
GDAP1 Involvement in Mitochondrial Function and Oxidative Stress, Investigated in a Charcot-Marie-Tooth Model of hiPSCs-Derived Motor Neurons

 , ,
, ,  , and
, and 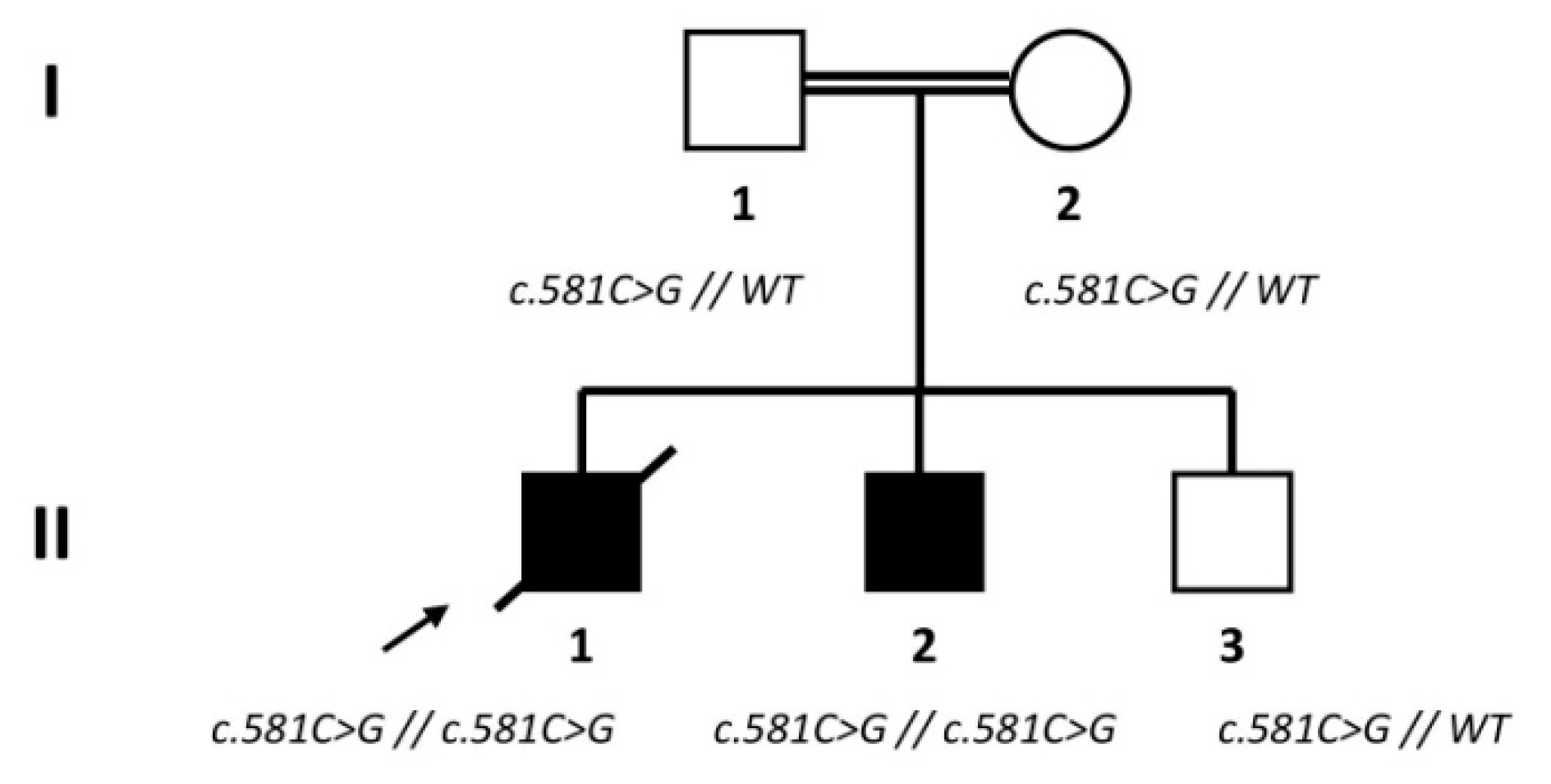{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Skin Biopsies and Fibroblasts Cell Culture
2.3. HiPSCs Generation and Characterization
2.4. Motor Neurons Generation and Culture
2.5. RNA Analysis
2.6. Immunocytochemistry (ICC)
2.7. Electron Microscopy
2.8. Adenosine Triphosphate (ATP) Quantification
2.9. Succinate Dehydrogenase (Complex II) Activity
2.10. Mitochondrial Superoxide Quantification
2.11. Statistical Analysis
3. Results
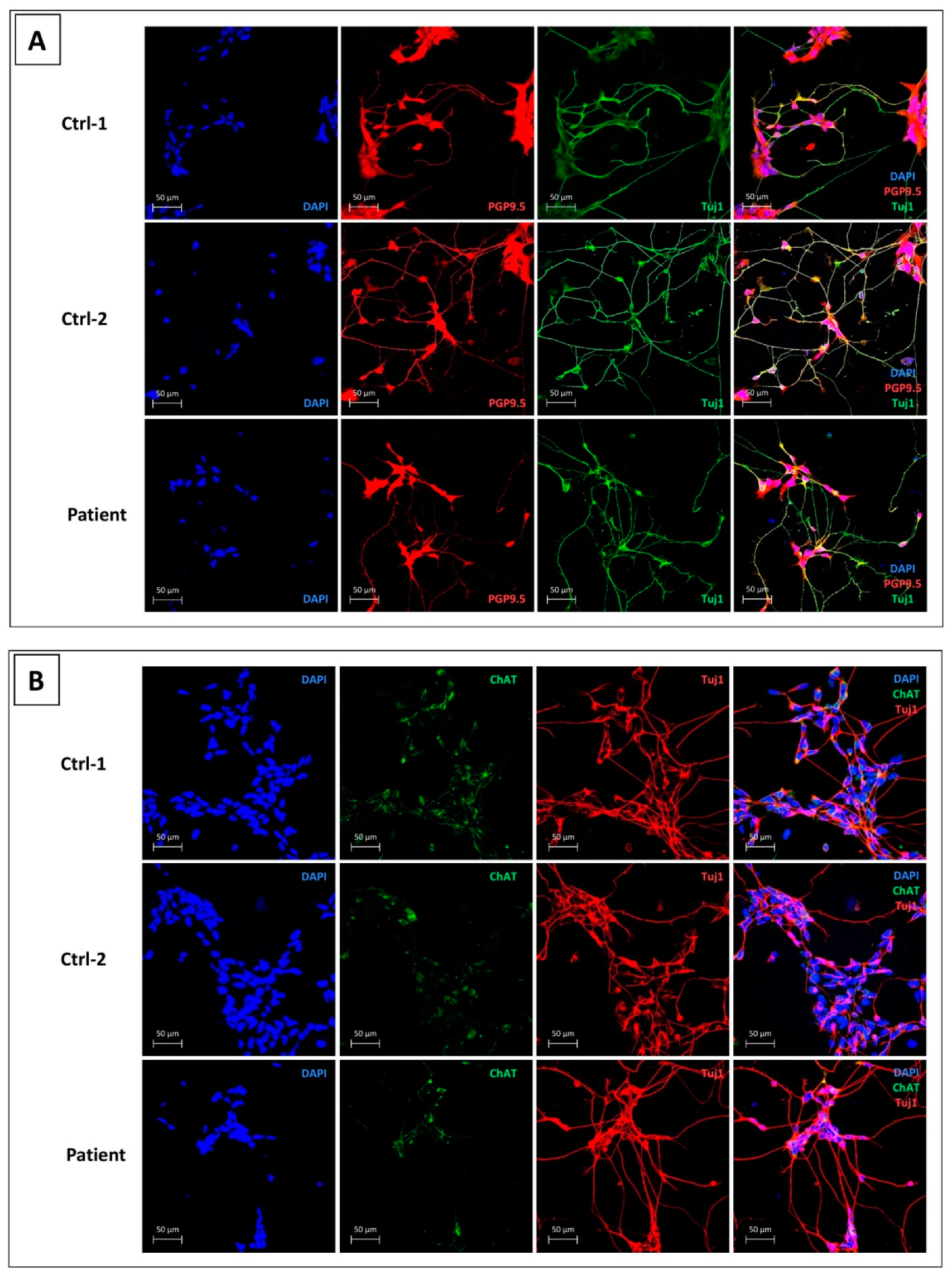
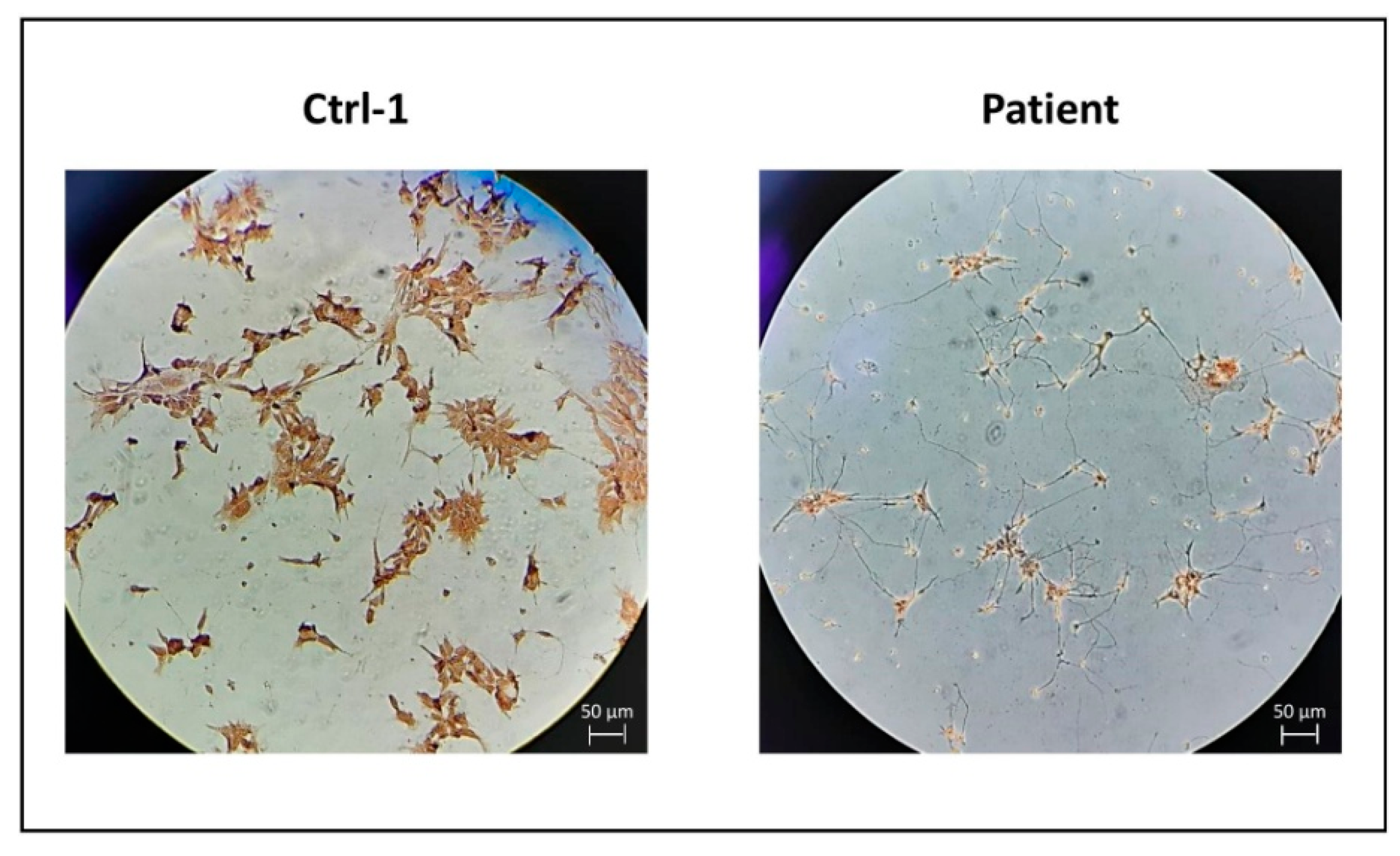
3.1. Control and CMT2H hiPSCs Efficiently Differentiate into MNs
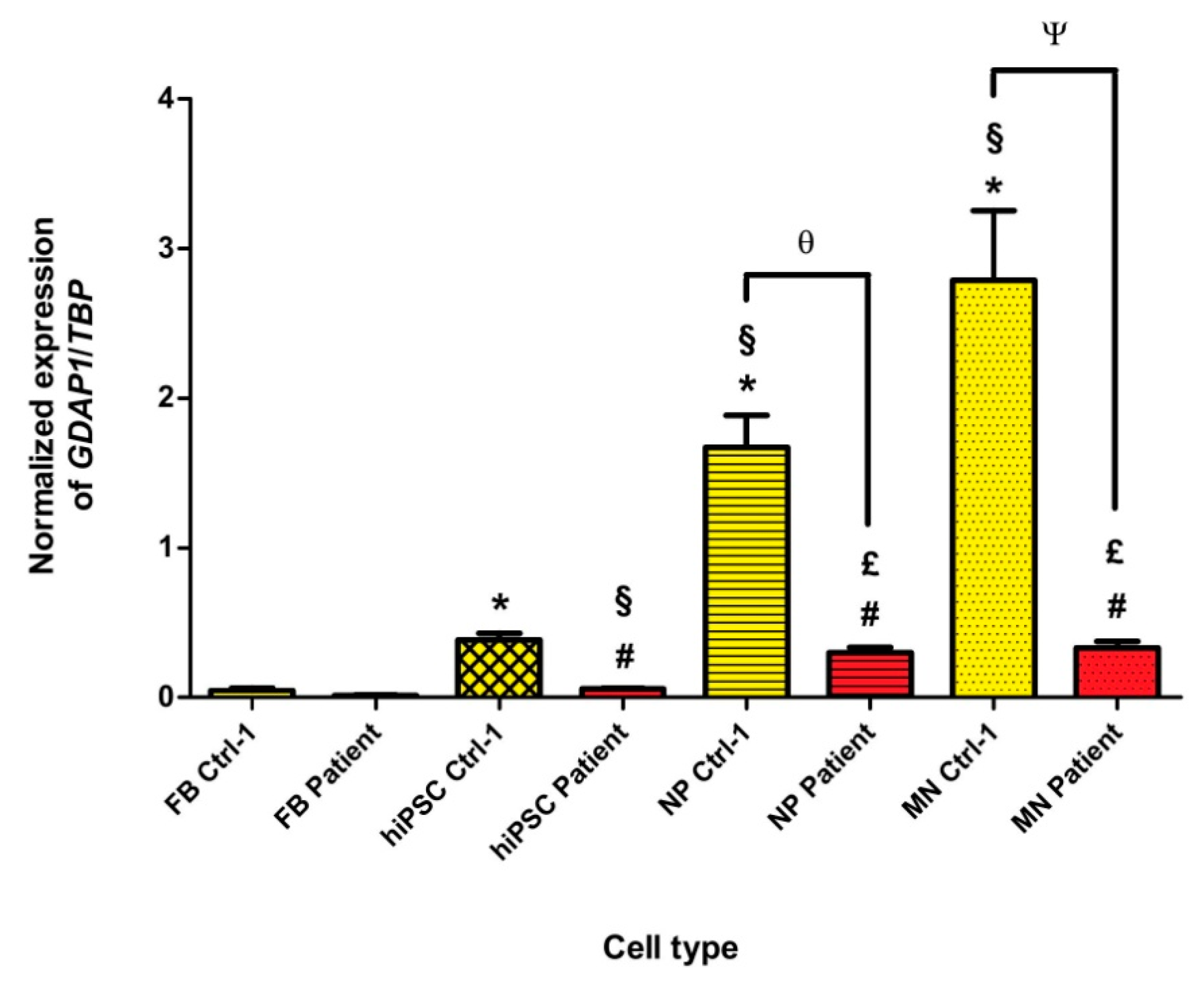
3.2. GDAP1 mRNA Is Expressed in Neural Cells of Controls, but It Is Absent in p.Ser194* Cells
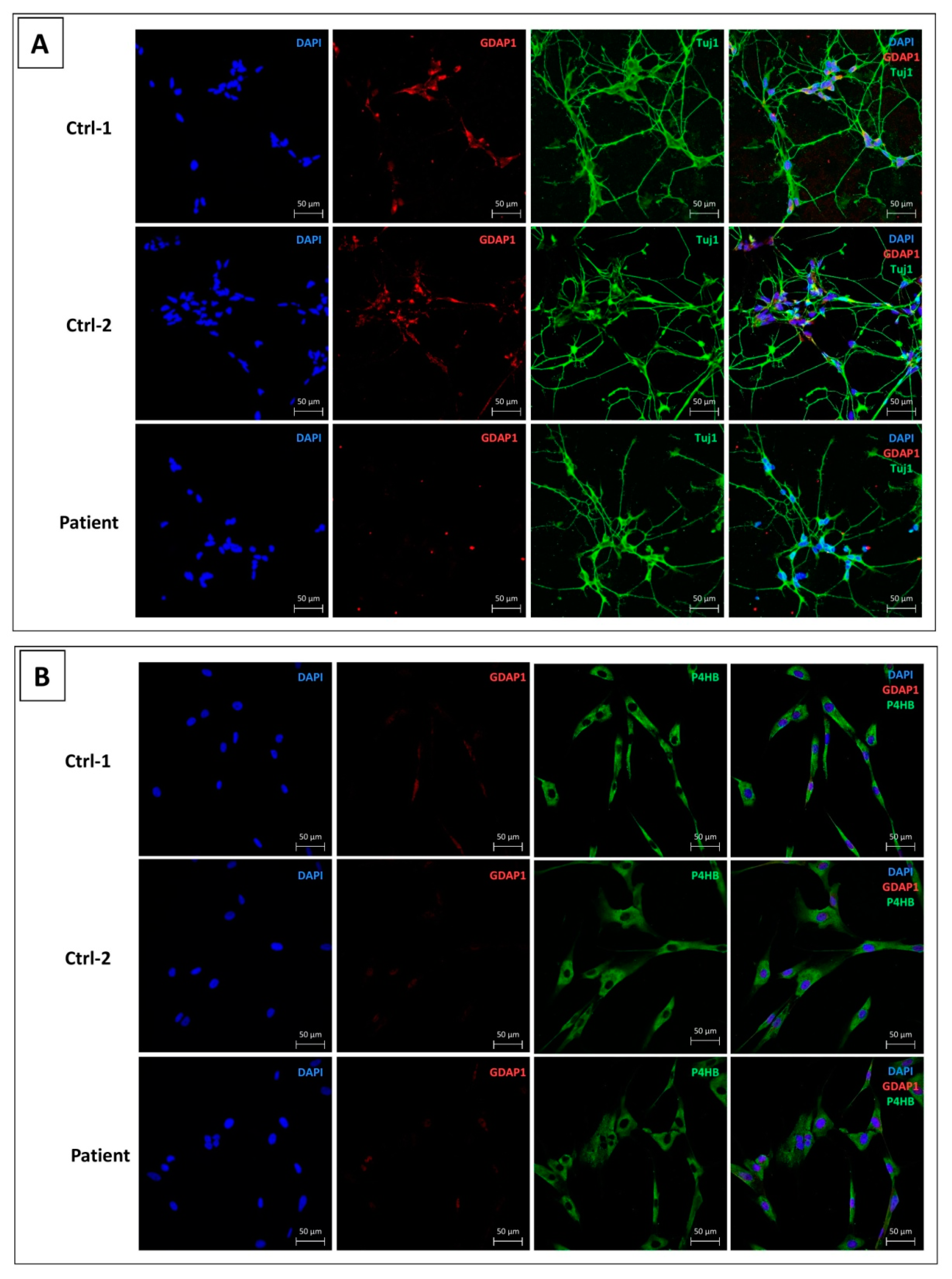
3.3. GDAP1 Protein Is Expressed in MNs of Controls and Absent in p.Ser194* MNs
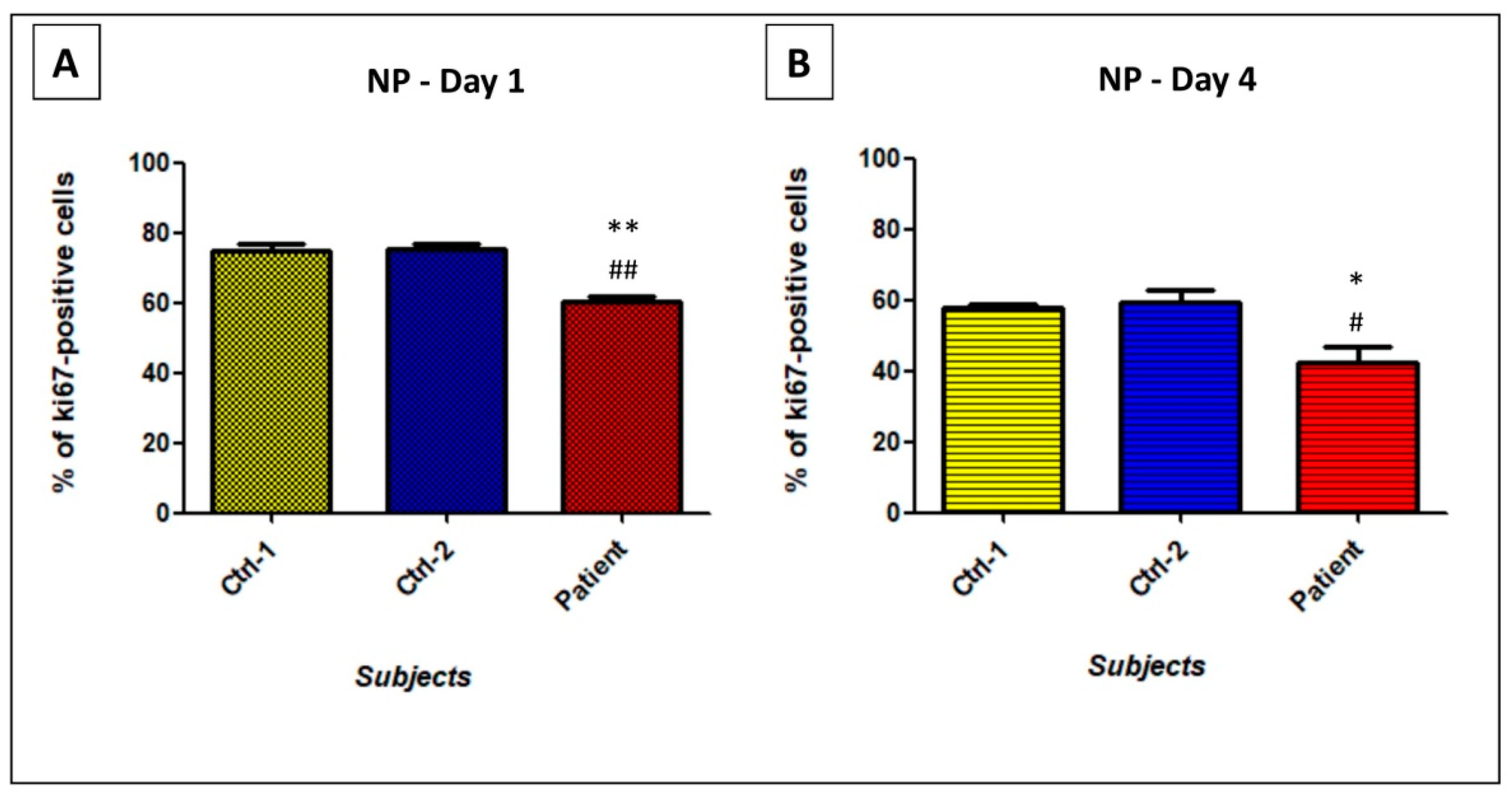
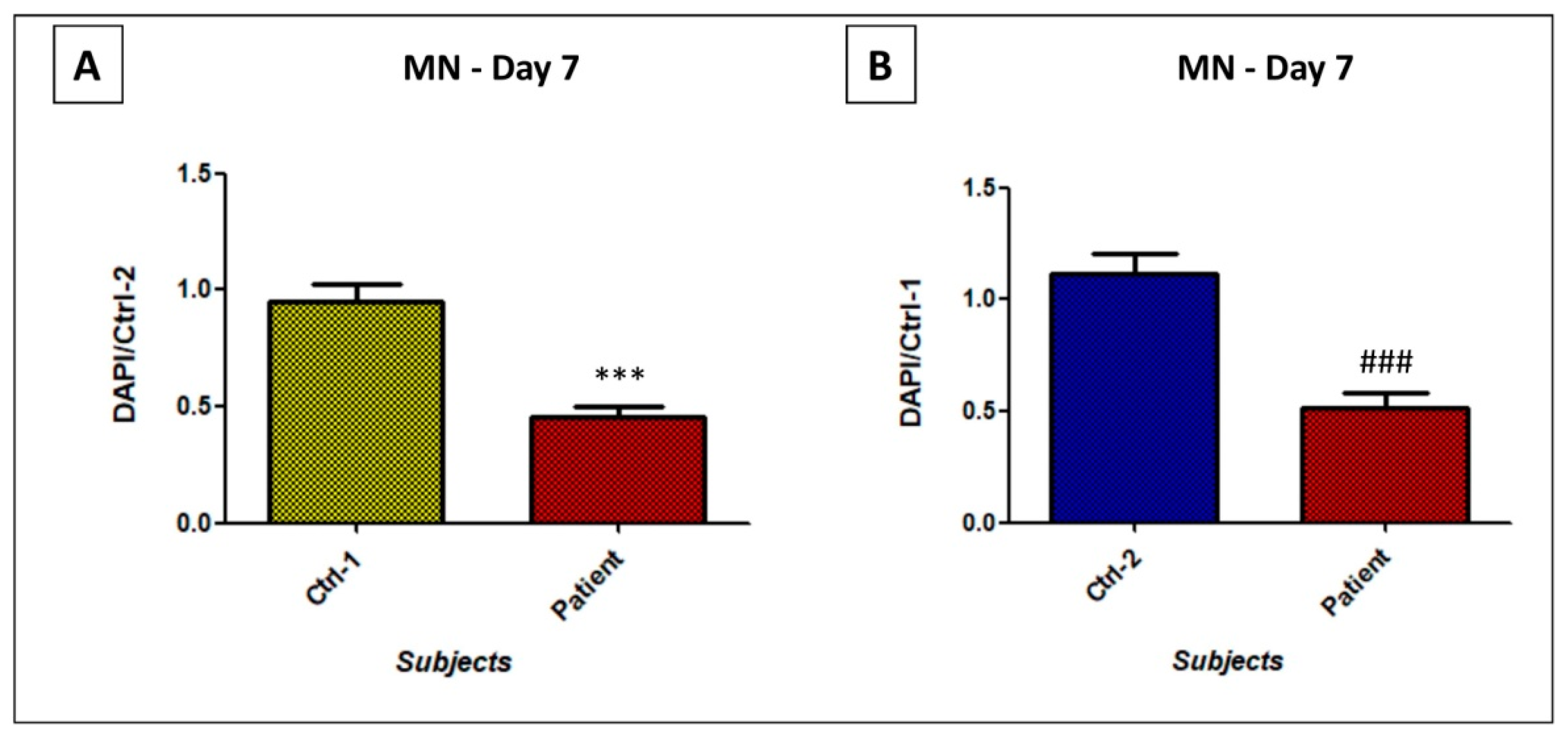
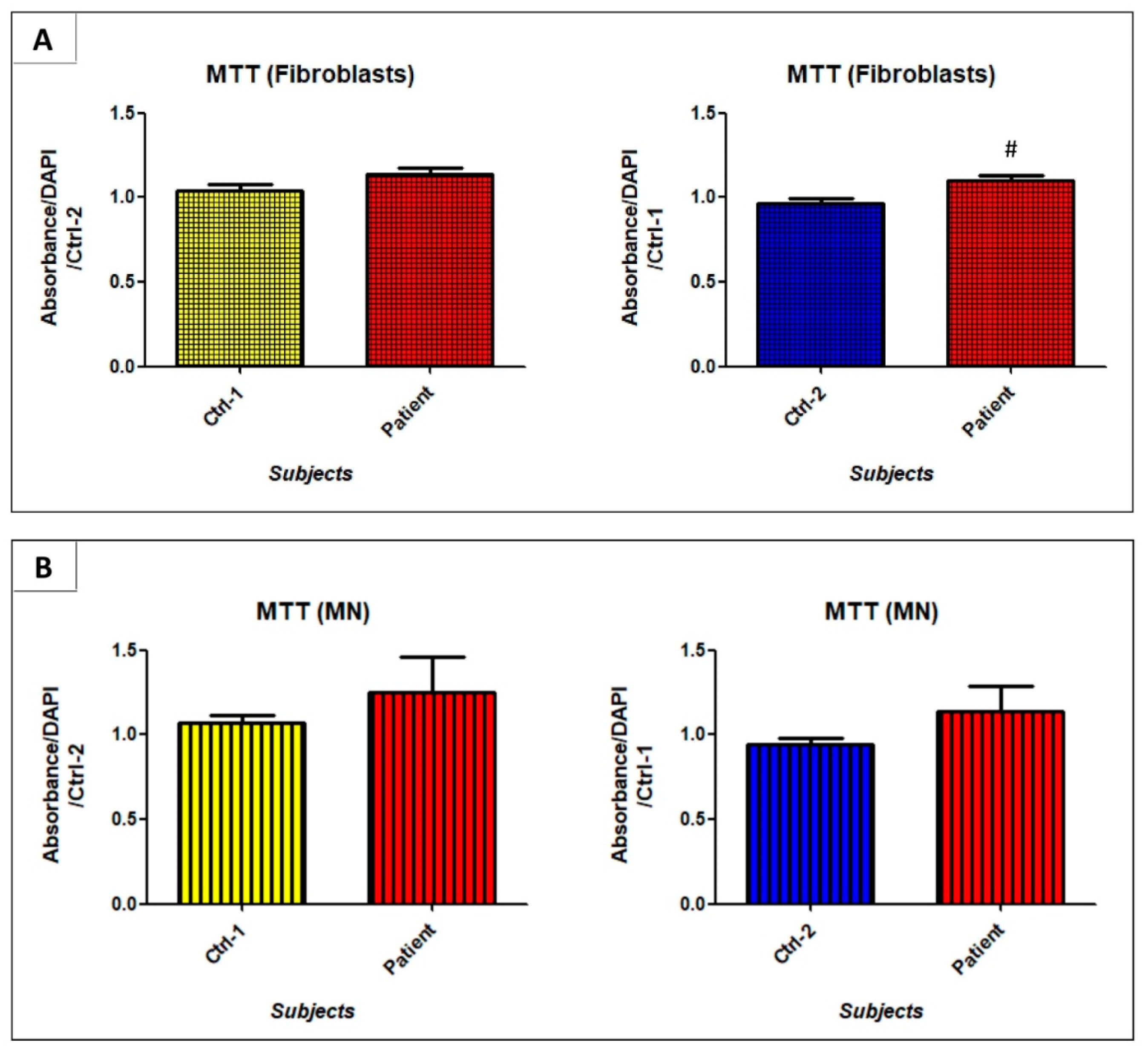
3.4. GDAP1 Mutation Impacts Cell Proliferation and Viability of Neural Cells
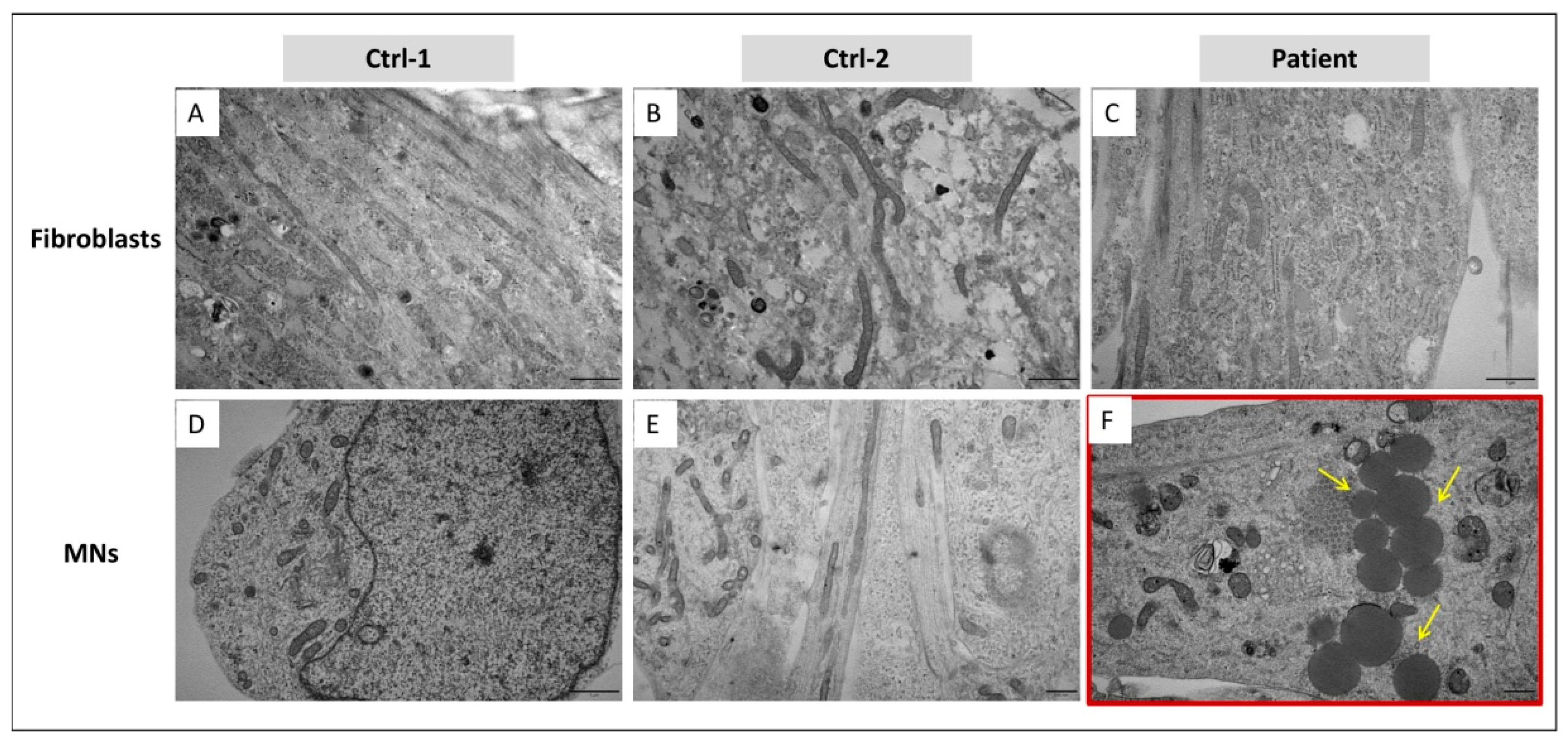
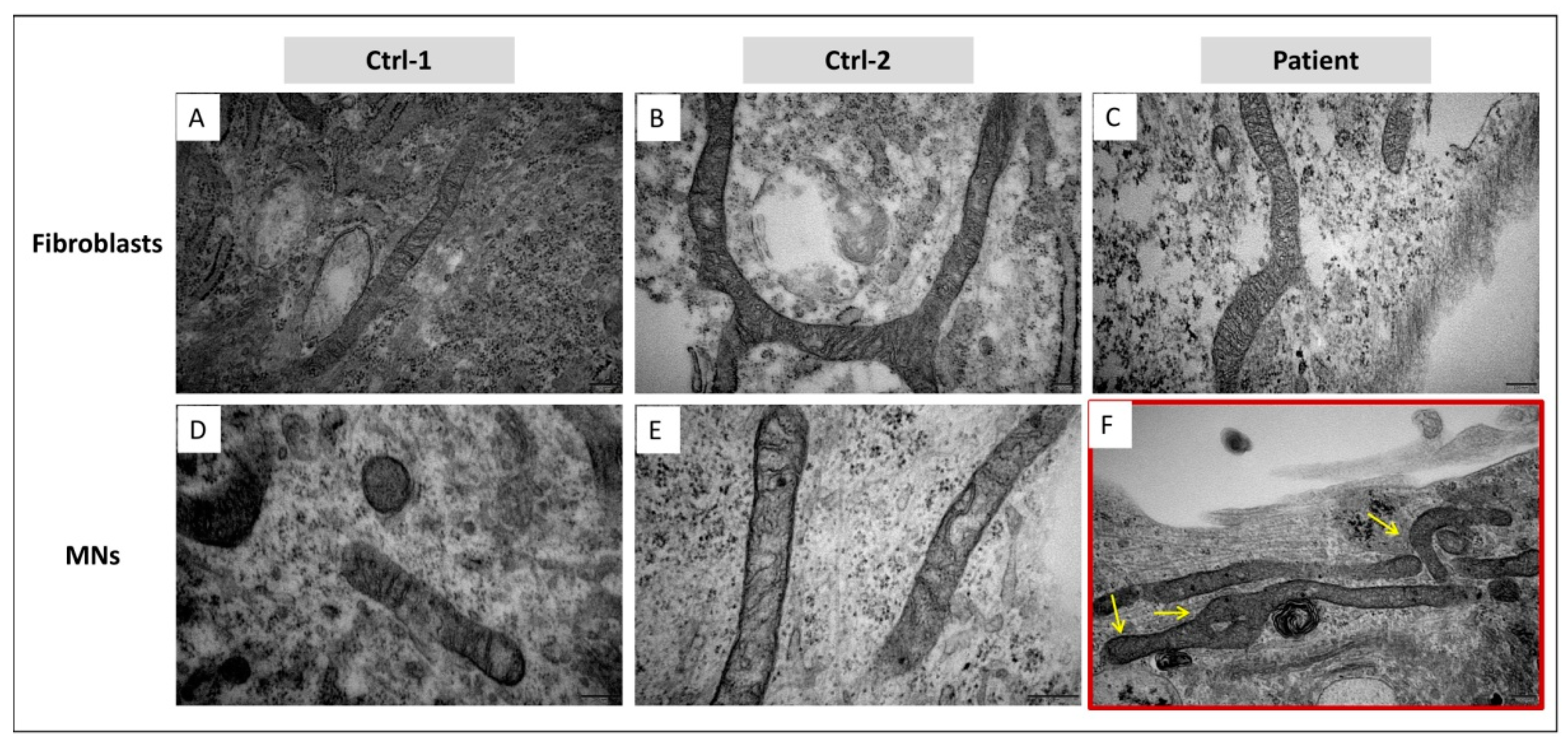
3.5. In MNs, GDAP1 Mutation Is Associated with Cytosolic Lipid Droplets and Perturbed Mitochondrial Morphology
3.6. GDAP1 Mutation Does Not Strongly Alter Oxidative Phosphorylation
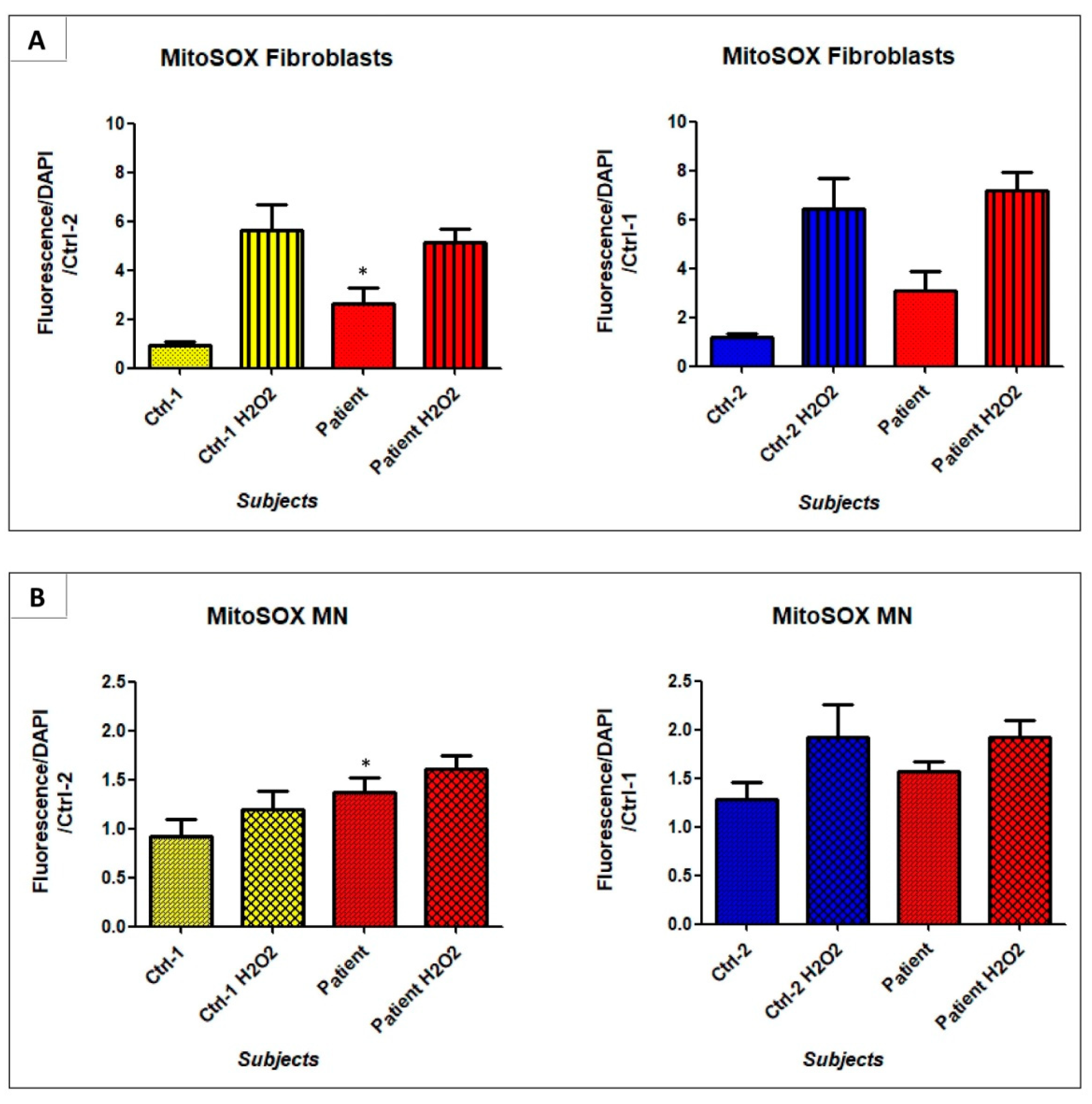
3.7. GDAP1 Mutation Could Promote Mitochondrial Oxidative Stress
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bird, T.D. Charcot-Marie-Tooth (CMT) Hereditary Neuropathy Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2020. [Google Scholar]
- Liu, L.; Zhang, R. Intermediate Charcot-Marie-Tooth Disease. Neurosci. Bull. 2014, 30, 999–1009. [Google Scholar] [CrossRef]
