Y human neuroblastoma cell lines (
Table 35) [
156].
Within the recent years, sphingosines are gaining recognition as important signaling mediator of apoptosis. Possible mechanisms involved in sphingosine-mediated cancer cell death are related to its kinase modulating potential, mainly, regarding protein kinase C (PKC), protein kinase A (PKA), and protein kinase Cδ (PKCδ). PKA activation and PKCδ cleaving are associated with regulation of the dimeric 14-3-3 protein function displaying a vital clue of the control pro-apoptotic mediators such as BAD and signal-regulating kinase 1 (ASK-1). Activation of the Jun N-terminal kinases (JNK) is subsequently regulated by ASK-1 [
157].
Second suggested mechanism of action may be related to the sphingolipid biosynthetic pathway, which is known to be activated in response factors resulting in accumulation of ceramide and sphingosine in apoptotic cells [
157]. Halisphingosines A and B may be a substrate for ceramide synthase and sphingosine kinase resulting in sphingosine converting either into ceramide or sphingosine-1-phosphate (S1P). Ceramide exert antiproliferative effects inducing apoptotic mediators, whereas S1P promotes cell survival and inhibition of apoptosis [
158]. Based on the experimental results as well as on knowledge about sphingolipid synthesis and signaling role in debilitation of cell proliferation in U87MG glioma cells [
159], authors came to a conclusion that possible cytotoxic mechanism of the sphingosines from
H. tubifera marine sponge is elucidated by induction of apoptotic mediators and ceramide production. In addition, ethyl acetate fraction was shown to be able to inhibit the production of peroxyl radicals [
156].
11. Psammaplins
Natural compound named psammaplin A possessing a wide spectrum of biological activities was initially isolated by several independent research teams from marine sponge
Psammaplysilla (revised to
Pseudoceratina) sp. or from unidentified sponges back in 1987 [
160]. Actually, psammaplin A is found in marine microalgae, cyanobacteria, and in the heterotrophic bacteria living in association with invertebrates (e.g., sponges, tunicates, and soft corals). Psammaplin A was found to have unique symmetrical structure of the disulfide bromine tyrosine dimers with phenol properties and became a starting substance for the whole psammaplin family. Later, psammaplin A was synthesized by Hoshino et al. [
161] and that allowed to produce a wide variety of its derivatives and then investigate their antitumor and other activities [
162]. Later, psammaplins C, E, F, G, and K were also obtained [
163].
Psammaplin A was confirmed to possess antiproliferative effects against various cancer cell lines including triple-negative breast (MDA-MB-231), doxorubicin-resistant human breast (MCF-7/adr), colon (HCT15), ovarian (SK-OV-3), lung (A549, LM4175), bone (BoM1833), endometria, brain (BrM-2a), skin (SK-MEL-2), and central nervous system (XF498) cancer cell lines [
162,
164]. Cytotoxic effects of psammaplin A are related to the multiple enzyme inhibition such as topoisomerase II, farnesyl protein transferase, mycothiol-S-conjugate amidase, leucine aminopeptidase, DNA polymerase α-primase, aminopeptidase N. In addition, psammaplin A activates peroxisome proliferator-activated receptor gamma (PPARγ) and induces apoptosis in MCF-7 cells [
165]. Psammaplin A and its derivatives, psammaplin F and psammaplin G were discovered as highly potent inhibitors of DNA methyltransferase (DNMT) and histone deacetylases (HDAC) playing critical roles in the epigenetic regulation of gene expression [
163]. Structural modification with the following investigations of structure–activity relationships demonstrate that disulfide bonds and the oxime moieties are indispensable for the antibacterial and antiproliferative activities of psammaplin A.
Psammaplin A (Sigma Chemical Co., St. Louis, MO, USA) in the in vitro experimental setting suppressed cell viability in the glioblastoma U373MG with IC
50 values 5 μg/mL after 18 h agitation period (
Table 36) [
166].
One of the important effects of psammaplin A with high clinical value is related to its influence on the radiosensitivity of the human cancer cells. Investigation of the psammaplin A influence on the radiosensitivity of the glioblastoma U373MG cells have shown that pretreatment with psammaplin A resulted in increased radiosensitization U373MG cells and psammaplin A significantly enhanced radiation induced cell death in U373MG. The dose enhancement ratio (DER) was defined using experimentally adjusted dose necessary for the survival fraction. DER for psammaplin A in U373MG cells was 1.29. It is known that DNMT1, DNMT3A, and DNMT3B are the main functional methyl transferases responsible for setting and maintaining DNA methylation in mammas. Psammaplin A was shown to provoke dramatic reduction of the DNMT1 and DNMT3A expression in the U373MG cells and does not affect DNMT3B expression (at least in a dose 5 μg/mL) [
166].
Previously apoptosis was considered as a potential radiosensitization mechanism. Different results were reported indicating the role of apoptosis as a radiosensitizing mechanism induced by DNMT inhibitors. Some studies have shown that combination of radiation and DNMT inhibitor (zebularine) does not increase significantly sub-G1 population of apoptotic cells [
167]. On the other hand, DNMT inhibitor (5-aza-2′-deoxycytidine) was demonstrated to induce radiosensitization in the gastric cancer cell line via induction of accelerated rate of apoptosis that was indicated by increased expression of the p53, RASSF1, and DAPK gene expression [
168]. Psammaplin A was reported earlier to exert cytotoxic influence on the cancer cells via selective induction of the genes related to apoptosis [
168]. This effect of psammaplin A was assumed to lead to the increased radiation induced apoptosis. However, such radiation induced apoptosis was noted in the A549 cell line only and was not found in the U373MG line. That may be explained, at least partially, by the state of the p53 expression. Cell line U373MG contains mutated p53 and may be relatively more resistant to the radiation apoptosis because generally apoptosis is linked to p53 protein. DNA recovery (repair) is another process occurring in the cellular radiosensitizing determination, and DNA reparation activation in the cancer cell after sublethal DNA damage induced by radiation may be one of the resistance factors. Gamma-H2AX been identified as a marker of DNA double-strand break and immune cytochemical analysis with anti-γH2AX-antibodies showed that γH2AX expression zones in cells treated with radiation only goes down in time but can be prolonged (remains unchanged) within 24 h period in the U373MG cells treated with DNMT inhibitor before radiation procedure [
166]. These results suggest that inhibition of the DNA damage recovery process is a mechanism that makes a base for radiosesitisizing effects of the DNMT inhibitors in glioma cells. Therefore, psammaplin A possesses a potential to increase radiosensitivity in U373MG glioblastoma cells probably via suppression of the DNA reparative processes.
One of the anti-glioblastoma mechanisms of psammaplins may be related to the induction of the autophagic flux in tumor cells. This phenomenon is of great importance because at present targeting autophagic pathways is thought might play a critical role in designing novel chemotherapeutic approaches in the treatment of human cancers, and the prevention of tumor-derived drug resistance. A number of pharmacological compounds including psammaplin A shown to induce autophagy in various human tumor cells, and some of these compounds affect expression of TP53 protein in cancer cells via cell cycle arrest in the G
0/G
1 phase and expression of cyclins [
169].
Psammaplin A isolated from the Psammaplinaplysilla sponge decreased cell viability of glioblastoma U87MG cells in a dose-dependent and time-dependent manner with IC
50 ~7.5 μM for 24 h [
164]. This concentration was used for the subsequent molecular analysis of expression of the tumor protein (TP)-p53 family members and their autophagic target genes. Psammaplin A led to a marked increase (4.3 × fold) in the protein levels for TP73α and dramatically induced TP73α phosphorylation (8.7 × fold) that may be a mechanism reducing tumor cell survival and inducing cell death. The present data demonstrate that the autophagy pathway is one of the molecular mechanisms leading to the tumor cell survival modulation [
170]. Although autophagy can serve as a pro-survival phenomenon; it often delays tumor cell death via apoptosis, essentially contributing to the demise of tumor cells upon treatment with the anticancer compounds of various origins. TP53 family proteins were found to play a critical role in autophagy signaling [
171] and induce the multiple molecular pathways including transcriptional activation of genes targeting autophagic machinery [
172]. It was found out that psammaplin A activates transcription of autophagic genes through TP53 family member′s transcriptional function. It upregulates expression levels of ATG5 and UVRAG in U87MG cells and promotes ATG5 and UVRAG promoter activities. Psammaplin A have been demonstrated also to be able to stimulate expression of autophagic proteins involved into autophagy signaling in human glioblastoma tumor cells in vitro. Treatment of U87MG cells with 7.5 μM psammaplin A led to a substantial increase in the ATG5 and UVRAG protein expression [
164]. Therefore, psammaplin A has a capacity to upregulate expression of autophagic signaling intermediates in human glioblastoma cells in vitro through a transcriptional regulation by TP53 family members.
Low efficiency of the chemotherapy of the brain malignancies due to the high chemoresistance of the glioblastoma stem cells remains problematic for modern neuro-oncopharmacology. This particular subpopulation of the tumor cells governs tumor initiation and recurrence, they are particularly difficult to be eradicated with chemotherapy. One of the causes of that phenomenon is an elevated expression of P-glycoprotein (Pgp), which is an efflux pump that recognizes chemotherapeutics including temozolomide and other anti-glioma drugs as substrates and pushes them out of the cells [
173,
174]. Implementation of the direct Pgp inhibitors in combinatorial therapy was rather not successful because of serious adverse events and toxicity. This toxicity is attributed to inhibition of Pgp present in healthy tissues and unexpected drug–drug interactions. Indirect inhibition of Pgp via influencing the carbonic anhydrase activity was thought an alternative approach. Tumor acidosis is known as a hallmark of cancer. Membrane-bound carbonic anhydrases IX (CAIX) and/or XII (CAXII) colocalized with the membrane drug efflux protein, Pgp, in a range of drug resistant cancer cells including glioblastoma, maintain the intracellular/extracellular pH for efficient Pgp activity, and optimal tumor growth, invasion, and metastasis [
175]. Nowadays, CAIX- and CAXII-specific inhibitors are considered as potential antitumor agents that indirectly reduce Pgp activity and resensitize solid tumors to Pgp substrates [
176].
