
Epilepsy Phenotypes of Vitamin B6-Dependent Diseases: An Updated Systematic Review

 , and
, and
Abstract
:
1. Background
2. Search Methods
3. Results
3.1. Patient Characteristics
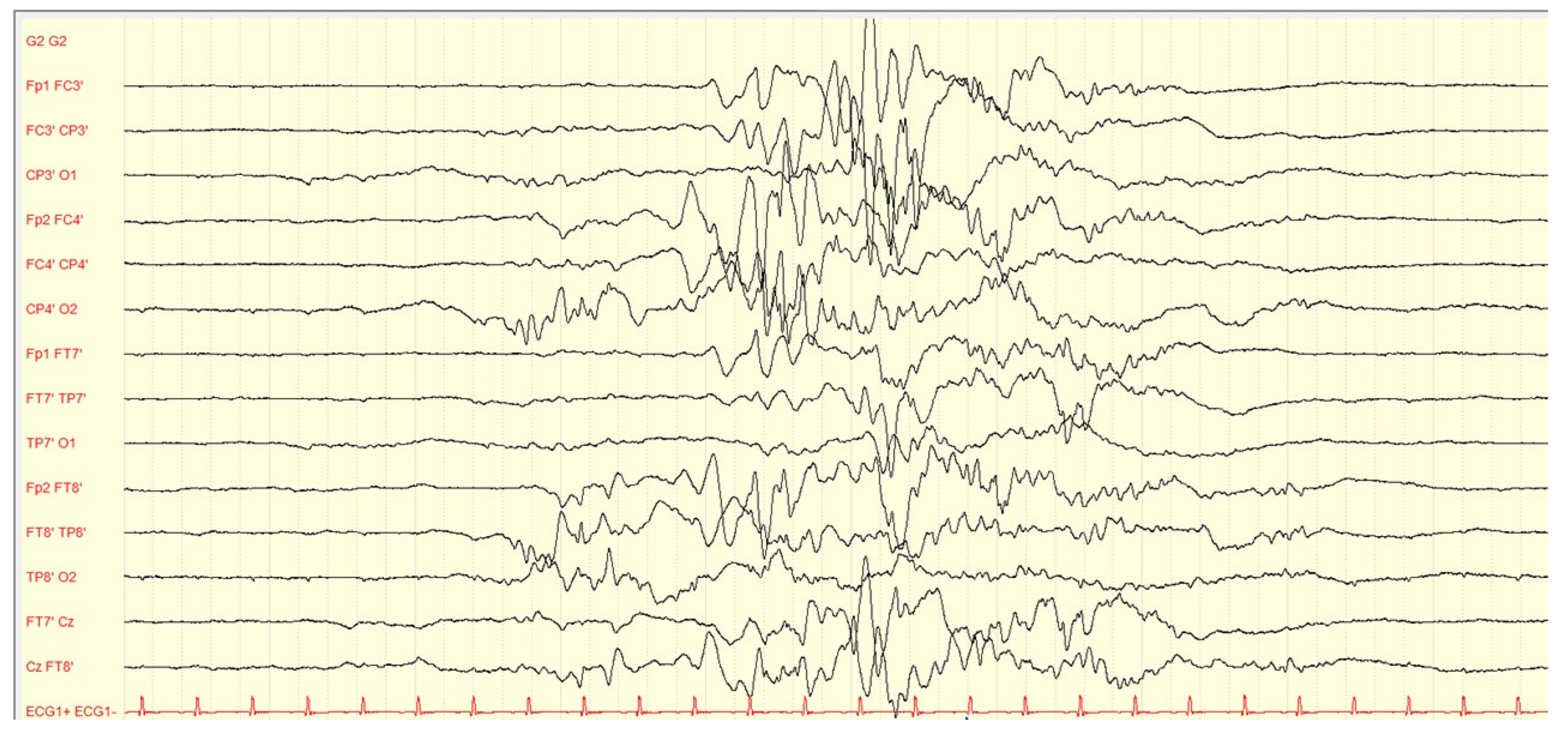
3.2. Epilepsy Phenotype
3.3. Treatment
4. Discussion
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Stockler, S.; Plecko, B.; Gospe, S.M., Jr.; Coulter-Mackie, M.; Connolly, M.; van Karnebeek, C.; Mercimek-Mahmutoglu, S.; Hartmann, H.; Scharer, S.; Struijs, E.; et al. Pyridoxine dependent epilepsy and antiquitin deficiency: Clinical and molecular characteristics and recommendations for diagnosis, treatment and follow-up. Mol. Genet. Metab. 2011, 104, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, M. Actual insights into treatable inborn errors of metabolism causing epilepsy. J. Pediatr. Neurosci. 2018, 13, 13–23. [Google Scholar] [PubMed]
- Wilson, M.P.; Plecko, B.; Mills, P.B.; Clayton, P. Disorders affecting vitamin B6 metabolism. J. Inherit. Metab. Dis. 2019, 42, 629–646. [Google Scholar] [CrossRef]
- Mastrangelo, M.; Cesario, S. Update on the treatment of vitamin B6 dependent epilepsies. Exp. Rev. Neurother. 2019, 19, 1135–1147. [Google Scholar] [CrossRef] [PubMed]
- Toldo, I.; Bonardi, C.M.; Bettella, E.; Polli, R.; Talenti, G.; Burlina, A.; Sartori, S.; Murgia, A. Brain Malformations Associated to Aldh7a1 Gene Mutations: Report of a Novel Homozygous Mutation and Literature Review. Eur. J. Paediatr. Neurol. 2018, 22, 1042–1053. [Google Scholar] [CrossRef] [PubMed]
- Coci, E.G.; Codutti, L.; Fink, C.; Bartsch, S.; Grüning, G.; Lücke, T.; Kurth, I.; Riedel, J. Novel Homozygous Missense Mutation in ALDH7A1 Causes Neonatal Pyridoxine Dependent Epilepsy. Mol. Cell Probes 2017, 32, 18–23. [Google Scholar] [CrossRef] [PubMed]
- di Salvo, M.L.; Mastrangelo, M.; Nogués, I.; Tolve, M.; Paiardini, A.; Carducci, C.; Mei, D.; Montomoli, M.; Tramonti, A.; Guerrini, R.; et al. Pyridoxine-5′-Phosphate Oxidase (Pnpo) Deficiency: Clinical and Biochemical Alterations Associated with the C.347g > A (P.·Arg116gln) Mutation. Mol. Genet. Metab. 2017, 122, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, R.M.; Patel, A.A.; Walsh, B.; Baumer, F.M.; Shah, A.S.; Peters, J.M.; Rodan, L.H.; Agrawal, P.B.; Pearl, P.L.; Takeoka, M. Systemic Manifestations in Pyridox(Am)Ine 5′-Phosphate Oxidase Deficiency. Pediatr. Neurol. 2017, 76, 47–53. [Google Scholar] [CrossRef]
- Xue, J.; Chang, X.; Zhang, Y.; Yang, Z. Novel Phenotypes of Pyridox(Am)Ine-5’-Phosphate Oxidase Deficiency and High Prevalence of c.445_448del Mutation in Chinese Patients. Metab. Brain Dis. 2017, 32, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Darin, N.; Reid, E.; Prunetti, L.; Samuelsson, L.; Husain, R.A.; Wilson, M.; el Yacoubi, B.; Footitt, E.; Chong, W.K.; Wilson, L.C.; et al. Mutations in PROSC Disrupt Cellular Pyridoxal Phosphate Homeostasis and Cause Vitamin-B6-Dependent Epilepsy. Am. J. Hum. Genet. 2016, 99, 1325–1337. [Google Scholar] [CrossRef]
- Cirillo, M.; Venkatesan, C.; Millichap, J.J.; Stack, C.V.; Nordli, D.R. Case Report: Intravenous and Oral Pyridoxine Trial for Diagnosis of Pyridoxine-Dependent Epilepsy. Pediatrics 2015, 136, e257–e261. [Google Scholar] [CrossRef]
- Veeravigrom, M.; Damrongphol, P.; Ittiwut, R.; Ittiwut, C.; Suphapeetiporn, K.; Shotelersuk, V. Pyridoxal 5’-Phosphate-Responsive Epilepsy with Novel Mutations in the PNPO Gene: A Case Report. Genet. Mol. Res. 2015, 14, 14131–14135. [Google Scholar] [CrossRef]
- Marguet, F.; Barakizou, H.; Tebani, A.; Abily-Donval, L.; Torre, S.; Bayoudh, F.; Jebnoun, S.; Brasseur-Daudruy, M.; Marret, S.; Laquerriere, A.; et al. Pyridoxine-Dependent Epilepsy: Report on Three Families with Neuropathology. Metab. Brain Dis. 2016, 31, 1435–1443. [Google Scholar] [CrossRef]
- de Rooy, R.L.P.; Halbertsma, F.J.; Struijs, E.A.; van Spronsen, F.J.; Lunsing, R.J.; Schippers, H.M.; van Hasselt, P.M.; Plecko, B.; Wohlrab, G.; Whalen, S.; et al. Pyridoxine Dependent Epilepsy: Is Late Onset a Predictor for Favorable Outcome? Eur. J. Paediatr. Neurol. 2018, 22, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, B.; Abeling, N.G.; Salomons, G.S.; Struys, E.A.; Simas-Mendes, M.; Geukers, V.G.; Poll-The, B.T. Pyridoxine Responsive Epilepsy Caused by a Novel Homozygous PNPO Mutation. Mol. Genet. Metab. Rep. 2016, 6, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Falsaperla, R.; Vari, M.S.; Toldo, I.; Murgia, A.; Sartori, S.; Vecchi, M.; Suppiej, A.; Burlina, A.; Mastrangelo, M.; Leuzzi, V.; et al. Pyridoxine-Dependent Epilepsies: An Observational Study on Clinical, Diagnostic, Therapeutic and Prognostic Features in a Pediatric Cohort. Metab. Brain Dis. 2018, 33, 261–269. [Google Scholar] [CrossRef]