- Baxter, R.V.; Ben Othmane, K.; Rochelle, J.M.; Stajich, J.E.; Hulette, C.; Dew-Knight, S.; Hentati, F.; Ben Hamida, M.; Bel, S.; Stenger, J.E.; et al. Ganglioside-Induced Differentiation-Associated Protein-1 Is Mutant in Charcot-Marie-Tooth Disease Type 4A/8q21. Nat. Genet. 2002, 30, 21–22. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, A.; Pedrola, L.; Sevilla, T.; García-Planells, J.; Chumillas, M.J.; Mayordomo, F.; LeGuern, E.; Marín, I.; Vílchez, J.J.; Palau, F. The Gene Encoding Ganglioside-Induced Differentiation-Associated Protein 1 Is Mutated in Axonal Charcot-Marie-Tooth Type 4A Disease. Nat. Genet. 2002, 30, 22–25. [Google Scholar] [CrossRef]
- Mathis, S.; Goizet, C.; Tazir, M.; Magdelaine, C.; Lia, A.-S.; Magy, L.; Vallat, J.-M. Charcot–Marie–Tooth Diseases: An Update and Some New Proposals for the Classification. J. Med. Genet. 2015, 52, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Sivera, R.; Frasquet, M.; Lupo, V.; García-Sobrino, T.; Blanco-Arias, P.; Pardo, J.; Fernández-Torrón, R.; de Munain, A.L.; Márquez-Infante, C.; Villarreal, L.; et al. Distribution and Genotype-Phenotype Correlation of GDAP1 Mutations in Spain. Sci. Rep. 2017, 7, 6677. [Google Scholar] [CrossRef] [Green Version]
- Niemann, A.; Ruegg, M.; La Padula, V.; Schenone, A.; Suter, U. Ganglioside-Induced Differentiation Associated Protein 1 Is a Regulator of the Mitochondrial Network. J. Cell Biol. 2005, 170, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Pedrola, L.; Espert, A.; Wu, X.; Claramunt, R.; Shy, M.E.; Palau, F. GDAP1, the Protein Causing Charcot–Marie–Tooth Disease Type 4A, Is Expressed in Neurons and Is Associated with Mitochondria. Hum. Mol. Genet. 2005, 14, 1087–1094. [Google Scholar] [CrossRef]
- Pedrola, L.; Espert, A.; Valdés-Sánchez, T.; Sánchez-Piris, M.; Sirkowski, E.E.; Scherer, S.S.; Fariñas, I.; Palau, F. Cell Expression of GDAP1 in the Nervous System and Pathogenesis of Charcot-Marie-Tooth Type 4A Disease. J. Cell. Mol. Med. 2008, 12, 679–689. [Google Scholar] [CrossRef]
- López Del Amo, V.; Seco-Cervera, M.; García-Giménez, J.L.; Whitworth, A.J.; Pallardó, F.V.; Galindo, M.I. Mitochondrial Defects and Neuromuscular Degeneration Caused by Altered Expression of Drosophila Gdap1: Implications for the Charcot–Marie–Tooth Neuropathy. Hum. Mol. Genet. 2015, 24, 21–36. [Google Scholar] [CrossRef] [Green Version]
- López del Amo, V.; Palomino-Schätzlein, M.; Seco-Cervera, M.; García-Giménez, J.L.; Pallardó, F.V.; Pineda-Lucena, A.; Galindo, M.I. A Drosophila Model of GDAP1 Function Reveals the Involvement of Insulin Signalling in the Mitochondria-Dependent Neuromuscular Degeneration. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2017, 1863, 801–809. [Google Scholar] [CrossRef]