One of the most potent inhibitors of CAIX and CAXII enzymes today is one of the psammaplin A derivatives, namely, psammaplin C isolated from the marine sponge
Pseudoceratina purpurea in 1991 [
177]. The structure of psammaplin C comprises several structural elements: (I) a 3-bromo-4-hydroxy benzylidene moiety; (II) an oxime group; and (III) an aminoethane sulfonamide chain (
Figure 18). Alteration of the carbon anhydrase inhibiting activity of psammaplins on the human Ca isoenzyme panel showed that psammaplin C had exceptionally strong inhibition activity for hCA XII, with an inhibition constant (Ki) 0.79 nM. For comparison: acetazolamide, the par excellence therapeutically established CA inhibitor, had shown inhibition activity for hCA XII, with a Ki 5.7 nM [
178]. Subnanomolar CA inhibition is uncommon. Moreover, one of the synthesized psammaplin C derivatives, which is a free oxime derived from the thiadiazoyl sulfonamide scaffold, showed very strong inhibition of all the CA isozymes, in particular CA XII with a Ki of 0.56 nM [
179].
Experimental studies in mice with orthotopically implanted glioblastoma neurospheres derived tumors (with coexpression of Pgp and CAXII) or xenografts derived from the patients showed that co-therapy of temozolomide with a CA XII inhibitors may more effectively affect glioblastoma by suppressing an important temozolomide resistance mechanism. CA XII inhibitors themselves did not affect tumor growth in the mouse model studies and did not prolong life span of experimental animals, but significantly enhanced efficacy of temozolomide. Simultaneously, application of the CA XII inhibitors was particularly effective against the glioblastoma with high anti-TMZ resistance. The combination of TMZ with CA XII inhibitors rescued the antiproliferative and proapoptotic effects of TMZ, as verified by reduced intratumor-positive immunostaining for Ki67 and increased activation of caspase-3 [
175,
179]. Thus, although TMZ alone reduced Pgp expression and activity in neurospheres, this reduction was not sufficient to yield antitumor efficacy in vivo. The greater inhibition of Pgp, as achieved by combining the potent CA XII inhibitor (psammaplin C derivative) with TMZ, was however able to restore the intracellular cytotoxic levels of TMZ and facilitate enhanced drug efficacy. It should be emphasized that psammaplin C shows no toxicity and is only effective if used in combination with a chemotherapy [
175].
Therefore, a new combination therapy, based on a CA XII inhibitor with TMZ may be considered as a potentially viable clinical tool to overcome Pgp-mediated TMZ-resistance in glioblastoma stem cells as well as more effective therapy of the brain tumors [
179].
12. Xyloketals
The xyloketal group of natural compounds was isolated from the mangrove fungus
Xylaria sp. This group of related ketals is structurally unique [
180]. Xyloketal A has C3 symmetry with a cis-junction between the tetrahydropyran and tetrahydrofuran rings (
Figure 19), the other members are missing axial symmetry. The methyl groups at C-5 of C rings are also oriented syn to other methyl groups at C-2 placed between the oxygens in the spiroketal functions. The angular skeleton of xyloketal B, a bis adduct analogue of the tris adduct xyloketal A, is more stable than the linearly condensed xyloketal C, which spontaneously rearranges in solution to the more stable angular structure of xyloketal B. Xyloketal D is an acetylated mono adduct structure, and xyloketal E is a tetrahydrofuran-linked angular bis adduct related to xyloketal B. Some studies reported that xyloketal B exerts several bioactive functions including anti-stress and anti-ageing, neuroprotective effects, and antioxidant activity [
181].
24 h long treatment with xyloketal B in various concentrations of (from 31.25 to 1000 μM) reduced U251 cell viability in a concentration-dependent manner. Cell viability was significantly decreased by 85.4% ± 2.9%, 61.4% ± 4.3%, 12.2% ± 2.6%, and 1.3% ± 0.1% in comparison to control for 125, 250, 500, and 1000 μM xyloketal B concentration, respectively (
p < 0.05). Nonlinear curve fit was carried out to evaluate the dose–response of xyloketal B with IC
50 287.1 ± 1.0 μM (
Table 37). Xyloketal B applied in the concentration range 37.5–300 μM exert sell proliferation inhibiting activity depending on the period of application and concentration. It should be emphasized that xyloketal B in concentrations <300 μM generally exerted only cell proliferation inhibition activity but not a cytotoxic effect. In concentration greater than 300 μM xyloketal B significantly inhibited cell migration in U251 line [
182].
Experiment devoted to investigation of the mechanisms providing antiproliferative activity xyloketal B have shown that p-Akt and p-ERK1/2 protein expression in the U251 cells was significantly decreased after xyloketal B treatment in concentration 300 μM for 24 h. Therefore, reduced cell viability, proliferation, and cell migration in glioblastoma is caused by suppression of the signaling pathways PI3K/Akt and MEK/ERK as well as by the TRPM7 current blockade without alteration of the TRPM7 protein expression in glioma cells. Authors suppose that the marine compound xyloketal B may be a promising drug candidate for anti-glioblastoma therapy [
182].
13. Pheophorbide a
Pheophorbide α is a typical representative of the organic compound class tetrapyrroles, which as suggested by the name, consists of four pyrrole-derived substances linked either in linear or cyclic fashion via methine bridges. There are well known and studies members in this class of molecules such as hemes giving blood its red color and the chlorophylls, which are responsible for the green color of plants, algae, and some bacteria, as well as cobalamin, siroheme, coenzyme F430, heme d1, and the photopigments bilins [
183]. Molecular formula of pheophorbide a is C
35H
36N
4O
5 and its IUPAC name is (3S,4S)-9-ethenyl-14-ethyl-21-(methoxycarbonyl)-4,8,13,18-tetramethyl-20-oxo-3-phorbinepropanic acid. Pheophorbide a purified from an edible red seaweed
Grateloupia elliptica was shown to exhibit strong anticancer activity with no direct photo-irradiation against various cancer cell lines including mouse melanoma cells (B16-BL6), human epithelial carcinoma cells (HeLa), human cervical cancer cells (SiHa), and human ovarian cancer cells (SK-OV-3) with IC
50 values 18.3 ± 2.9, 9.5 ± 2.3, 13.2 ± 2.6, and 7.0 ± 2.0 μg/mL, respectively. However, pheophorbide a exerted the strongest anticancer effect against U87MG glioblastoma cells that was comparable with activity of the positive control drug paclitaxel [
184]. Cytotoxic effect of pheophorbide a was not observed in the normal HUVECs whereas the positive control paclitaxel was found to demonstrate significant and notable cytotoxic activity (
Table 38).
The glioblastoma growth inhibition activity exerted by pheophorbide a was related to the cell cycle arrest in the G
0/G
1 phase and apoptosis as well as with genomic DNA degradation. Pheophorbide a specifically inhibited only growing U87MG cells but did not affect resting ones. The results obtained through the studies suggest that pheophorbide a isolated from
G. elliptica could be a good source of glioblastoma-specific therapeutics with no clinically notable side effects [
184].
14. Phorboxazoles
In 1995, two unique compounds named phorboxazole A and its C13 epimer phorboxazole B were isolated from the marine sponge Phorbas sp. Their complex chemical structure had given a base to consider these phorboxazoles as a new class of natural products containing an unprecedented array of oxane, oxazole, macrolide, and polyene moieties. In addition to the pronounced antifungal activity against
Candida albicans shown previously in the in vitro experiments, phorboxazoles were reported to provide extremely high cytostatic effects towards the National Cancer Institute’s panel of 60 tumor cell lines with the mean GI
50 values less than 1.6 × 10
−9 M [
185]. Later, a complete synthesis of phorboxazole A was performed [
186] that gave a start to a series of studies devoted to fabrication of the synthetic phorboxazole derivatives. Thus, phorboxazoles were suggested to be the most potent natural cytostatic agents that are discovered to date [
187].
Experimental studies comparing antiproliferative activity of the phorboxazole derivatives against human glioblastoma cell line U373 have shown these tumor cells in a concentration-dependent fashion with low nanomolar IC
50 values were inhibited by synthetic phorboxazole A as well as its analogues 45,46-dehydrobromo-phorboxazole A bearing an alkyne part in C45–C46 position of the terminal bromide, and 33-O-methyl-phorboxazole A containing mixed methyl ketal instead of the C33 positioned hemiketal (
Table 39). IC
50 values for other synthetic phorboxazole analogues, especially, for 32-methyl-phorboxolide A (C1–C32 analog), 31-methyl-phorboxylate (C31–C46 analog), 29-phorboxamide A (hydrated analog), C1–C38 of phorboxazole A, and 18-methyl-phorboxylate (C18–C46) were experimentally shown to be greater than 2 mM.
Structure–activity relationship studies demonstrated that just simple modifications like replacement of the terminal vinyl bromide of phorboxazole A with an alkyne, or C33 hemiketal with a mixed methyl ketal did not lead to substantial loss of anticancer activity. Neither C1–C32 macrolide-containing domain, nor C31–C46 side-chain portion were separately sufficient to sustain the potent anticancer activity of phorboxazole A. The simple covalent joining of the macrolide and side-chain parts of phorboxazole A via an amide at C29–C31 instead of the planar vinyl-substituted oxazole at this position of phorboxazole A was not sufficient to regain appreciable activity. Maintaining the central oxazole but truncating the side chain by omission of the lipophilic C39–C46 polyene domain resulted in abolished antitumor activity. Deletion of the C2–C17 portion of the phorboxazoles containing oxazole, acrylate, and bispyran moieties similarly resulted in activity loss. These findings suggest that macrolide, central oxazole, and polyene side chain portions of phorboxazoles are necessary for a potent anticancer activity [
187].
15. Phlorotannins
Phlorotannins are the tannin derivatives existing in a form of polyphenols that built as a result of the phloroglucinol unit polymerization. The most widely known phlorotannins are phloroglucinol, eckol, dieckol, 8,8′-bieckol, 6,6′-bieckol, dioxinodehydroeckol, phlorofucofuroeckol, and a few others [
188].
Eckol is a precursor compound illustrating the dibenzo-1,4-dioxin class of phlorotannins and containing phloroglucinol components linked to each other in multiple fashion (
Figure 20). Eckol is known to be produced in several marine organisms, in particular, in brown algae including
Ecklonia cava (Laminariaceae), and red algae [
189].