- Wang, S.; Sun, J.; Tu, Y.; Zhu, L.; Feng, Z. Clinical and Genetic Characteristics of Pyridoxine-Dependent Epilepsy: Case Series Report of Three Chinese Patients with Phenotypic Variability. Exp. Med. 2017, 14, 1989–1992. [Google Scholar] [CrossRef] [PubMed]
- Porri, S.; Fluss, J.; Plecko, B.; Paschke, E.; Korff, C.M.; Kern, I. Positive Outcome Following Early Diagnosis and Treatment of Pyridoxal-5′-Phosphate Oxidase Deficiency: A Case Report. Neuropediatrics 2014, 45, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Oesch, G.; Maga, A.M.; Friedman, S.D.; Poliachik, S.L.; Budech, C.B.; Wright, J.N.; Bok, L.A.; Gospe, S.M. Geometric Morphometrics Reveal Altered Corpus Callosum Shape in Pyridoxine-Dependent Epilepsy. Neurology 2018, 91, e78–e86. [Google Scholar] [CrossRef]
- Mercimek-Mahmutoglu, S.; Cordeiro, D.; Cruz, V.; Hyland, K.; Struys, E.A.; Kyriakopoulou, L.; Mamak, E. Novel Therapy for Pyridoxine Dependent Epilepsy Due to ALDH7A1 Genetic Defect: L-Arginine Supplementation Alternative to Lysine-Restricted Diet. Eur. J. Paediatr. Neurol. 2014, 18, 741–746. [Google Scholar] [CrossRef]
- Riikonen, R.; Mankinen, K.; Gaily, E. Long-Term Outcome in Pyridoxine-Responsive Infantile Epilepsy. Eur. J. Paediatr. Neurol. 2015, 19, 647–651. [Google Scholar] [CrossRef]
- Coman, D.; Lewindon, P.; Clayton, P.; Riney, K. PNPO Deficiency and Cirrhosis: Expanding the Clinical Phenotype. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2016; Volume 25, pp. 71–75. [Google Scholar]
- Tincheva, S.; Todorov, T.; Todorova, A.; Georgieva, R.; Stamatov, D.; Yordanova, I.; Kadiyska, T.; Georgieva, B.; Bojidarova, M.; Tacheva, G.; et al. First Cases of Pyridoxine-Dependent Epilepsy in Bulgaria: Novel Mutation in the ALDH7A1 Gene. Neurol. Sci. 2015, 36, 2209–2212. [Google Scholar] [CrossRef]
- Mercimek-Mahmutoglu, S.; Patel, J.; Cordeiro, D.; Hewson, S.; Callen, D.; Donner, E.J.; Hahn, C.D.; Kannu, P.; Kobayashi, J.; Minassian, B.A.; et al. Diagnostic Yield of Genetic Testing in Epileptic Encephalopathy in Childhood. Epilepsia 2015, 56, 707–716. [Google Scholar] [CrossRef] [PubMed]
- van de Ven, S.; Gardeitchik, T.; Kouwenberg, D.; Kluijtmans, L.; Wevers, R.; Morava, E. Long-Term Clinical Outcome, Therapy and Mild Mitochondrial Dysfunction in Hyperprolinemia. J. Inherit. Metab. Dis. 2014, 37, 383–390. [Google Scholar] [CrossRef]
- Sudarsanam, A.; Singh, H.; Wilcken, B.; Stormon, M.; Arbuckle, S.; Schmitt, B.; Clayton, P.; Earl, J.; Webster, R. Cirrhosis Associated with Pyridoxal 5′-Phosphate Treatment of Pyridoxamine 5′-Phosphate Oxidase Deficiency. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2014; Volume 17, pp. 67–70. [Google Scholar]
- Khayat, M.; Korman, S.H.; Frankel, P.; Weintraub, Z.; Hershckowitz, S.; Sheffer, V.F.; Elisha, M.B.; Wevers, R.A.; Falik-Zaccai, T.C. PNPO Deficiency: An under Diagnosed Inborn Error of Pyridoxine Metabolism. Mol. Genet. Metab. 2008, 94, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Goyal, M.; Fequiere, P.R.; McGrath, T.M.; Hyland, K. Seizures with Decreased Levels of Pyridoxal Phosphate in Cerebrospinal Fluid. Pediatr. Neurol 2013, 48, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Mercimek-Mahmutoglu, S.; Horvath, G.A.; Coulter-Mackie, M.; Nelson, T.; Waters, P.J.; Sargent, M.; Struys, E.; Jakobs, C.; Stockler-Ipsiroglu, S.; Connolly, M.B. Profound Neonatal Hypoglycemia and Lactic Acidosis Caused by Pyridoxine-Dependent Epilepsy. Pediatrics 2012, 129, e1368–e1372. [Google Scholar] [CrossRef]
- Demirbilek, H.; Alanay, Y.; Alikaşifoǧlu, A.; Topçu, M.; Mornet, E.; Gönç, N.; Özön, A.; Kandemir, N. Hypophosphatasia Presenting with Pyridoxine-Responsive Seizures, Hypercalcemia, and Pseudotumor Cerebri: Case Report. JCRPE J. Clin. Res. Pediatr. Endocrinol. 2012, 4, 34–38. [Google Scholar] [CrossRef]
- van Karnebeek, C.D.M.; Hartmann, H.; Jaggumantri, S.; Bok, L.A.; Cheng, B.; Connolly, M.; Coughlin, C.R.; Das, A.M.; Gospe, S.M.; Jakobs, C.; et al. Lysine Restricted Diet for Pyridoxine-Dependent Epilepsy: First Evidence and Future Trials. Mol. Genet. Metab. 2012, 107, 335–344. [Google Scholar] [CrossRef]
- de Roo, M.G.A.; Abeling, N.G.G.M.; Majoie, C.B.; Bosch, A.M.; Koelman, J.H.T.M.; Cobben, J.M.; Duran, M.; Poll-The, B.T. Infantile Hypophosphatasia without Bone Deformities Presenting with Severe Pyridoxine-Resistant Seizures. Mol. Genet. Metab. 2014, 111, 404–407. [Google Scholar] [CrossRef]
- Ruiz, A.; García-Villoria, J.; Ormazabal, A.; Zschocke, J.; Fiol, M.; Navarro-Sastre, A.; Artuch, R.; Vilaseca, M.A.; Ribes, A. A New Fatal Case of Pyridox(Am)Ine 5′-Phosphate Oxidase (PNPO) Deficiency. Mol. Genet. Metab. 2008, 93, 216–218. [Google Scholar] [CrossRef] [PubMed]