- Niemann, A.; Huber, N.; Wagner, K.M.; Somandin, C.; Horn, M.; Lebrun-Julien, F.; Angst, B.; Pereira, J.A.; Halfter, H.; Welzl, H.; et al. The Gdap1 Knockout Mouse Mechanistically Links Redox Control to Charcot–Marie–Tooth Disease. Brain 2014, 137, 668–682. [Google Scholar] [CrossRef]
- Barneo-Muñoz, M.; Juárez, P.; Civera-Tregón, A.; Yndriago, L.; Pla-Martin, D.; Zenker, J.; Cuevas-Martín, C.; Estela, A.; Sánchez-Aragó, M.; Forteza-Vila, J.; et al. Lack of GDAP1 Induces Neuronal Calcium and Mitochondrial Defects in a Knockout Mouse Model of Charcot-Marie-Tooth Neuropathy. PLOS Genet. 2015, 11, e1005115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rzepnikowska, W.; Kaminska, J.; Kabzińska, D.; Kochański, A. Pathogenic Effect of GDAP1 Gene Mutations in a Yeast Model. Genes 2020, 11, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, N.; Guimaraes, S.; Schrader, M.; Suter, U.; Niemann, A. Charcot-Marie-Tooth Disease-associated Mutants of GDAP1 Dissociate Its Roles in Peroxisomal and Mitochondrial Fission. EMBO Rep. 2013, 14, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Cassereau, J.; Chevrollier, A.; Gueguen, N.; Malinge, M.-C.; Letournel, F.; Nicolas, G.; Richard, L.; Ferre, M.; Verny, C.; Dubas, F.; et al. Mitochondrial Complex I Deficiency in GDAP1-Related Autosomal Dominant Charcot-Marie-Tooth Disease (CMT2K). Neurogenetics 2009, 10, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Cassereau, J.; Chevrollier, A.; Codron, P.; Goizet, C.; Gueguen, N.; Verny, C.; Reynier, P.; Bonneau, D.; Lenaers, G.; Procaccio, V. Oxidative Stress Contributes Differentially to the Pathophysiology of Charcot-Marie-Tooth Disease Type 2K. Exp. Neurol. 2020, 323, 113069. [Google Scholar] [CrossRef]
- Chen, C.X.; Li, J.Q.; Dong, H.L.; Bai, G.; Wu, Z.Y. Identification and Functional Characterization of Novel GDAP1 Variants in Chinese Patients with Charcot-Marie-Tooth Disease. Authorea 2020. [Google Scholar] [CrossRef]