Hyun et al. [
190] investigated the effect of eckol on stem cells and malignancies in glioma stem-like cells. The study was performed with the use of glioma cancer cell lines U87MG and U373MG. Patient-derived glioma stem-like cells X01GB and X03AOA were established from acutely resected human tumor tissues. The X01GB line was derived from a patient with a glioblastoma multiforme, and X03AOA was derived from a patient with anaplastic oligoastrocytoma.
It was shown before that glioma subpopulation expressing CD133 protein is enriched in cancer stem-like cells showing greater tumorigenic potential than CD133 negative cells. In a similar way to neural stem/progenitor culture, those glioma stem-like cells expressing CD133 can be enriched in a serum-free medium supplemented with growth factors, where glioma cell fraction continue to proliferate and form spheres instead of a monolayer [
191]. Treatment of the sphere-cultured glioma cells with eckol in concentrations from 50 to 90 μM also decreased the expression of the glioma stem-like cell markers CD133, Nestin, and Musashi-1. Although cell death was not significantly triggered by the eckol treatment at the concentration range 10 to 80 μM, there was an increase of cell kill rate noted after application of eckol in concentration 90 μM. Eckol treatment led to a marked reduction of the Sox2 levels in glioma-initiating cells. Sox2 is a transcription factor essential for maintaining the self-renewal of several types of undifferentiated stem cells, in particular, neural stem cells. Sox2 is also key component for maintaining self-renewal capacity of the brain cancer stem cells [
192]. Eckol suppressed expression of Notch2 and β-catenin in the cancer stem-like cells in several human cancers. Results of the in vivo experiments showed that eckol treatment contributes to marked tumor growth inhibition in xenograft mice [
190]. Importantly, eckol treatment also effectively reduced the resistance of the glioma stem-like cells to ionizing radiation and temozolomide.
It is also known that the key signaling pathways PI3K-Akt and Ras-Raf-1-Erk activated in cancer stem-like cells are involved in the survival and maintaining the stemness in cancer stem-like cells. PI3K-Akt pathway contributes to the resistance of cancer cells to ionizing radiation as well [
193]. Hyun et al. have discovered that eckol treatment effectively inhibits both PI3K-Akt and Ras-Raf-1-Erk pathways in the glioma stem-like cells. Treatment with eckol was also noted to induce a marked suppression of the PI3K and Akt activities, and completely inhibit Ras-Raf-1 interaction and Raf-1 and Erk activations in the sphere-forming glioma stem-like cells. On the base of presented results, authors made a conclusion that eckol may enhance the sensitivity of glioma stem-like cells to anticancer therapies such as ionizing radiation or chemical drugs via inhibition of PI3K-Akt and Ras-Raf-1-Erk pathways [
190].
The synthesized phloroglucinol derivative 2,4-bis(4-fluorophenylacetyl)phloroglucinol (BFP) induced cell death and slowed proliferation rate in three glioma cell lines U251, U87, and C6 in a concentration-dependent manner. At the same time, it did not affect primary human astrocytes (
Table 40) [
194].
Treatment with BFP (3 µM) for 4 h induced a nuclear shrinkage and nuclear condensation. Nuclear fragmentation was noted after 8 h of treatment. BFP also induced concentration-dependent sub-G
1 arrest in U251 glioma cells. Furthermore, treatment of U251 cells with BFP induced significantly upregulated Bax protein expression but did not affect expression of Bcl-2 protein. BFP application also increased procaspase-3 degradation and caspase-3 cleaved form expression in glioma cells. Upstream procaspase-9 also degraded, and cleaved-caspase-9 increased upon BFP treatment in U251 cells. BFP also increased cleaved-PARP expression time-dependently. Treatment of U251 glioma cells with BFP induced generation of the reactive oxygen species (ROS) and also increased expression of the endoplasmic reticulum (ER) stress markers such glucose-regulated protein (GRP)-78, GRP-94, IRE1, phosphorylation of eukaryotic translation initiation factor 2 alpha (eIF-2) and induced upregulation of the CAAT/enhancer-binding protein homologous protein (CHOP). In addition, BFP treatment provoked down-stream caspase activation such as pro-caspase-7 and procaspase-12 degradation suggesting the induction of ER stress [
194]. These results indicate that the phloroglucinol derivative BFP induces glioma cell death via mediation of the ROS generation, which subsequently enhances GPR78 and CHOP expression, increases activity of caspase-9 and caspase-3 leading to apoptosis.
16. Carotenoids
Carotenoids along with chlorophylls and phycobiliproteins build a large group of organic pigments. They represent a class of tetraterpene pigments commonly found in bacteria, fungi, algae, as well as high plants and animals. The most known and studied carotenoids include fucoxanthin, astaxanthin, sifonaxanthin, violaxanthin, neoxanthin, β-carotene, capsanthin, lutein, and others [
195,
196].
Fucoxanthin is an orange pigment possessing unique structure that contains allenic bond, conjugated carbonyl, 5,6-monoepoxide, and acetyl groups (
Figure 21) [
197]. This compound is produced by both microalgae Bacillariophyta and macroalgae Phaeophyceae. Besides, it can be extracted from almost all photosynthetic and nonphotosynthetic organisms such as bacteria and fungi [
198].
Seaweeds are rich with fucoxanthin, which is considered as effective anticancer, antioxidant, antiangiogenic, antidiabetic, antiobese, anti-inflammatory, and photo-protection pharmacological agent due to its powerful antioxidant properties [
198]. Antiproliferative and antiapoptosis effects of fucoxanthin were confirmed in the human cancer cell lines including hepatic carcinoma HepG2, gastric adenocarcinoma MGC-803, and non-small-cell lung cancer cells [
199].
Regarding the effects of fucoxanthin on the glioma cell growth, it was shown that fucoxanthin in concentrations 25, 50, 75, and 100 µM significantly reduced cell viability in the glioma U87 and U251cell lines in dose-dependent and time-dependent manner, but did not affect viability of healthy neurons. Applied in concentrations 25 and 50 µM it induced apoptosis due to the disruption of mitochondrial potential (∆ψm) and destroyed mitochondrial function. It was also found that fucoxanthin provides an increment in Bax expression and a decrement in Bcl-2 expression in both U87 and U251 cells [
200]. The balance of Bcl-2/Bax dominates the switch which turns on or off the apoptosis. Enhanced activation of cleaved-PARP, caspase-9 and caspase-3 was noted in the fucoxanthin-treated tumor cells. Electron microscopy studies had confirmed that fucoxanthin promotes cell death by induction apoptosis via mitochondrial pathway. It was further figured out that expression of phosphorylation in Akt and mTOR was dramatically decreased by fucoxanthin treatment of the glioma cells in concentration of 25 and 50 µM. Moreover, the level of phosphorylated mTOR in the glioma cells was notably reduced at higher concentration of fucoxanthin. The expressions of active Akt/mTOR form were lessened due to the glioma cell exposure with fucoxanthin or PI3K inhibitor, LY294002, individually.
In was shown in the scratch wound healing assay and trans-well assays that fucoxanthin can inhibit invasion and migration rates of the human glioblastoma cells. It was found out also that fucoxanthin possess a capacity of human glioblastoma cell inhibition and the protein levels of MMP-2 and MMP-9 was significantly suppressed after incubation of glioma cells with fucoxanthin at the concentration of 25 and 50 µM. The degree of the phosphorylated-p38 expression was significantly decreased in a concentration-dependent manner after treatment of the cells with fucoxanthin. However, fucoxanthin abnormally increased the protein levels of phosphorylated ERK and did not affect expression of p-JNK/JNK. In addition, the MMPs was significantly reduced by the p38 inhibitor (SB203580) treatment. Results of the tumor xenograft studies in the immune compromised nude mice (BALB/c-nude) used as a test model had shown that tumor volume and weight were obviously lowered that demonstrate the tumor growth inhibiting capacity of fucoxanthin in U87 cells. Such molecules as p-Akt, p-mTOR, p-p38, MMP-2/9, and BCL-2 were all downregulated, whereas Bax and cleaved-caspases-9 were upregulated after fucoxanthin treatment of the tumor tissues. These findings were consistent with in vitro results [
200]. Therefore, there are convincing experimental evidences indicating that fucoxanthin exerts antiglioma activity, promotes apoptosis via PI3K/Akt/mTOR pathway inhibition and suppresses tumor invasion and migration due to the restriction of the p38/MMPs signaling pathway in human glioblastoma cells. Authors of the research studies suppose that fucoxanthin may represent a new anticancer drug prototype that can damage glioma cells and prevent metastasis. It may find its way as an emerging therapeutic agent in the future.
Hydratoperidinin
Carotenoid hydratoperidinin isolated from the sea anemone
Anthopleura midori showed moderate antiproliferative activity against rat glioma C6 and human glioma U251 cells [
111]. Antitumor activity of hydratoperidinin was significantly higher than temozolomide activity (
Table 41).
17. Conclusions
Quantitative analysis of the data presented in the previous sections of this review shows that more than 100 structurally diverse marine natural compounds possess significant capacity of suppressing proliferative activity of the various glioma cell lines and often even reduce their viability. Some of those compounds are capable to inhibit activity of the cancer cells themselves as well as the tumor stem cells. Authors of the experimental investigations suppose that those compounds may be considered as prototypes for the development of novel anti-glioma pharmaceuticals. In the most studies average inhibiting concentrations of the tested compounds under in vitro conditions were comparable with cytotoxic concentration of antitumor drugs including temozolomide, which is the first line drug of the standard antiglioblastoma chemotherapy. A group of compounds such as ecteinascidin-770, renieramycin M, papuamine, actinomycins D, V and X0β, fradimycin B, valinomycin, streptodepsipeptides P11A and P11B had demonstrated extremely high cytotoxicity inducing glioma cell death in the nanomolar concentration range where half inhibiting concentrations of the officinal antitumor drugs are usually in the range between a few micromoles and several dozen micromoles. For example, IC
50 for temozolomide is approximately 85.8 and 69.5 µM for the glioma cell lines SHG-44 and C6, respectively, and more than 100 µM for U87-MG and U251 cells [
113]. At the same time, phorboxazoles isolated from marine sponges and exerted anti-glioma effects in nanomolar concentrations have a property of the most potent cytostatic agents among all pharmaceuticals studied so far [
187]. Despite cytotoxic activity is not the only parameter of the anticancer potential of chemical compounds, we would like to point out that the substances with the IC
50 values greater than 100–150 µM rather do not have a perspective as the potential anti-glioma drugs.