- Ville, D.; Ginguene, C.; Marignier, S.; Portes, V.d.; de Bellescize, J. Early Diagnosis of Pyridoxine-Dependent Epilepsy: Video-EEG Monitoring and Biochemical and Genetic Investigation. Eur. J. Paediatr. Neurol. 2013, 17, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Segal, E.B.; Grinspan, Z.M.; Mandel, A.M.; Gospe, S.M. Biomarkers Aiding Diagnosis of Atypical Presentation of Pyridoxine-Dependent Epilepsy. Pediatr. Neurol. 2011, 44, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Millet, A.; Salomons, G.S.; Cneude, F.; Corne, C.; Debillon, T.; Jakobs, C.; Struys, E.; Hamelin, S. Novel Mutations in Pyridoxine-Dependent Epilepsy. Eur. J. Paediatr. Neurol. 2011, 15, 74–77. [Google Scholar] [CrossRef]
- Tlili, A.; Hamida Hentati, N.; Chaabane, R.; Gargouri, A.; Fakhfakh, F. Pyridoxine-Dependent Epilepsy in Tunisia Is Caused by a Founder Missense Mutation of the ALDH7A1 Gene. Gene 2013, 518, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, C.; Liese, J.; Schwarz, T.; Kunzmann, S.; Wirbelauer, J.; Nowak, J.; Hamann, J.; Girschick, H.; Graser, S.; Dietz, K.; et al. Compound Heterozygosity of Two Functional Null Mutations in the ALPL Gene Associated with Deleterious Neurological Outcome in an Infant with Hypophosphatasia. Bone 2013, 55, 150–157. [Google Scholar] [CrossRef]
- Bok, L.A.; Maurits, N.M.; Willemsen, M.A.; Jakobs, C.; Teune, L.K.; Poll-The, B.T.; de Coo, I.F.; Toet, M.C.; Hagebeuk, E.E.; Brouwer, O.F.; et al. The EEG Response to Pyridoxine-IV Neither Identifies nor Excludes Pyridoxine-Dependent Epilepsy. Epilepsia 2010, 51, 2406–2411. [Google Scholar] [CrossRef]
- Baumgartner-Sigl, S.; Haberlandt, E.; Mumm, S.; Scholl-Bürgi, S.; Sergi, C.; Ryan, L.; Ericson, K.L.; Whyte, M.P.; Högler, W. Pyridoxine-Responsive Seizures as the First Symptom of Infantile Hypophosphatasia Caused by Two Novel Missense Mutations (c.677T > C, p.M226T.; c.1112C > T, p.T371I) of the Tissue-Nonspecific Alkaline Phosphatase Gene. Bone 2007, 40, 1655–1661. [Google Scholar] [CrossRef]
- Bennett, C.L.; Chen, Y.; Hahn, S.; Glass, I.A.; Gospe, S.M. Prevalence of ALDH7A1 Mutations in 18 North American Pyridoxine-Dependent Seizure (PDS) Patients. Epilepsia 2009, 50, 1167–1175. [Google Scholar] [CrossRef]
- Striano, P.; Battaglia, S.; Giordano, L.; Capovilla, G.; Beccaria, F.; Struys, E.A.; Salomons, G.S.; Jakobs, C. Two Novel ALDH7A1 (Antiquitin) Splicing Mutations Associated with Pyridoxine-Dependent Seizures. Epilepsia 2009, 50, 933–936. [Google Scholar] [CrossRef]
- Oliveira, R.; Pereira, C.; Rodrigues, F.; Alfaite, C.; Garcia, P.; Robalo, C.; Fineza, I.; Gonçalves, O.; Struys, E.; Salomons, G.; et al. Pyridoxine-Dependent Epilepsy Due to Antiquitin Deficiency: Achieving a Favourable Outcome. Epileptic Disord. 2013, 15, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Pérez, B.; Gutiérrez-Solana, L.G.; Verdú, A.; Merinero, B.; Yuste-Checa, P.; Ruiz-Sala, P.; Calvo, R.; Jalan, A.; Marín, L.L.; Campos, O.; et al. Clinical, Biochemical, and Molecular Studies in Pyridoxine-Dependent Epilepsy. Antisense Therapy as Possible New Therapeutic Option. Epilepsia 2013, 54, 239–248. [Google Scholar] [CrossRef]
- Ware, T.L.; Earl, J.; Salomons, G.S.; Struys, E.A.; Peters, H.L.; Howell, K.B.; Pitt, J.J.; Freeman, J.L. Typical and Atypical Phenotypes of PNPO Deficiency with Elevated CSF and Plasma Pyridoxamine on Treatment. Dev. Med. Child Neurol. 2014, 56, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Mills, P.B.; Footitt, E.J.; Mills, K.A.; Tuschl, K.; Aylett, S.; Varadkar, S.; Hemingway, C.; Marlow, N.; Rennie, J.; Baxter, P.; et al. Genotypic and Phenotypic Spectrum of Pyridoxine-Dependent Epilepsy (ALDH7A1 Deficiency). Brain 2010, 133, 2148–2159. [Google Scholar] [CrossRef] [PubMed]
- Bok, L.A.; Halbertsma, F.J.; Houterman, S.; Wevers, R.A.; Vreeswijk, C.; Jakobs, C.; Struys, E.; van der Hoeven, J.H.; Sival, D.A.; Willemsen, M.A. Long-Term Outcome in Pyridoxine-Dependent Epilepsy. Dev. Med. Child Neurol. 2012, 54, 849–854. [Google Scholar] [CrossRef]
- Bagci, S.; Zschocke, J.; Hoffmann, G.F.; Bast, T.; Klepper, J.; Müller, A.; Heep, A.; Bartmann, P.; Franz, A.R. Pyridoxal Phosphate-Dependent Neonatal Epileptic Encephalopathy. Arch. Dis. Child.–Fetal Neonatal Ed. 2008, 93, F151–F152. [Google Scholar] [CrossRef]
- Gallagher, R.C.; van Hove, J.L.K.; Scharer, G.; Hyland, K.; Plecko, B.; Waters, P.J.; Mercimek-Mahmutoglu, S.; Stockler-Ipsiroglu, S.; Salomons, G.S.; Rosenberg, E.H.; et al. Folinic Acid-Responsive Seizures Are Identical to Pyridoxine-Dependent Epilepsy. Ann. Neurol. 2009, 65, 550–556. [Google Scholar] [CrossRef]
- Baumgart, A.; von Spiczak, S.; Verhoeven-Duif, N.M.; Møller, R.S.; Boor, R.; Muhle, H.; Jähn, J.A.; Klitten, L.L.; Hjalgrim, H.; Lindhout, D.; et al. Atypical Vitamin B6 Deficiency: A Rare Cause of Unexplained Neonatal and Infantile Epilepsies. J. Child Neurol. 2014, 29, 704–707. [Google Scholar] [CrossRef]