- Niemann, A.; Wagner, K.M.; Ruegg, M.; Suter, U. GDAP1 Mutations Differ in Their Effects on Mitochondrial Dynamics and Apoptosis Depending on the Mode of Inheritance. Neurobiol. Dis. 2009, 36, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.M.; Rüegg, M.; Niemann, A.; Suter, U. Targeting and Function of the Mitochondrial Fission Factor GDAP1 Are Dependent on Its Tail-Anchor. PLoS ONE 2009, 4, e5160. [Google Scholar] [CrossRef] [Green Version]
- Kabzińska, D.; Niemann, A.; Drac, H.; Huber, N.; Potulska-Chromik, A.; Hausmanowa-Petrusewicz, I.; Suter, U.; Kochański, A. A New Missense GDAP1 Mutation Disturbing Targeting to the Mitochondrial Membrane Causes a Severe Form of AR-CMT2C Disease. Neurogenetics 2011, 12, 145–153. [Google Scholar] [CrossRef]
- Noack, R.; Frede, S.; Albrecht, P.; Henke, N.; Pfeiffer, A.; Knoll, K.; Dehmel, T.; Meyer zu Hörste, G.; Stettner, M.; Kieseier, B.C.; et al. Charcot–Marie–Tooth Disease CMT4A: GDAP1 Increases Cellular Glutathione and the Mitochondrial Membrane Potential. Hum. Mol. Genet. 2012, 21, 150–162. [Google Scholar] [CrossRef] [Green Version]
- Pla-Martín, D.; Rueda, C.B.; Estela, A.; Sánchez-Piris, M.; González-Sánchez, P.; Traba, J.; de la Fuente, S.; Scorrano, L.; Renau-Piqueras, J.; Alvarez, J.; et al. Silencing of the Charcot–Marie–Tooth Disease-Associated Gene GDAP1 Induces Abnormal Mitochondrial Distribution and Affects Ca2+ Homeostasis by Reducing Store-Operated Ca2+ Entry. Neurobiol. Dis. 2013, 55, 140–151. [Google Scholar] [CrossRef]
- González-Sánchez, P.; Pla-Martín, D.; Martínez-Valero, P.; Rueda, C.B.; Calpena, E.; del Arco, A.; Palau, F.; Satrústegui, J. CMT-Linked Loss-of-Function Mutations in GDAP1 Impair Store-Operated Ca2+ Entry-Stimulated Respiration. Sci. Rep. 2017, 7, 42993. [Google Scholar] [CrossRef] [Green Version]
- Cantarero, L.; Juárez-Escoto, E.; Civera-Tregón, A.; Rodríguez-Sanz, M.; Roldán, M.; Benítez, R.; Hoenicka, J.; Palau, F. Mitochondria–Lysosome Membrane Contacts Are Defective in GDAP1-Related Charcot–Marie–Tooth Disease. Hum. Mol. Genet. 2021, 29, 3589–3605. [Google Scholar] [CrossRef]