Important factor for the successful development of natural compounds towards novel anti-glioma agents is an index of selectivity degree of compounds regarding cancer cells. Usually, it is expressed as a ratio of IC
50 for the normal cells (for example, for astrocytes, fibroblasts, or HUVECs) to the IC
50 for glioma cells. Unfortunately, this parameter was provided only in few numbers of publications. Nevertheless, it should be mentioned that such compounds pheophorbide a isolated from read algae [
184], triterpene and steroid glycosides from the starfish and holothurians [
103,
110], sesquiterpene aplysin discovered in gastropod molluscs [
120], anthracycline antibiotic SZ-685C from marine fungi [
97], as well as macrolide antibiotics flavofungin II and spectinabilin isolated from marine streptomyces bacteria [
102] demonstrated substantial inhibition of the glioma cells along with mild toxic influence on the normal cells. At the same time, a series of compounds such as, for example, meriolins and fascaplysins apart of the extremely strong antiglioma activity exert pronounced toxic effect on the normal cells [
51,
64]. Structures of such compounds can be considered only as a base for further discovery of the new agents that would be less toxic for the normal cells.
Results of analysis of the taxonomical diversity of the marine species that were used as a source of the compounds with anti-glioma activity pose some interest (
Table 42). Despite an enormous list of those compounds, about half of the active compounds were isolated from a few actinomyces bacterial strains belonging to the one genus Streptomyces (about 40 compounds) and from marine sponges (about 20 compounds) belonging to the class Demospongiae (phylum Porifera). Also, significant number of those compounds were isolated from star fish (class Asteroidea), sea cucumbers (class Holothuroidea) and ascidians (phylum Chordata, subphylum Tunicata). Compounds exerting anti-glioma activity in small amounts was found in cyanobacteria, marine fungi, red and brown alga, actinians (sea anemones), crustaceans, and mollusks. Therefore, now we have a substantially limited number of taxons that were marked with