- Kaczorowska, M.; Kmiec, T.; Jakobs, C.; Kacinski, M.; Kroczka, S.; Salomons, G.S.; Struys, E.A.; Jozwiak, S. Pyridoxine-Dependent Seizures Caused by Alpha Amino Adipic Semialdehyde Dehydrogenase Deficiency: The First Polish Case with Confirmed Biochemical and Molecular Pathology. J. Child Neurol. 2008, 23, 1455–1459. [Google Scholar] [CrossRef]
- Belachew, D.; Kazmerski, T.; Libman, I.; Goldstein, A.C.; Stevens, S.T.; DeWard, S.; Vockley, J.; Sperling, M.A.; Balest, A.L. Infantile Hypophosphatasia Secondary to a Novel Compound Heterozygous Mutation Presenting with Pyridoxine-Responsive Seizures. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2013; Volume 11, pp. 17–24. [Google Scholar]
- Proudfoot, M.; Jardine, P.; Straukiene, A.; Noad, R.; Parrish, A.; Ellard, S.; Weatherby, S. Long-Term Follow-up of a Successfully Treated Case of Congenital Pyridoxine-Dependent Epilepsy. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2013; Volume 10, pp. 103–106. [Google Scholar]
- Pearl, P.L.; Hyland, K.; Chiles, J.; McGavin, C.L.; Yu, Y.; Taylor, D. Partial Pyridoxine Responsiveness in PNPO Deficiency. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2013; Volume 9, pp. 139–142. [Google Scholar]
- Kluger, G.; Blank, R.; Paul, K.; Paschke, E.; Jansen, E.; Jakobs, C.; Wörle, H.; Plecko, B. Pyridoxine-Dependent Epilepsy: Normal Outcome in a Patient with Late Diagnosis after Prolonged Status Epilepticus Causing Cortical Blindness. Neuropediatrics 2008, 39, 276–279. [Google Scholar] [CrossRef]
- Scharer, G.; Brocker, C.; Vasiliou, V.; Creadon-Swindell, G.; Gallagher, R.C.; Spector, E.; van Hove, J.L.K. The Genotypic and Phenotypic Spectrum of Pyridoxine-Dependent Epilepsy Due to Mutations in ALDH7A1. J. Inherit. Metab. Dis. 2010, 33, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, S.; Bowling, F.; Carpenter, K.; Earl, J.; Chaitow, J.; Pitt, J.; Mornet, E.; Sillence, D.; Ellaway, C. Perinatal Hypophosphatasia Presenting as Neonatal Epileptic Encephalopathy with Abnormal Neurotransmitter Metabolism Secondary to Reduced Co-Factor Pyridoxal-5′-Phosphate Availability. J. Inherit. Metab. Dis. 2010, 33, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, G.F.; Schmitt, B.; Windfuhr, M.; Wagner, N.; Strehl, H.; Bagci, S.; Franz, A.R.; Mills, P.B.; Clayton, P.T.; Baumgartner, M.R.; et al. Pyridoxal 5′-Phosphate May Be Curative in Early-Onset Epileptic Encephalopathy. J. Inherit. Metab. Dis. 2007, 30, 96–99. [Google Scholar] [CrossRef]
- Bok, L.A.; Been, J.v.; Struys, E.A.; Jakobs, C.; Rijper, E.A.M.; Willemsen, M.A. Antenatal Treatment in Two Dutch Families with Pyridoxine-Dependent Seizures. Eur. J. Pediatr. 2010, 169, 297–303. [Google Scholar] [CrossRef]
- Ahmed, S.; DeBerardinis, R.J.; Ni, M.; Afroze, B. Vitamin B6-Dependent Epilepsy Due to Pyridoxal Phosphate-Binding Protein (PLPBP) Defect–First Case Report from Pakistan and Review of Literature. Ann. Med. Surg. 2020, 60, 721–727. [Google Scholar] [CrossRef]
- Efthymiou, S.; Dutra-Clarke, M.; Maroofian, R.; Kaiyrzhanov, R.; Scala, M.; Reza Alvi, J.; Sultan, T.; Christoforou, M.; Tuyet Mai Nguyen, T.; Mankad, K.; et al. Expanding the Phenotype of PIGS-Associated Early Onset Epileptic Developmental Encephalopathy. Epilepsia 2021, 62, e35–e41. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.M.; Murakami, Y.; Wigby, K.M.; Baratang, N.v.; Rousseau, J.; St-Denis, A.; Rosenfeld, J.A.; Laniewski, S.C.; Jones, J.; Iglesias, A.D.; et al. Mutations in PIGS, encoding a GPI Transamidase, Cause a Neurological Syndrome Ranging from Fetal Akinesia to Epileptic Encephalopathy. Am. J. Hum. Genet. 2018, 103, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Namavar, Y.; Duineveld, D.J.; Both, G.I.A.; Fiksinski, A.M.; Vorstman, J.A.S.; Verhoeven-Duif, N.M.; Zinkstok, J.R. Psychiatric Phenotypes Associated with Hyperprolinemia: A Systematic Review. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2021, 186, 289–317. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Paria, P.; Saini, A.G.; Suthar, R.; Bhatia, V.; Attri, S.V. Metabolic Epilepsy in Hyperprolinemia Type II Due to a Novel Nonsense ALDH4A1 Gene Variant. Metab. Brain Dis. 2021, 36, 1413–1417. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Mao, X.; Long, H.; Xiao, B.; Luo, Z.; Xiao, W.; Jin, X. Compound Heterozygous PIGS Variants Associated with Infantile Spasm, Global Developmental Delay, Hearing Loss, Visual Impairment, and Hypotonia. Front. Genet. 2020, 11, 564. [Google Scholar] [CrossRef] [PubMed]
- Pavitt, S.; Sandoval Karamian, A.G.; Chattree, G.; Klotz, J.; Beres, S. Teaching Video NeuroImages: Atypical Abnormal Eye Movements in PNPO-Related Epilepsy. Neurology 2021, 96, e1927. [Google Scholar] [CrossRef]