- Binięda, K.; Rzepnikowska, W.; Kolakowski, D.; Kaminska, J.; Szczepankiewicz, A.A.; Nieznańska, H.; Kochański, A.; Kabzińska, D. Mutations in GDAP1 Influence Structure and Function of the Trans-Golgi Network. Int. J. Mol. Sci. 2021, 22, 914. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faye, P.-A.; Vedrenne, N.; Miressi, F.; Rassat, M.; Romanenko, S.; Richard, L.; Bourthoumieu, S.; Funalot, B.; Sturtz, F.; Favreau, F.; et al. Optimized Protocol to Generate Spinal Motor Neuron Cells from Induced Pluripotent Stem Cells from Charcot Marie Tooth Patients. Brain Sci. 2020, 10, 407. [Google Scholar] [CrossRef] [PubMed]
- Saporta, M.A.; Dang, V.; Volfson, D.; Zou, B.; Xie, X.S.; Adebola, A.; Liem, R.K.; Shy, M.; Dimos, J.T. Axonal Charcot–Marie–Tooth Disease Patient-Derived Motor Neurons Demonstrate Disease-Specific Phenotypes Including Abnormal Electrophysiological Properties. Exp. Neurol. 2015, 263, 190–199. [Google Scholar] [CrossRef] [Green Version]
- Van Lent, J.; Verstraelen, P.; Asselbergh, B.; Adriaenssens, E.; Mateiu, L.; Verbist, C.; De Winter, V.; Eggermont, K.; Van Den Bosch, L.; De Vos, W.H.; et al. Induced Pluripotent Stem Cell-Derived Motor Neurons of CMT Type 2 Patients Reveal Progressive Mitochondrial Dysfunction. Brain 2021, awab226. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Woo, S.-Y.; Hong, Y.B.; Choi, H.; Kim, J.; Choi, H.; Mook-Jung, I.; Ha, N.; Kyung, J.; Koo, S.K.; et al. HDAC6 Inhibitors Rescued the Defective Axonal Mitochondrial Movement in Motor Neurons Derived from the Induced Pluripotent Stem Cells of Peripheral Neuropathy Patients with HSPB1 Mutation. Stem Cells Int. 2016, 2016, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Huang, L.; He, R.; Huang, W.; Wang, H.; Lai, X.; Zou, Z.; Sun, J.; Ke, Q.; Zheng, M.; et al. Modeling the Pathogenesis of Charcot-Marie-Tooth Disease Type 1A Using Patient-Specific IPSCs. Stem Cell Rep. 2018, 10, 120–133. [Google Scholar] [CrossRef] [Green Version]
- Martí, S.; León, M.; Orellana, C.; Prieto, J.; Ponsoda, X.; López-García, C.; Vílchez, J.J.; Sevilla, T.; Torres, J. Generation of a Disease-Specific IPS Cell Line Derived from a Patient with Charcot-Marie-Tooth Type 2K Lacking Functional GDAP1 Gene. Stem Cell Res. 2017, 18, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Miressi, F.; Faye, P.-A.; Pyromali, I.; Bourthoumieux, S.; Derouault, P.; Husson, M.; Favreau, F.; Sturtz, F.; Magdelaine, C.; Lia, A.-S. A Mutation Can Hide Another One: Think Structural Variants! Comput. Struct. Biotechnol. J. 2020, 18, 2095–2099. [Google Scholar] [CrossRef] [PubMed]