the presence of anti-glioma activity. That does not mean that the further search of active compounds should be limited with only those taxons. That is the opposites, taking to account the fact that taxonomical diversity of the marine species is substantially greater than the terrestrial species diversity. Also, chemical diversity of the marine compounds is probably exceeding the one of terrestrial species. Therefore, it would be reasonable to expand the search of the anticancer agents in representatives of other marine taxons.


























{kind=link}
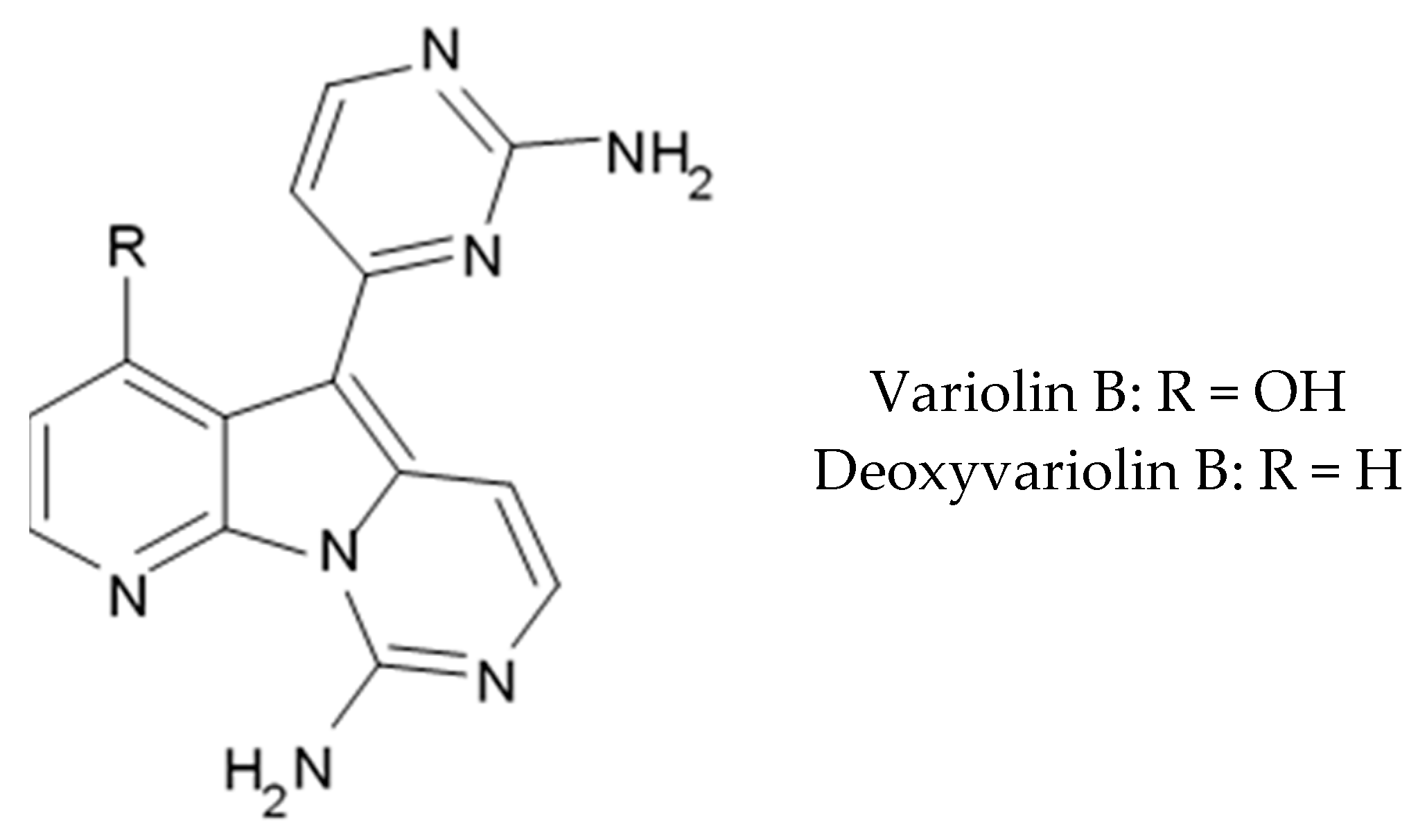{kind=link}
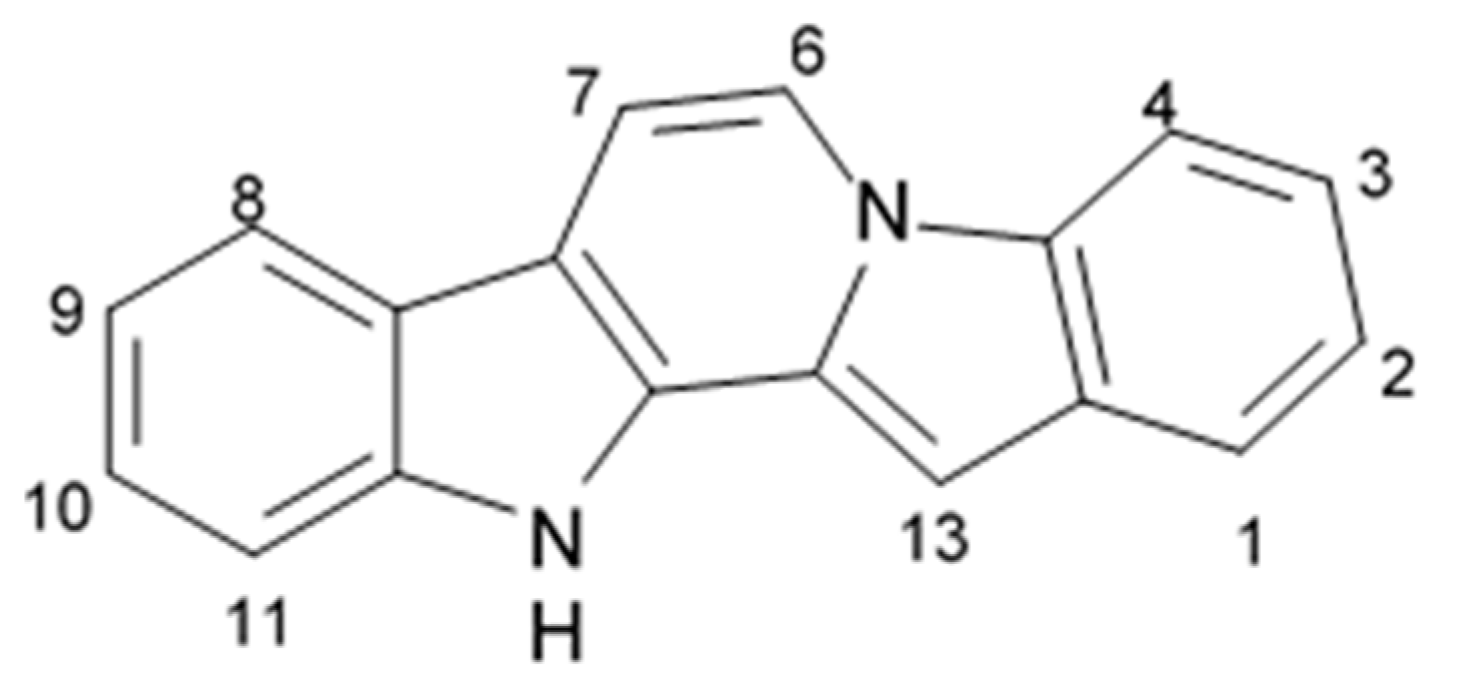{kind=link}
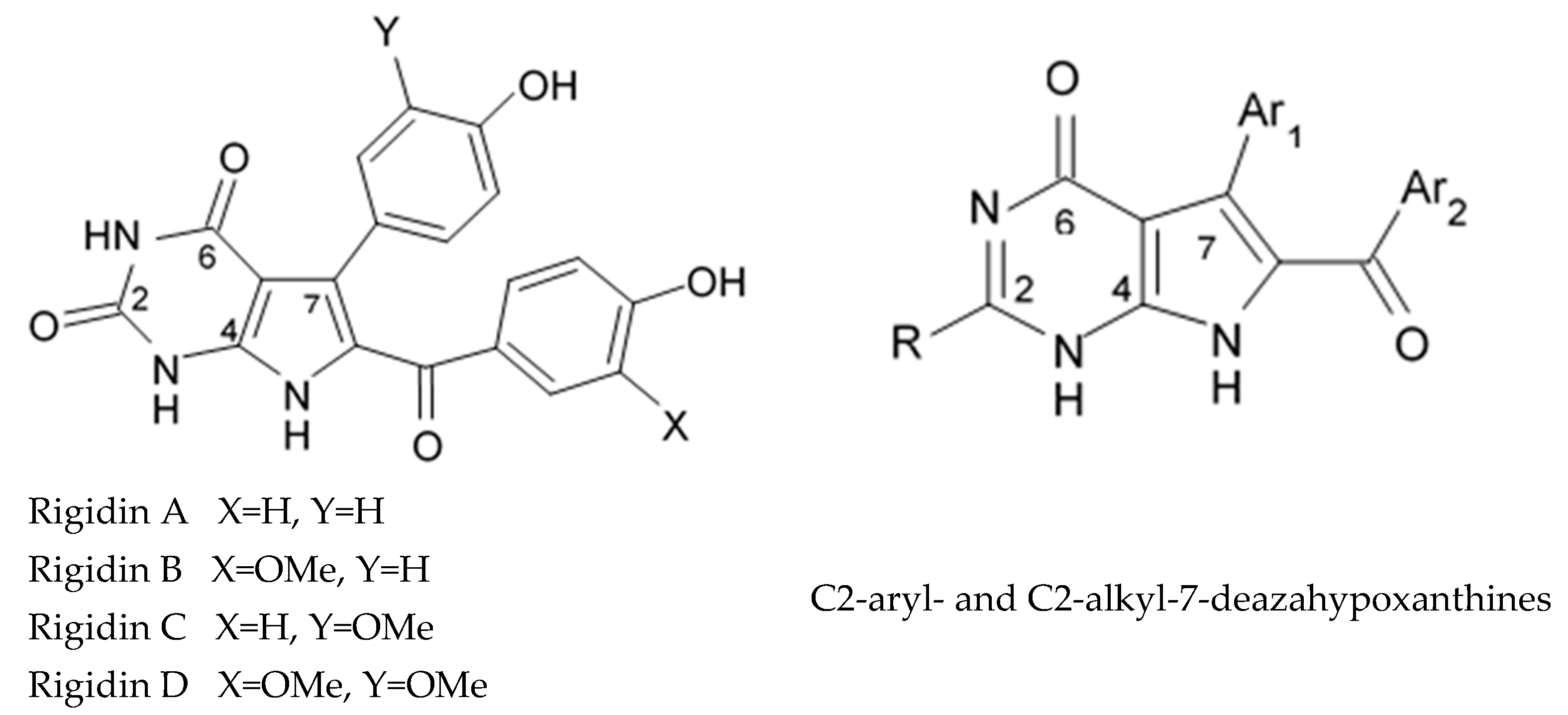{kind=link}