- Kundap, U.P.; Paudel, Y.N.; Shaikh, M.F. Animal Models of Metabolic Epilepsy and Epilepsy Associated Metabolic Dysfunction: A Systematic Review. Pharmaceuticals 2020, 13, 106. [Google Scholar] [CrossRef]
- Akiyama, T.; Hyodo, Y.; Hasegawa, K.; Oboshi, T.; Imai, K.; Ishihara, N.; Dowa, Y.; Koike, T.; Yamamoto, T.; Shibasaki, J.; et al. Pyridoxal in the Cerebrospinal Fluid May Be a Better Indicator of Vitamin B6–Dependent Epilepsy Than Pyridoxal 5′-Phosphate. Pediatr. Neurol. 2020, 113, 33–41. [Google Scholar] [CrossRef]
- Espinoza, A.C.; Wright, M.A.; Candee, M.S.; Trandafir, C.; Nelson, G.R. Child Neurology: Late-Onset Vitamin B6-Dependent Epilepsy Identified by Rapid Genome Sequencing. Neurology 2021, 96, 911–914. [Google Scholar] [CrossRef] [PubMed]
- Johannsen, J.; Bierhals, T.; Deindl, P.; Hecher, L.; Hermann, K.; Hempel, M.; Kloth, K.; Denecke, J. Excessive Seizure Clusters in an Otherwise Well-Controlled Epilepsy as a Possible Hallmark of Untreated Vitamin B6-Responsive Epilepsy Due to a Homozygous PLPBP Missense Variant. J. Pediatr. Genet. 2019, 08, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Farmania, R.; Gupta, A.; Ankur, K.; Chetry, S.; Sharma, S. Complexities of Pyridoxine Response in PNPO Deficiency. Epilepsy Behav. Rep. 2021, 16, 100443. [Google Scholar] [CrossRef]
- Tsao, H.S.; Case, S.D. Pyridoxine-Dependent Epilepsy as a Cause of Neonatal Seizures. Rhode Isl. Med. J. 2022, 105, 17–21. [Google Scholar]
- Vossler, D.G.; Bainbridge, J.L.; Boggs, J.G.; Novotny, E.J.; Loddenkemper, T.; Faught, E.; Amengual-Gual, M.; Fischer, S.N.; Gloss, D.S.; Olson, D.M.; et al. Treatment of Refractory Convulsive Status Epilepticus: A Comprehensive Review by the American Epilepsy Society Treatments Committee. Epilepsy Curr. 2020, 20, 245–264. [Google Scholar] [CrossRef]
- Tseng, L.A.; Abdenur, J.E.; Andrews, A.; Aziz, V.G.; Bok, L.A.; Boyer, M.; Buhas, D.; Hartmann, H.; Footitt, E.J.; Grønborg, S.; et al. Timing of Therapy and Neurodevelopmental Outcomes in 18 Families with Pyridoxine-Dependent Epilepsy. Mol. Genet. Metab. 2022, 135, 350–356. [Google Scholar] [CrossRef]
- Pal, M.; Lace, B.; Labrie, Y.; Laflamme, N.; Rioux, N.; Setty, S.T.; Dugas, M.A.; Gosselin, L.; Droit, A.; Chrestian, N.; et al. A Founder Mutation in the PLPBP Gene in Families from Saguenay-Lac-St-Jean Region Affected by a Pyridoxine-Dependent Epilepsy. JIMD Rep. 2021, 59, 32–41. [Google Scholar] [CrossRef]
- Strijker, M.; Tseng, L.A.; van Avezaath, L.K.; Oude Luttikhuis, M.A.M.; Ketelaar, T.; Coughlin, C.R.; Coenen, M.A.; van Spronsen, F.J.; Williams, M.; de Vries, M.C.; et al. Cognitive and Neurological Outcome of Patients in the Dutch Pyridoxine-Dependent Epilepsy (PDE-ALDH7A1) Cohort, a Cross-Sectional Study. Eur. J. Paediatr. Neurol. 2021, 33, 112–120. [Google Scholar] [CrossRef]
- Heath, O.; Pitt, J.; Mandelstam, S.; Kuschel, C.; Vasudevan, A.; Donoghue, S. Early-Onset Vitamin B6-Dependent Epilepsy Due to Pathogenic PLPBP Variants in a Premature Infant: A Case Report and Review of the Literature. JIMD Rep. 2021, 58, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Osman, C.; Foulds, N.; Hunt, D.; Jade Edwards, C.; Prevett, M. Diagnosis of Pyridoxine-Dependent Epilepsy in an Adult Presenting with Recurrent Status Epilepticus. Epilepsia 2020, 61, e1–e6. [Google Scholar] [CrossRef] [PubMed]
- Işikay, S. Late Diagnosis of Pyridoxine-Dependent Epilepsy in Two Adolescent Siblings. Ann. Indian Acad. Neurol. 2021, 24, 770–771. [Google Scholar] [CrossRef]
- Johnstone, D.L.; Al-Shekaili, H.H.; Tarailo-Graovac, M.; Wolf, N.I.; Ivy, A.S.; Demarest, S.; Roussel, Y.; Ciapaite, J.; van Roermund, C.W.T.; Kernohan, K.D.; et al. PLPHP Deficiency: Clinical, Genetic, Biochemical, and Mechanistic Insights. Brain 2019, 142, 542–559. [Google Scholar] [CrossRef] [PubMed]
- Mohanlal, S.; Bindu, P.S.; Sureshbabu, S.; Kumar, S. Variable Treatment Response in a Patient with Pyridoxal N Phosphate Oxidase (PNPO) Deficiency- Understanding the Paradox. Epilepsy Behav. Rep. 2020, 14, 100357. [Google Scholar] [CrossRef] [PubMed]
- Lugli, L.; Bariola, M.C.; Ori, L.; Lucaccioni, L.; Berardi, A.; Ferrari, F. Further Delineation of Pyridoxine-Responsive Pyridoxine Phosphate Oxidase Deficiency Epilepsy: Report of a New Case and Review of the Literature with Genotype-Phenotype Correlation. J. Child Neurol. 2019, 34, 937–943. [Google Scholar] [CrossRef]
- Minet, P.; Sarret, C.; Miret, A.; Mention, K.; Benoist, J.F.; Remerand, G. Clinical and Biochemical Outcome of a Patient with Pyridoxine-Dependent Epilepsy Treated by Triple Therapy (Pyridoxine Supplementation, Lysine-Restricted Diet, and Arginine Supplementation). Acta Neurol. Belg. 2021, 121, 1669–1675. [Google Scholar] [CrossRef]