- Wetzig, A.; Alaiya, A.; Al-Alwan, M.; Pradez, C.; Pulicat, M.S.; Al-Mazrou, A.; Shinwari, Z.; Sleiman, G.; Ghebeh, H.; Al-Humaidan, H.; et al. Differential Marker Expression by Cultures Rich in Mesenchymal Stem Cells. BMC Cell Biol. 2013, 14, 54. [Google Scholar] [CrossRef] [Green Version]
- Itoh, M.; Umegaki-Arao, N.; Guo, Z.; Liu, L.; Higgins, C.A.; Christiano, A.M. Generation of 3D Skin Equivalents Fully Reconstituted from Human Induced Pluripotent Stem Cells (IPSCs). PLoS ONE 2013, 8, e77673. [Google Scholar] [CrossRef] [Green Version]
- Rzepnikowska, W.; Kochański, A. A Role for the GDAP1 Gene in the Molecular Pathogenesis of Charcot-Marie-Tooth Disease. Acta Neurobiol. Exp. (Warsz.) 2018, 78, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Civera-Tregón, A.; Domínguez, L.; Martínez-Valero, P.; Serrano, C.; Vallmitjana, A.; Benítez, R.; Hoenicka, J.; Satrústegui, J.; Palau, F. Mitochondria and Calcium Defects Correlate with Axonal Dysfunction in GDAP1-Related Charcot-Marie-Tooth Mouse Model. Neurobiol. Dis. 2021, 152, 105300. [Google Scholar] [CrossRef]
- Googins, M.R.; Woghiren-Afegbua, A.O.; Calderon, M.; St. Croix, C.M.; Kiselyov, K.I.; VanDemark, A.P. Structural and Functional Divergence of GDAP1 from the Glutathione S-transferase Superfamily. FASEB J. 2020, 34, 7192–7207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estela, A.; Pla-Martín, D.; Sánchez-Piris, M.; Sesaki, H.; Palau, F. Charcot-Marie-Tooth-Related Gene GDAP1 Complements Cell Cycle Delay at G2/M Phase in Saccharomyces Cerevisiae Fis1 Gene-Defective Cells. J. Biol. Chem. 2011, 286, 36777–36786. [Google Scholar] [CrossRef] [Green Version]
- Auer-Grumbach, M.; Fischer, C.; Papić, L.; John, E.; Plecko, B.; Bittner, R.; Bernert, G.; Pieber, T.; Miltenberger, G.; Schwarz, R.; et al. Two Novel Mutations in the GDAP1 and PRX Genes in Early Onset Charcot-Marie-Tooth Syndrome. Neuropediatrics 2008, 39, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Perkins, G.; Bossy-Wetzel, E.; Ellisman, M.H. New Insights into Mitochondrial Structure during Cell Death. Exp. Neurol. 2009, 218, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Vallat, J.