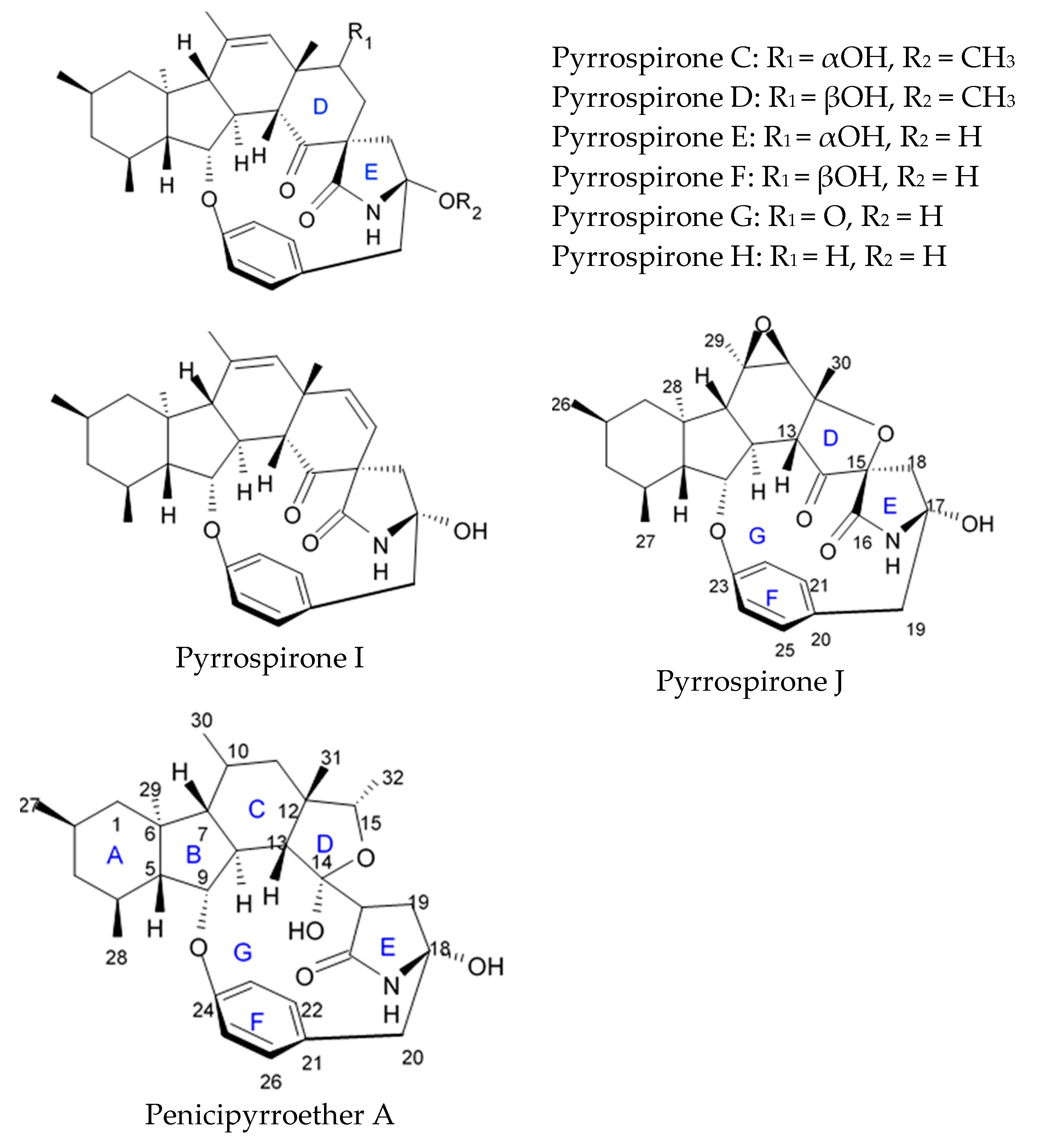{kind=link}
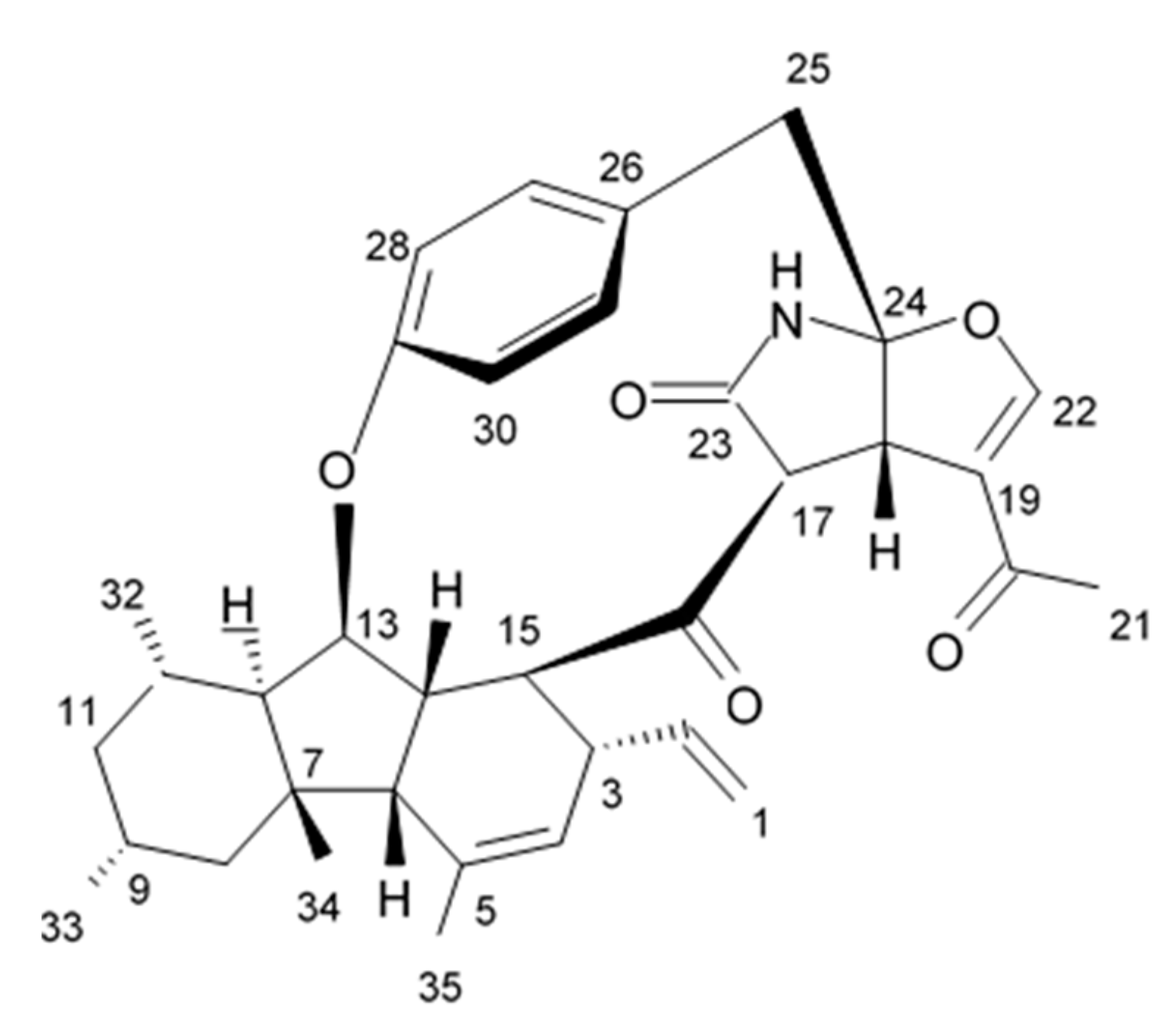{kind=link}
{kind=link}
{kind=link}
{kind=link}
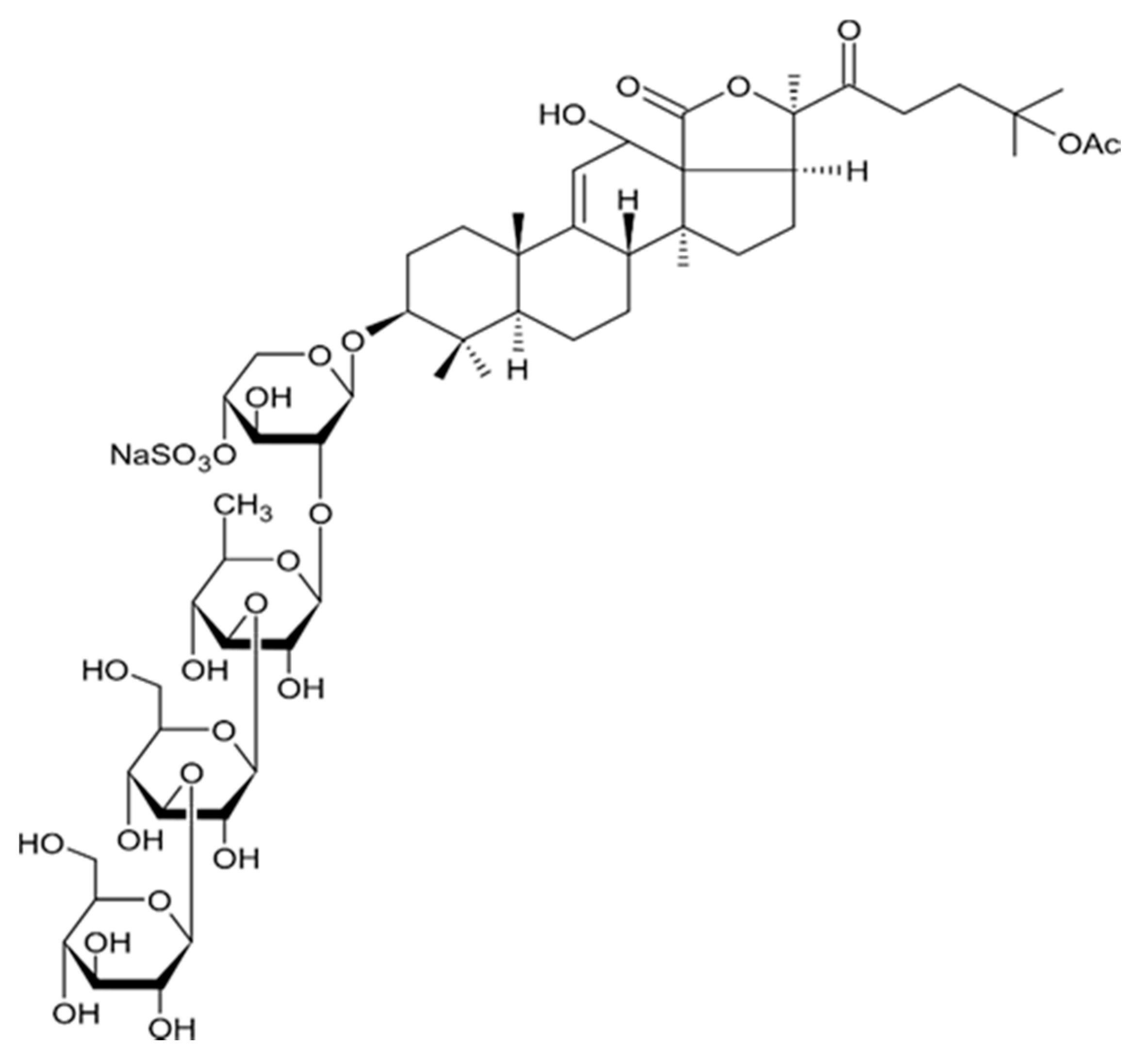{kind=link}
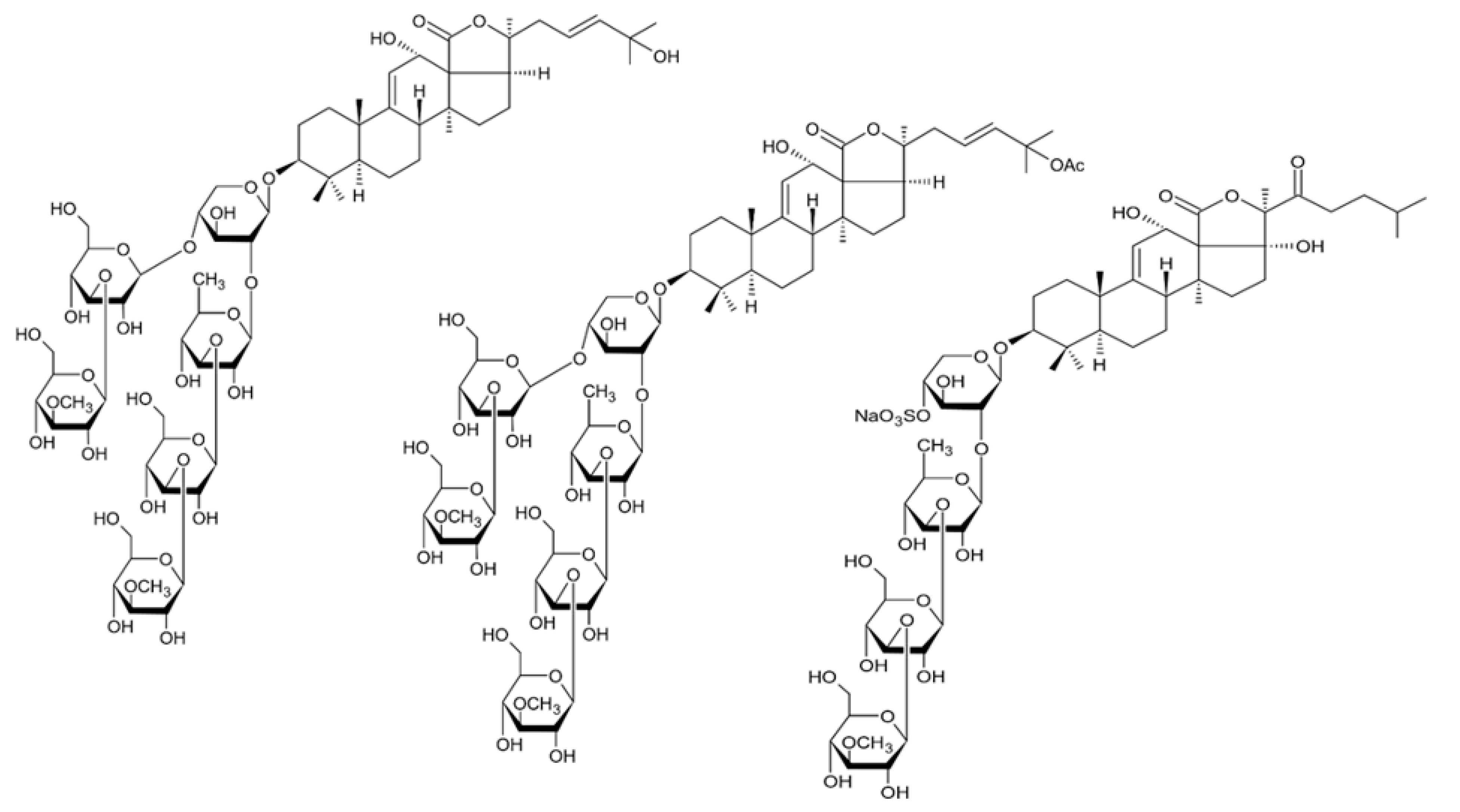{kind=link}
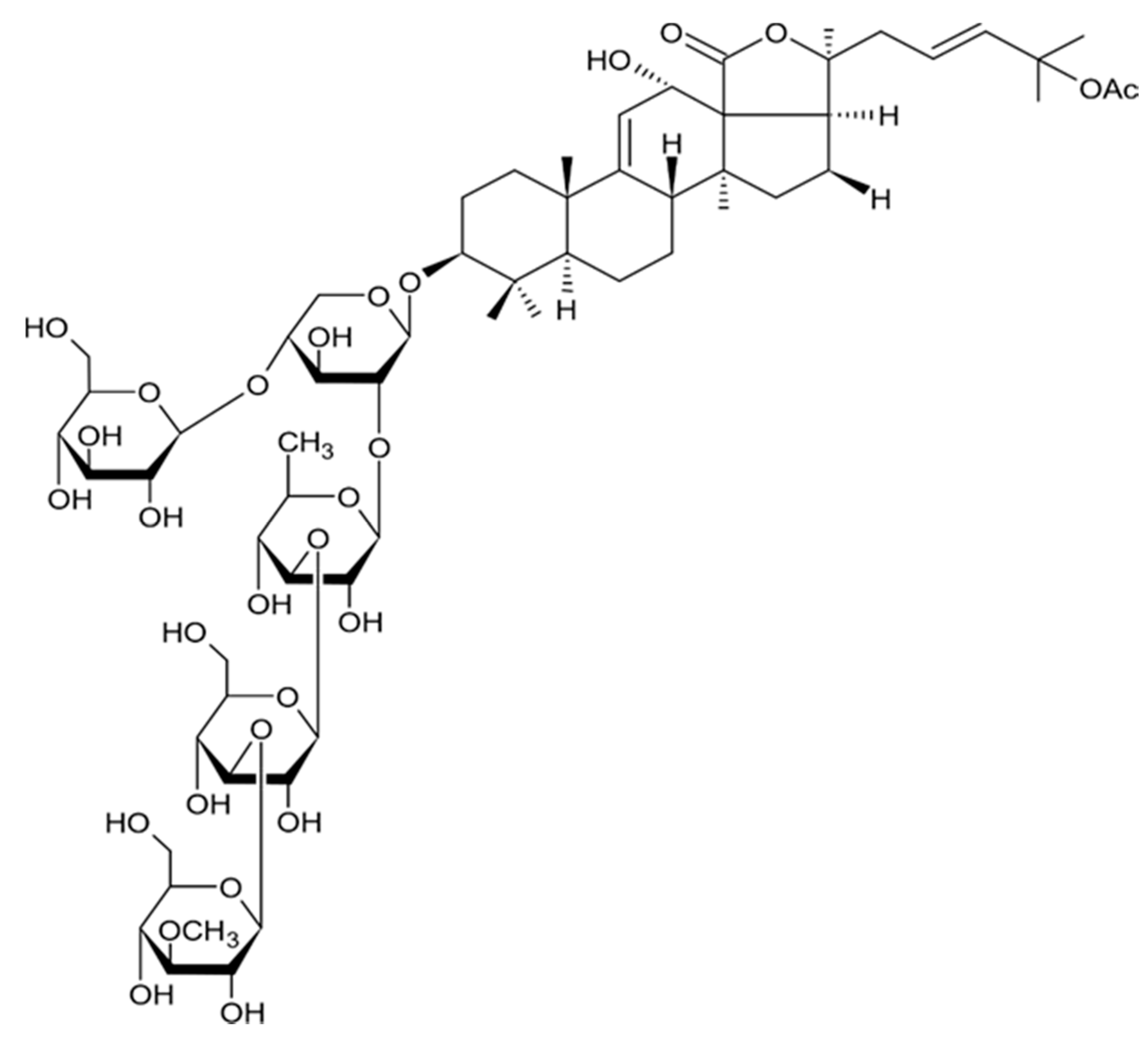{kind=link}
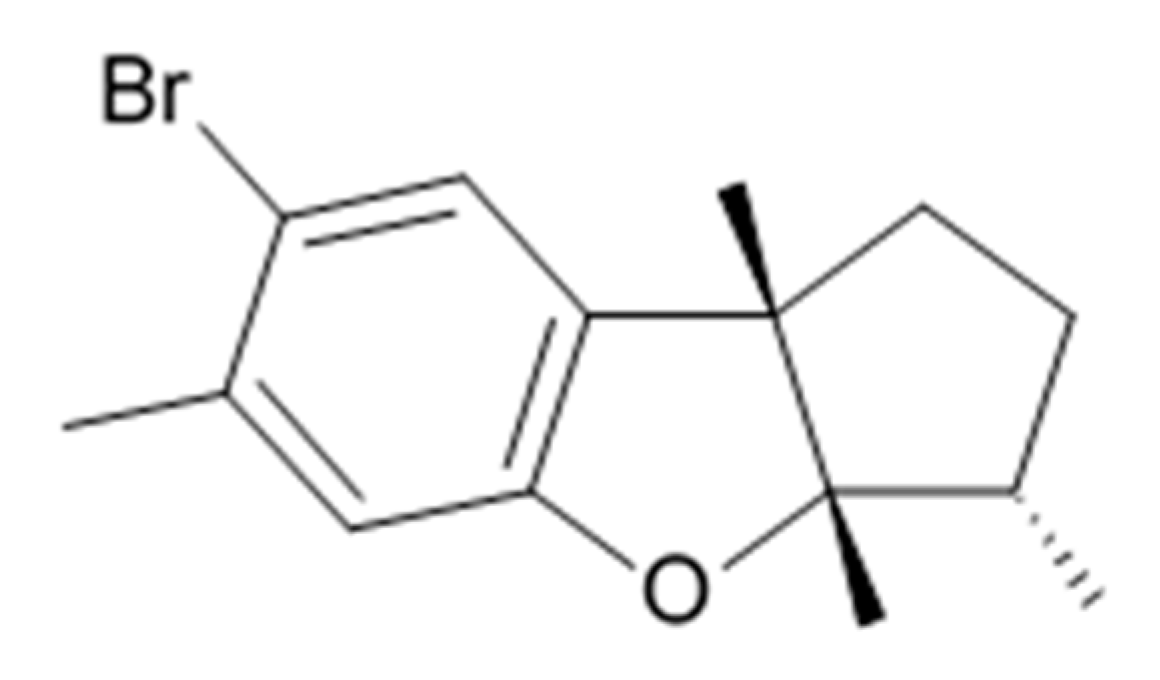{kind=link}
{kind=link}
{kind=link}
{kind=link}
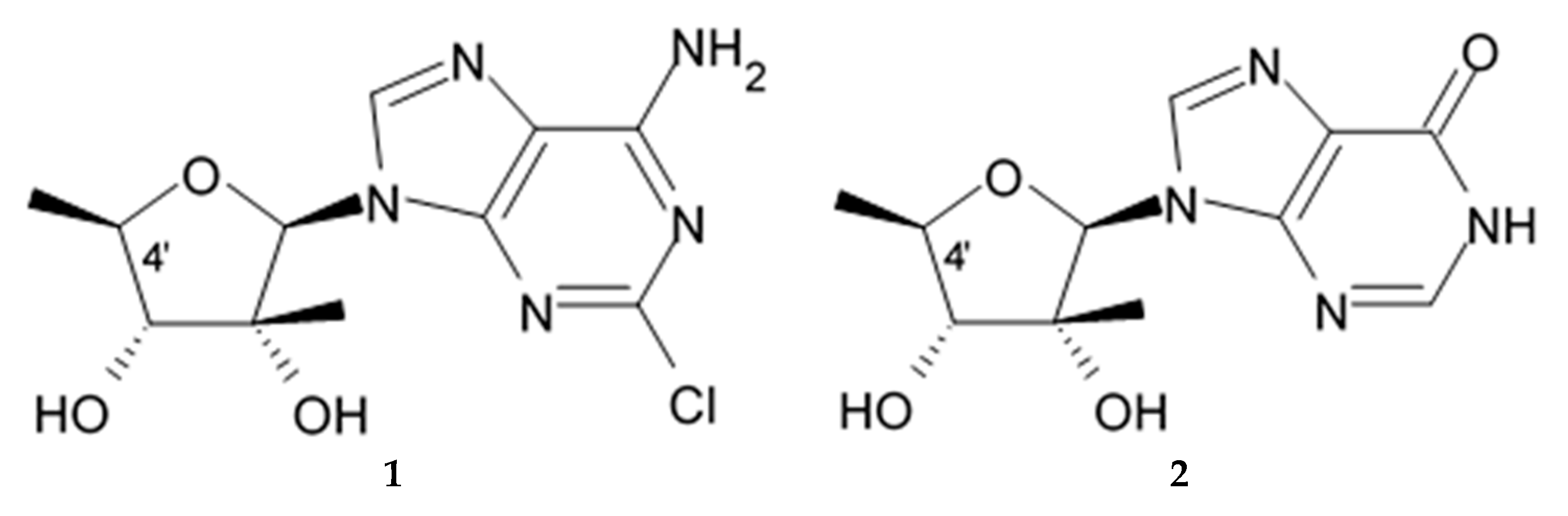{kind=link}
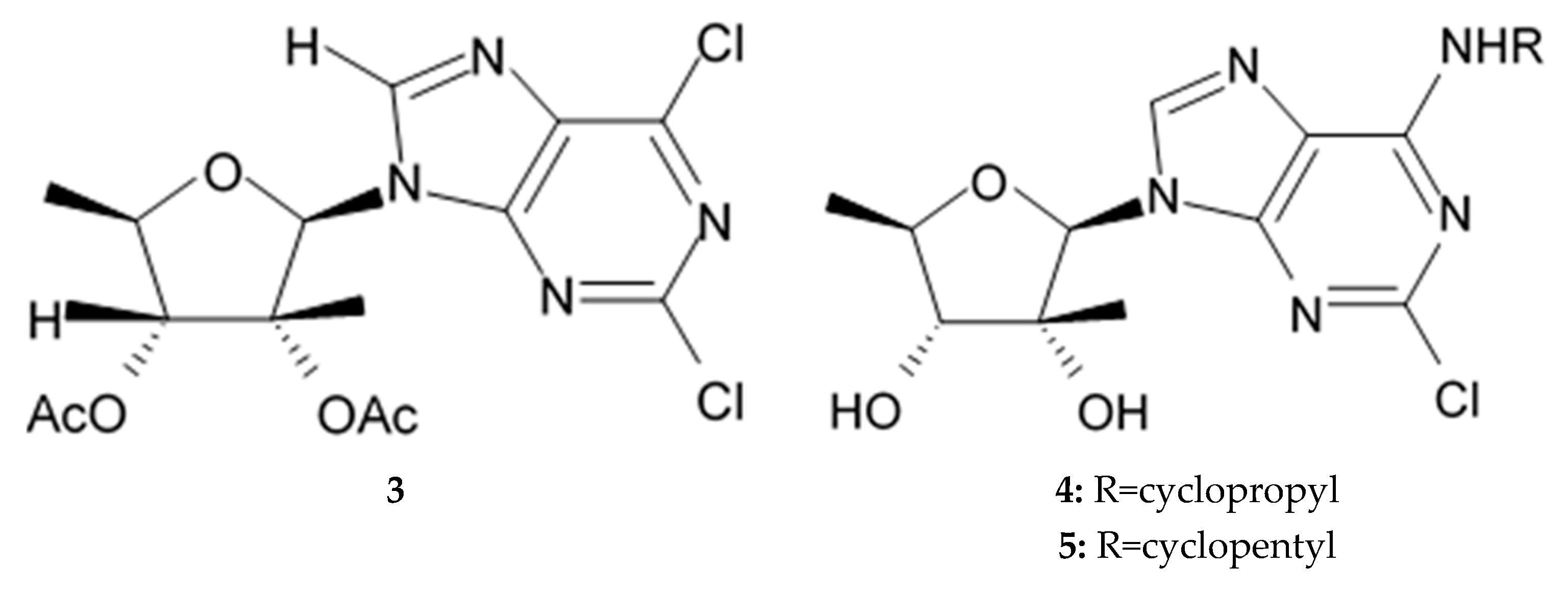{kind=link}
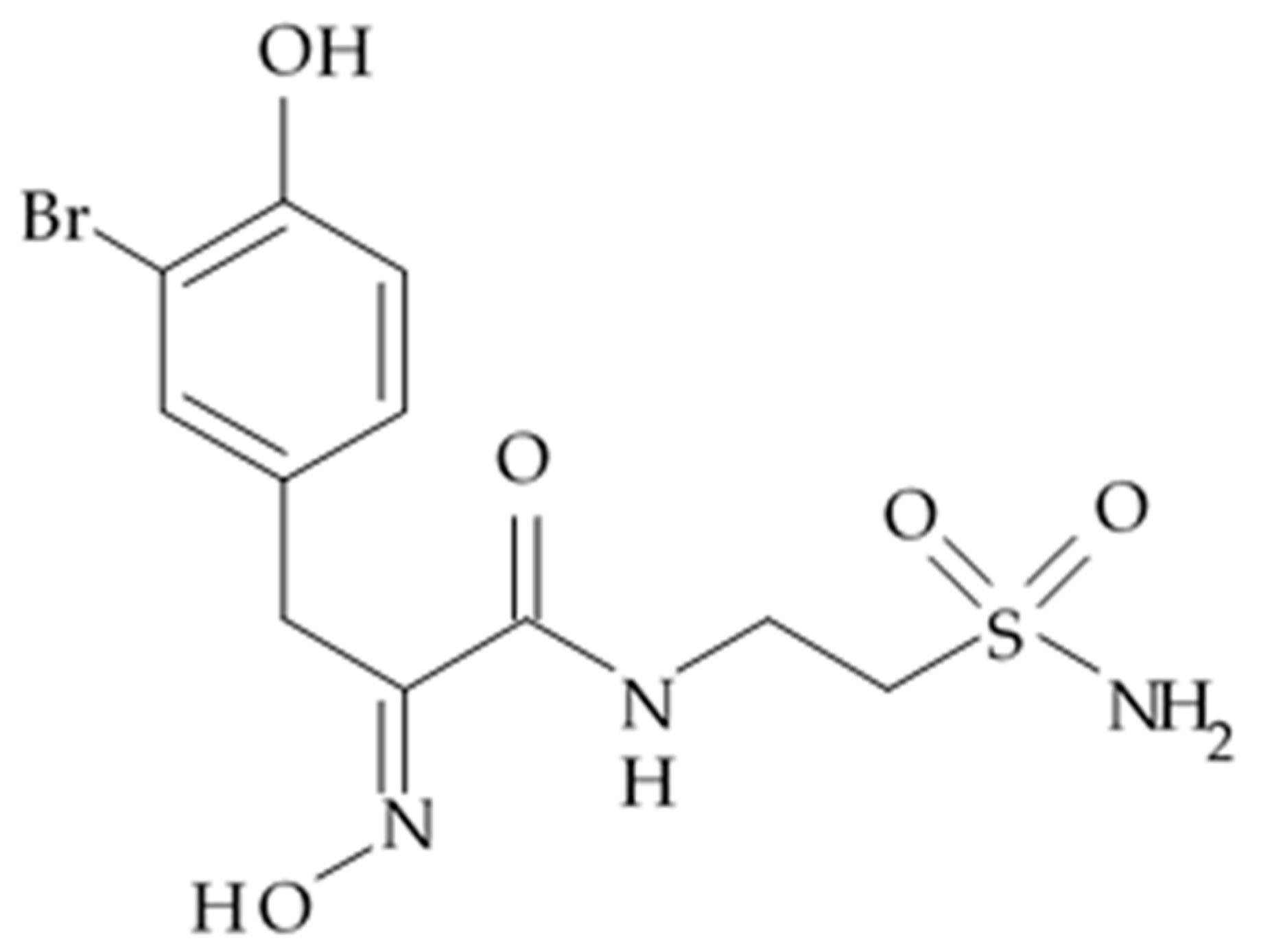{kind=link}
{kind=link}
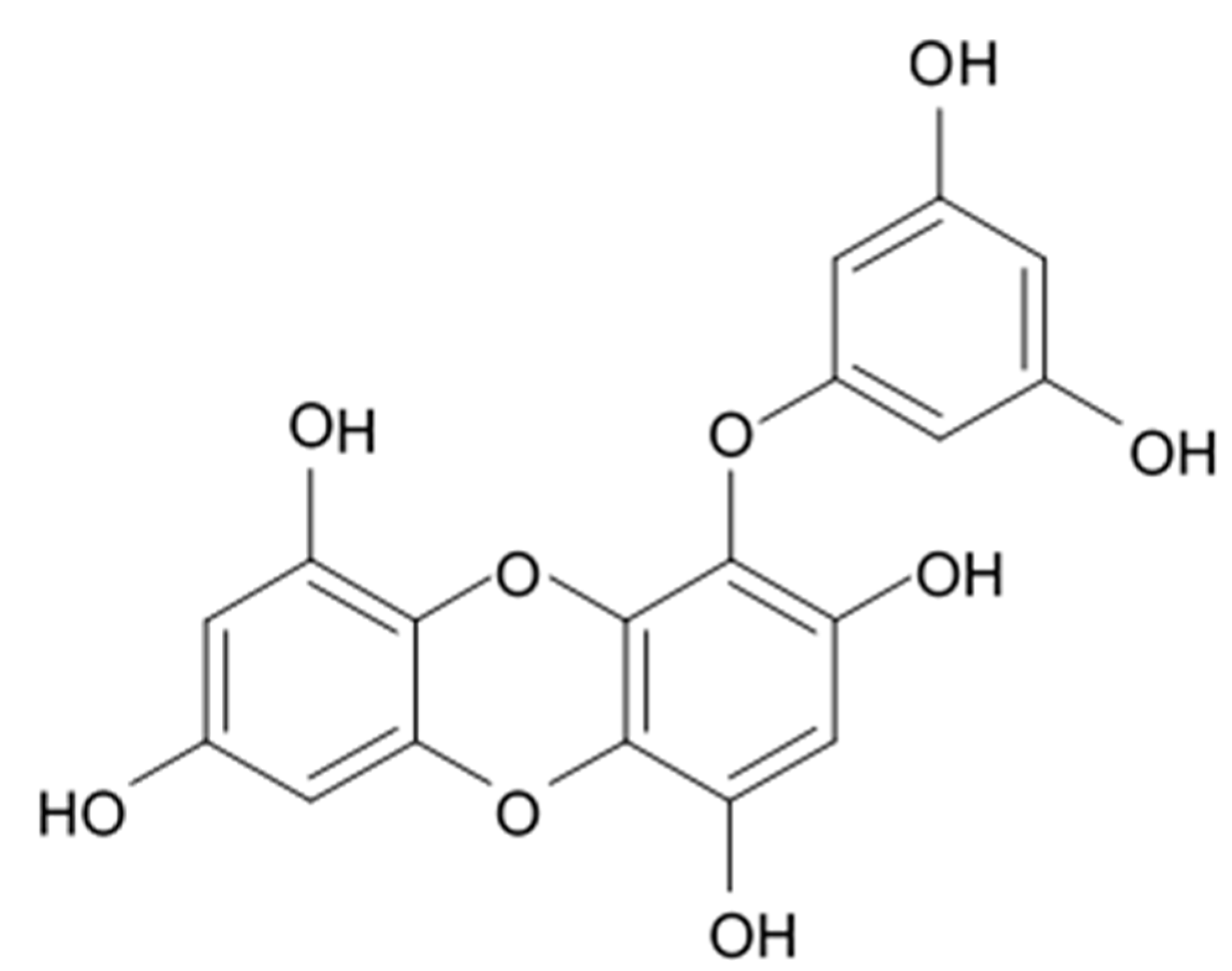{kind=link}
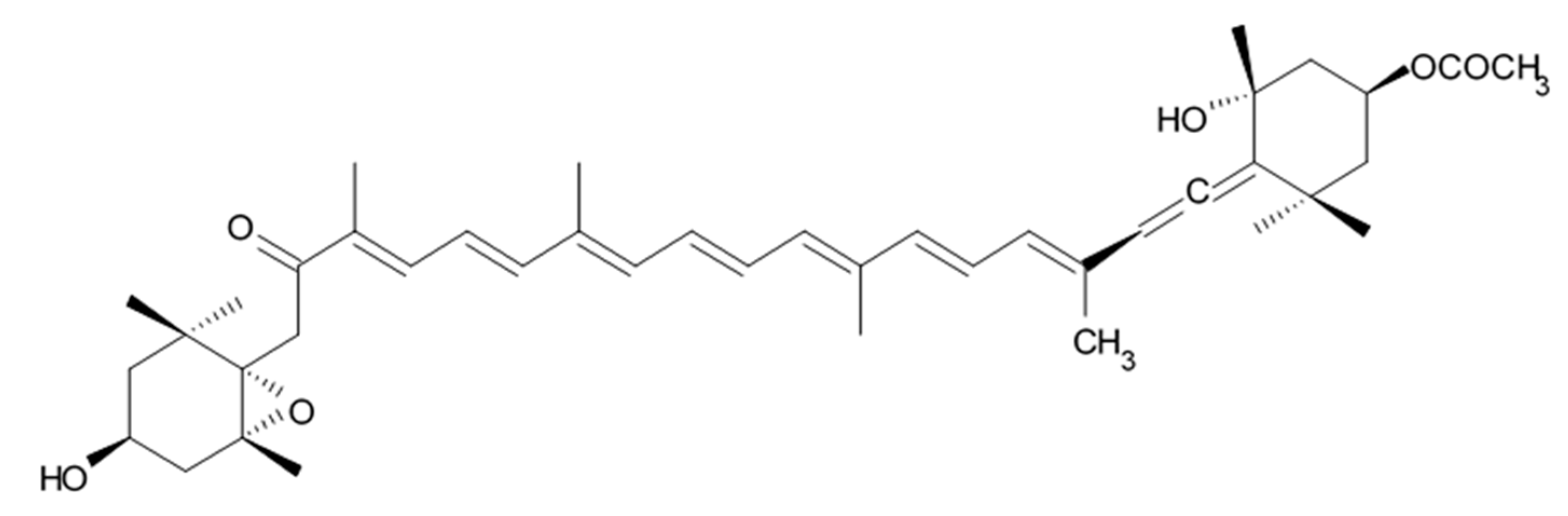{kind=link}















































