- Kava, M.P.; Bryant, L.; Rowe, P.; Lewis, B.; Greed, L.; Balasubramaniam, S. Beneficial Outcome of Early Dietary Lysine Restriction as an Adjunct to Pyridoxine Therapy in a Child with Pyridoxine Dependant Epilepsy Due to Antiquitin Deficiency. JIMD Rep. 2020, 54, 9–15. [Google Scholar] [CrossRef]
- Kesavan, S.; Singanamalla, B.; Krishna Sahitya, D.; Saini, A.; Vyas, S. Epilepsy and Hydrocephalus: Should Pyridoxine-Dependent Epilepsy Cross Our Minds? Ann. Indian Acad. Neurol. 2020, 23, 239–241. [Google Scholar]
- Jiao, X.; Gong, P.; Niu, Y.; Zhang, Y.; Yang, Z. A Rare Presentation Characterized by Epileptic Spasms in ALDH7A1, Pyridox(Am)Ine-5′-Phosphate Oxidase, and PLPBP Deficiency. Front. Genet. 2022, 13, 804461. [Google Scholar] [CrossRef]
- Alghamdi, M.; Bashiri, F.A.; Abdelhakim, M.; Adly, N.; Jamjoom, D.Z.; Sumaily, K.M.; Alghanem, B.; Arold, S.T. Phenotypic and Molecular Spectrum of Pyridoxamine-5′-Phosphate Oxidase Deficiency: A Scoping Review of 87 Cases of Pyridoxamine-5′-Phosphate Oxidase Deficiency. Clin. Genet. 2021, 99, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.V.; Frid, M.; Stödberg, T.; Barbaro, M.; Wedell, A.; Christensen, M.; Bak, M.; Ek, J.; Madsen, C.G.; Darin, N.; et al. Diagnostic Pitfalls in Vitamin B6-Dependent Epilepsy Caused by Mutations in the PLPBP Gene. JIMD Rep. 2019, 50, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Gong, P.; Wu, Y.; Zhang, Y.; Yang, Z. Analysis of the Phenotypic Variability as Well as Impact of Early Diagnosis and Treatment in Six Affected Families with ALDH7A1 Deficiency. Front. Genet. 2021, 12, 644447. [Google Scholar] [CrossRef] [PubMed]
- Gazeteci-Tekin, H.; Demir, M.; Aktan, G.; Tekgül, H.; Gökben, S. The Case of Pyridoxine Dependent Epilepsy Misdiagnosed as Non-Ketotic Hyperglycinemia. Turk. J. Pediatr. 2019, 61, 599–603. [Google Scholar] [CrossRef]
- Jiao, X.; Xue, J.; Gong, P.; Wu, Y.; Zhang, Y.; Jiang, Y.; Yang, Z. Clinical and Genetic Features in Pyridoxine-Dependent Epilepsy: A Chinese Cohort Study. Dev. Med. Child Neurol. 2020, 62, 315–321. [Google Scholar] [CrossRef]
- Gibaud, M.; Barth, M.; Lefranc, J.; Mention, K.; Villeneuve, N.; Schiff, M.; Maurey, H.; Barthez, M.A.; Caubel, I.; Chouchane, M.; et al. West Syndrome Is an Exceptional Presentation of Pyridoxine- and Pyridoxal Phosphate-Dependent Epilepsy: Data from a French Cohort and Review of the Literature. Front. Pediatr. 2021, 9, 621200. [Google Scholar] [CrossRef] [PubMed]
- Dowa, Y.; Shiihara, T.; Akiyama, T.; Hasegawa, K.; Inoue, F.; Watanabe, M. A Case of Pyridoxine-Dependent Epilepsy with Novel ALDH7A1 Mutations. Oxf. Med. Case Rep. 2020, 2020, 99–103. [Google Scholar] [CrossRef]
- Alghamdi, M.; Arold, S.T.; Hasan, H.; Bashiri, F. Pyridox(Am)Ine 5′-Phosphate Oxidase Deficiency: Severe Prenatal Presentation with Hypoxic Ischemic Encephalopathy. J. Pediatr. Epilepsy 2019, 8, 049–055. [Google Scholar] [CrossRef]
- Kaminiów, K.; Pająk, M.; Pająk, R.; Paprocka, J. Pyridoxine-Dependent Epilepsy and Antiquitin Deficiency Resulting in Neonatal-Onset Refractory Seizures. Brain Sci. 2022, 12, 65. [Google Scholar] [CrossRef]
- Motte, J.; Fisse, A.L.; Grüter, T.; Schneider, R.; Breuer, T.; Lücke, T.; Krueger, S.; Nguyen, H.P.; Gold, R.; Ayzenberg, I.; et al. Novel variants in a patient with late-onset hyperprolinemia type II: Diagnostic key for status epilepticus and lactic acidosis. BMC Neurol. 2019, 19, 345. [Google Scholar] [CrossRef]
- Basura, G.J.; Hagland, S.P.; Wiltse, A.M.; Gospe, S.M. Clinical features and the management of pyridoxine-dependent and pyridoxine responsive seizures: Review of 63 North American cases submitted to a patient Registry. Eur. J. Pediatr. 2009, 168, 697–704. [Google Scholar] [CrossRef]
- Naasan, G.; Yabroudi, M.; Rahi, A.; Mikati, M.A. Electroencephalographic changes in pyridoxine-dependant epilepsy: New observations. Epileptic Disord. 2009, 11, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Ramantani, G.; Bölsterli, B.K.; Abler, M.; Klepper, J.; Korinthenberg, R.; Kurlemann, G.; Tibussek, D.; Wolff, M.; Schmitt, B. Treatment of Infantile Spasm Syndrome: Update from the Interdisciplinary Guideline Committee Coordinated by the German-Speaking Society of Neuropediatrics. Neuropediatrics 2022, 53, 389–401. [Google Scholar] [CrossRef]
- Kalser, J.; Giuliano, F.; Peralta, M.; Plecko, B.; Bölsterli, B.K. Infantile spasms without hypsarrhythmia and paroxysmal eye-head movements in an infant with a pyridoxine dependent epilepsy due to PLPBP/PLPHP deficiency. Neuropediatrics 2022. [Google Scholar] [CrossRef]
- Ambegaonkar, G.; Cholidis, N. Pyridoxine Dependent Epilepsy presenting as recurrent status epilepticus associated with febrile illness. Eur. J. Paediatr. Neurol. 2017, 21, e35. [Google Scholar] [CrossRef]