-M.; Ouvrier, R.A.; Pollard, J.D.; Magdelaine, C.; Zhu, D.; Nicholson, G.A.; Grew, S.; Ryan, M.M.; Funalot, B. Histopathological Findings in Hereditary Motor and Sensory Neuropathy of Axonal Type with Onset in Early Childhood Associated With Mitofusin 2 Mutations. J. Neuropathol. Exp. Neurol. 2008, 67, 1097–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedetti, S.; Previtali, S.C.; Coviello, S.; Scarlato, M.; Cerri, F.; Di Pierri, E.; Piantoni, L.; Spiga, I.; Fazio, R.; Riva, N.; et al. Analyzing Histopathological Features of Rare Charcot-Marie-Tooth Neuropathies to Unravel Their Pathogenesis. Arch. Neurol. 2010, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Chan, D.C. Mitochondrial Dynamics in Regulating the Unique Phenotypes of Cancer and Stem Cells. Cell Metab. 2017, 26, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto, J.; León, M.; Ponsoda, X.; García-García, F.; Bort, R.; Serna, E.; Barneo-Muñoz, M.; Palau, F.; Dopazo, J.; López-García, C.; et al. Dysfunctional Mitochondrial Fission Impairs Cell Reprogramming. Cell Cycle 2016, 15, 3240–3250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarc, E.; Petan, T. Lipid Droplets and the Management of Cellular Stress. Yale J. Biol. Med. 2019, 18, 435–452. [Google Scholar]
- Boren, J.; Brindle, K.M. Apoptosis-Induced Mitochondrial Dysfunction Causes Cytoplasmic Lipid Droplet Formation. Cell Death Differ. 2012, 19, 1561–1570. [Google Scholar] [CrossRef] [Green Version]
- Farmer, B.C.; Walsh, A.E.; Kluemper, J.C.; Johnson, L.A. Lipid Droplets in Neurodegenerative Disorders. Front. Neurosci. 2020, 14, 742. [Google Scholar] [CrossRef]
- Bailey, A.P.; Koster, G.; Guillermier, C.; Hirst, E.M.A.; MacRae, J.I.; Lechene, C.P.; Postle, A.D.; Gould, A.P. Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila. Cell 2015, 163, 340–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panes, J.; Wendt, A.; Ramirez-Molina, O.; Castro, P.; Fuentealba, J. Deciphering the Role of PGC-1α in Neurological Disorders: From Mitochondrial Dysfunction to Synaptic Failure. Neural Regen. Res. 2022, 17, 237. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Calingasan, N.Y.; Yang, L.; Hennessey, T.; Johri, A.; Beal, M.F. Impairment of PGC-1alpha Expression, Neuropathology and Hepatic Steatosis in a Transgenic Mouse Model of Huntington’s Disease Following Chronic Energy Deprivation. Hum. Mol. Genet. 