- Yoshii, A.; Takeoka, M.; Kelly, P.J.; Krishnamoorthy, K.S. Focal status epilepticus as atypical presentation of pyridoxine-dependent epilepsy. J. Child Neurol. 2005, 20, 696–698. [Google Scholar] [CrossRef] [PubMed]
- Mills, P.B.; Camuzeaux, S.S.; Footitt, E.J.; Mills, K.A.; Gissen, P.; Fisher, L.; Das, K.B.; Varadkar, S.M.; Zuberi, S.; McWilliam, R.; et al. Epilepsy due to PNPO mutations: Genotype, environment and treatment affect presentation and outcome. Brain 2014, 137, 1350–1360. [Google Scholar] [CrossRef]
- Plecko, B.; Paul, K.; Mills, P.; Clayton, P.; Paschke, E.; Maier, O.; Hasselmann, O.; Schmiedel, G.; Kanz, S.; Connolly, M.; et al. Pyridoxine responsiveness in novel mutations of the PNPO gene. Neurology 2014, 82, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, C.R., 2nd; Tseng, L.A.; Bok, L.A.; Hartmann, H.; Footitt, E.; Striano, P.; Tabarki, B.M.; Lunsing, R.J.; Stockler-Ipsiroglu, S.; Gordon, S.; et al. International PDE Consortium. Association Between Lysine Reduction Therapies and Cognitive Outcomes in Patients With Pyridoxine-Dependent Epilepsy. Neurology 2022, 99, e2627–e2636. [Google Scholar] [CrossRef] [PubMed]
- Jamali, A.; Kristensen, E.; Tangeraas, T.; Arntsen, V.; Sikiric, A.; Kupliauskiene, G.; Myren-Svelstad, S.; Berland, S.; Sejersted, Y.; Gerstner, T.; et al. The spectrum of pyridoxine dependent epilepsy across the age span: A nationwide retrospective observational study. Epilepsy Res. 2023, 190, 107099. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DISEASE (OMIM) | ESTIMATED PREVALENCE | GENE | CLINICAL FEATURES | BIOCHEMICAL MARKERS | NEUROIMAGING FEATURES | THERAPEUTIC OPTIONS |
|---|---|---|---|---|---|---|
| ALDH7A1 DEFICIENCY | 1:20,000–600,000 | ALDH7A1 MIM #266100 | Neonatal seizure (focal and/or generalized, status epilepticus) only few cases of later onset Other features: Abnormal foetal movements, signs of hypoxic ischaemic encephalopathy, dystonia, increased startle response, irritability, intellectual disability, respiratory distress, abdominal distension, bilious vomiting, hepatomegaly, hypothermia, shock, and acidosis | Urinary α-AASA, urinary or plasma α- AASA/P6C ratio; | Decreased mielinization, periventricular leukomalacia, atrophy, dysplasia, and other white matter abnormalities, gliosis, arachnoid and subependimal cysts, mega cisterna magna, hydrocephalus, ventriculomegaly, wide extracerebral CSF spaces, cerebral atrophy, grey matter heterotropia, cortical displasies, mesial temporal sclerosis, cortical malformation, cerebellar hypoplasia/ atrophy, cerebellar wide posterior fossa, chiari I, hemorragies, hematoma, hipoxic-ischemic like encephalophaty signs, thinning/hypoplasia/ dysplasia/ agenesia of the corpus callosum | Acute: PN (iv): 50–300 mg Maintenance:PN (os): 25–900 mg/day (1.3−70 mg/kg/day) Lysine restricted diet Arginine supplementation |
| PNPO DEFICIENCY | 1–9/1,000,000 | PNPO MIM #610090 | Neonatal seizures (focal and/or generalized, status epilepticus) only few cases of later onset Other features: Dystonia, prematurity-related disorders, signs of metabolic derangement (including metabolic acidosis, hyperlactacidaemia, hypoglycaemia), anaemia, microcephaly and gastrointestinal symptoms (abdominal distension and hepatomegaly, autism | Ventriculomegalia, white matter abnormalities, decreased mielinization, periventricular leukomalacia, gliosis, cerebral atrophy, cortical malformation, hipoxic-ischemic like encephalophaty | Acute: PLP: 50–200 mg; PN: 50–600 mg Maintenance: PLP: 10–72 mg/kg/day; PN: 5.5–50 mg/kg/day | |
| PLPBP DEFICIENCY | unknown | PLPBP MIM #617290 | Neonatal seizure (focal and/or generalized, status epilepticus) only few cases of later onset Other features: Movement disorders, microcephaly, global developmental delay, autism | White matter abnormalities, decreased mielinization, cystic leukencephalopaty, ventriculomegalia, cerebral atrophy, mesial temporal sclerosis, cortical malformation, thinning of the corpus callosum | Acute: PN: 50–200 mg Maintenance: PLP: 10–58 mg/kg/day PN: 4.7–24 mg/kg/day | |
| HYPERPROLINAEMIA TYPE II | unknown | ALDH4A1 MIM #239500 | Seizure onset after neonatal period (generalized) other features: behavioral disturbances, intellectual disability; Possible asymptomatic forms | Plasma proline, urinary Δ-1-pyrroline- 5-carboxylate, Δ-1-pyrroline- 5-carboxylate dehydrogenase activity in leukocytes and skin fibroblast; | / | Acute: PN (iv): 110 mg Maintenance: PN (os): 50–150 mg |
| HYPOPHOSPHATASIA | unknown | ALPL MIM #241500 | Neonatal seizure (generalized) Other features: Developmental delay, Dysmorphisms, Sheletal abnormalities including brachy-telephalangy | cerebral atrophy, ventriculomegalia, white matter abnormalities, white matter atrophy | Acute: PN (iv): 25–200 mg Maintenance: PN: 10–30 mg/kg/day (25–160 mg/day) | |
| GPI ANCHOR SYNTHESIS DEFECTS | unknown | PIGO NGS) MIM 614730 PIGV MIM # 610274 PIGA MIM # 311770 PIGQ MIM # 605754 PIGC MIM # 601730 PIGH MIM # 600154 PIGP MIM # 605938 PIGY MIM # 610662 PIG E MIM 610274 | Seizure onset after neonatal period (focal and/or generalized) other features: developmental delay intellectual disability | decreased mielinization, white matter abnormalities, wide extracerebral CSF spaces, cerebral atrophy, cerebellar atrophy, thinning and hipoplasia of the corpus callosum | Acute:PN (IV): 100 mg Maintenance: PN: 100–400 mg (20–30 mg/kg/day) |
| ALDH7A1 DEFICIENCY | PNPO DEFICIENCY | PLPBP DEFICIENCY | HYPERPROLINAEMIA TYPE II | HYPOPHOSPHATASIA | GPI ANCHOR SYNTHESIS DEFECTS | |
|---|---|---|---|---|---|---|
| Number of patients (M and F) | 344 (126 and 119) | 66 (30 and 9) | 46 (12 and 10) | 4 (2 and 2) | 18 (8 and 10) | 19 (13 and 6) |
| Death during the follow-up | 5 | 5 | 7 | - | 6 | 4 |
| Mean age at the onset of seizures (days) | 63.13 | 53.92 | 22.67 | 450 | 14.35 | 96.87 |
| Patients with onset before the age of 1 month | 222 | 55 | 39 | - | 16 | 5 |
| Patients with onset after the age of 1 month | 62 | 10 | 5 | 3 | 1 | 11 |
| Patients with generalized motor seizures at onset | 92 | 20 | 20 | 1 | 10 | 5 |
| Patients with focal motor seizures at onset | 77 | 12 | 11 | - | 1 | 7 |
| Patients with epileptic spasms at the onset | 20 | 11 | 6 | - | - | 1 |
| Patients with febrile seizures at the onset | 27 | 5 | 2 | 1 | 1 | 2 |
| Patients with status epilepticus at the onset | 25 | 8 | 2 | 1 | - | 0 |
| Patients with generalized motor seizures during the follow-up | 15 | 8 | 4 | 1 | 4 | 8 |
| Patients with focal motor seizures during the follow-up | 10 | 7 | 1 | - | 1 | 3 |
| Patients with generalized non-motor seizures during the follow-up | - | - | - | 1 | 1 | 0 |
| Patients with focal non-motor seizures during the follow-up | - | - | - | - | - | 1 |
| Patients with epileptic spasms during the follow-up | 3 | 3 | - | - | - | 7 |
| Patients with febrile seizures during the follow-up | 11 | 2 | 7 | - | 1 | 0 |
| Patients with status epilepticus during the follow-up | 13 | 5 | 2 | - | 1 | 5 |
| Patients with global developmental delay | 103 | 19 | 19 | 1 | 1 | 9 |
| Patients with selective language disorder | 6 | 3 | 3 | - | 0 | |
| Patients with IQ higher than 85 | 64 | 19 | 10 | 1 | - | 0 |
| Patients with borderline IQ (71–84) | 13 | - | 1 | 3 | - | 0 |
| Patients with IQ below 70 | 35 | 9 | 5 | 0 | - | 0 |
| Patients with autism spectrum disorders | 2 | 2 | 0 | - | - | |
| Patients with suppression burst pattern at EEG | 21 | 18 | 27 | - | 4 | 2 |
| Patients with suppression hypsarrhythmia at EEG | 3 | 7 | 2 | - | - | 1 |
| Patients with paroxysmal abnormalities at EEG | 25 | 14 | 14 | - | 3 | 9 |
| Patients with non-paroxysmal abnormalities at EEG | 11 | 9 | 19 | - | 2 | 4 |
| Patients with no EEG abnormalities at the initial EEG | 14 | 1 | 1 | - | 2 | 0 |
| Patients with no EEG abnormalities during the follow up | 41 | 15 | 6 | 0 | 1 | 0 |
| Patients with MRI available | 151 | 30 | 34 | 0 | 9 | 17 |
| Patients with epileptogenic lesions at MRI | 27 | 4 | 8 | 0 | 0 | 0 |
| Patients responsive to pyridoxine | 122 | 23 | 20 | 2 | 4 | 2 |
| Patients responsive to PLP | 1 | 2 | 2 | - | - | - |
| Patients treated with lysine restricted diet | 20 | - | - | - | - | - |
| Patients treated with triple therapy | 12 | - | - | - | - | - |
| Patients treated with other antiseizure medications | 117 | 31 | 33 | 3 | 15 | 14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mastrangelo, M.; Gasparri, V.; Bernardi, K.; Foglietta, S.; Ramantani, G.; Pisani, F. Epilepsy Phenotypes of Vitamin B6-Dependent Diseases: An Updated Systematic Review. Children 2023, 10, 553. https://doi.org/10.3390/children10030553
Mastrangelo M, Gasparri V, Bernardi K, Foglietta S, Ramantani G, Pisani F. Epilepsy Phenotypes of Vitamin B6-Dependent Diseases: An Updated Systematic Review. Children. 2023; 10(3):553. https://doi.org/10.3390/children10030553
Chicago/Turabian StyleMastrangelo, Mario, Valentina Gasparri, Katerina Bernardi, Silvia Foglietta, Georgia Ramantani, and Francesco Pisani. 2023. "Epilepsy Phenotypes of Vitamin B6-Dependent Diseases: An Updated Systematic Review" Children 10, no. 3: 553. https://doi.org/10.3390/children10030553
APA StyleMastrangelo, M., Gasparri, V., Bernardi, K., Foglietta, S., Ramantani, G., & Pisani, F. (2023). Epilepsy Phenotypes of Vitamin B6-Dependent Diseases: An Updated Systematic Review. Children, 10(3), 553. https://doi.org/10.3390/children10030553