2010, 19, 3190–3205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Chandrasekaran, K.; Inoue, T.; Muragundla, A.; Russell, J.W. PGC-1α Regulation of Mitochondrial Degeneration in Experimental Diabetic Neuropathy. Neurobiol. Dis. 2014, 64, 118–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrasekaran, K.; Anjaneyulu, M.; Choi, J.; Kumar, P.; Salimian, M.; Ho, C.-Y.; Russell, J.W. Role of Mitochondria in Diabetic Peripheral Neuropathy: Influencing the NAD+-Dependent SIRT1–PGC-1α–TFAM Pathway. Int. Rev. Neurobiol. 2019, 145, 177–209. [Google Scholar] [CrossRef] [PubMed]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miressi, F.; Benslimane, N.; Favreau, F.; Rassat, M.; Richard, L.; Bourthoumieu, S.; Laroche, C.; Magy, L.; Magdelaine, C.; Sturtz, F.; et al. GDAP1 Involvement in Mitochondrial Function and Oxidative Stress, Investigated in a Charcot-Marie-Tooth Model of hiPSCs-Derived Motor Neurons. Biomedicines 2021, 9, 945. https://doi.org/10.3390/biomedicines9080945
Miressi F, Benslimane N, Favreau F, Rassat M, Richard L, Bourthoumieu S, Laroche C, Magy L, Magdelaine C, Sturtz F, et al. GDAP1 Involvement in Mitochondrial Function and Oxidative Stress, Investigated in a Charcot-Marie-Tooth Model of hiPSCs-Derived Motor Neurons. Biomedicines. 2021; 9(8):945. https://doi.org/10.3390/biomedicines9080945
Chicago/Turabian StyleMiressi, Federica, Nesrine Benslimane, Frédéric Favreau, Marion Rassat, Laurence Richard, Sylvie Bourthoumieu, Cécile Laroche, Laurent Magy, Corinne Magdelaine, Franck Sturtz, and et al. 2021. "GDAP1 Involvement in Mitochondrial Function and Oxidative Stress, Investigated in a Charcot-Marie-Tooth Model of hiPSCs-Derived Motor Neurons" Biomedicines 9, no. 8: 945. https://doi.org/10.3390/biomedicines9080945
APA StyleMiressi, F., Benslimane, N., Favreau, F., Rassat, M., Richard, L., Bourthoumieu, S., Laroche, C., Magy, L., Magdelaine, C., Sturtz, F., Lia, A. -S., & Faye, P. -A. (2021). GDAP1 Involvement in Mitochondrial Function and Oxidative Stress, Investigated in a Charcot-Marie-Tooth Model of hiPSCs-Derived Motor Neurons. Biomedicines, 9(8), 945. https://doi.org/10.3390/biomedicines9080945