
Oxygen for the Newborn: Friend or Foe?

 , , ,
, , ,  , ,
, , 
Abstract
:1. Introduction
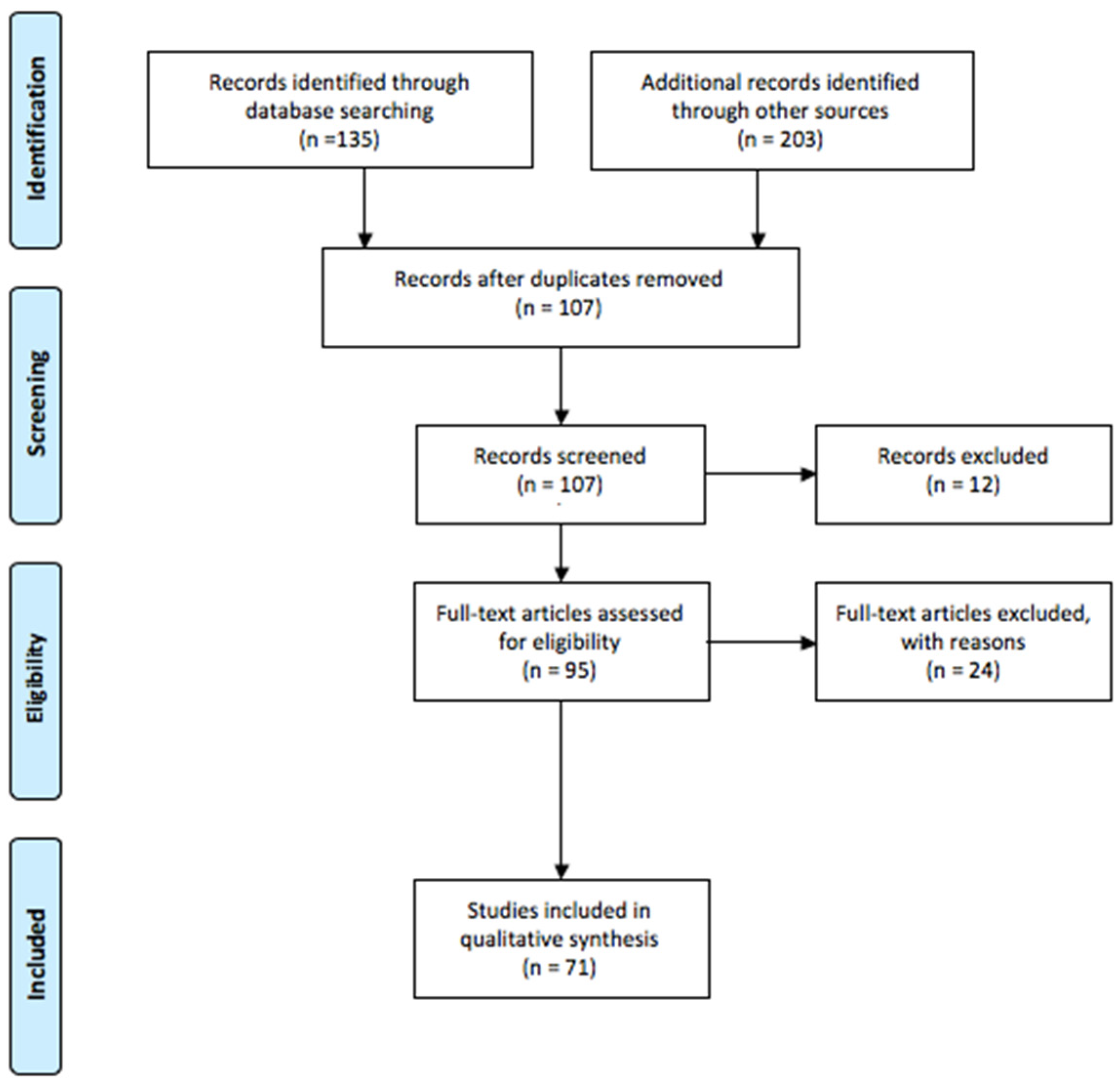
2. Methods
2.1. Research Strategy
2.2. Study Selection
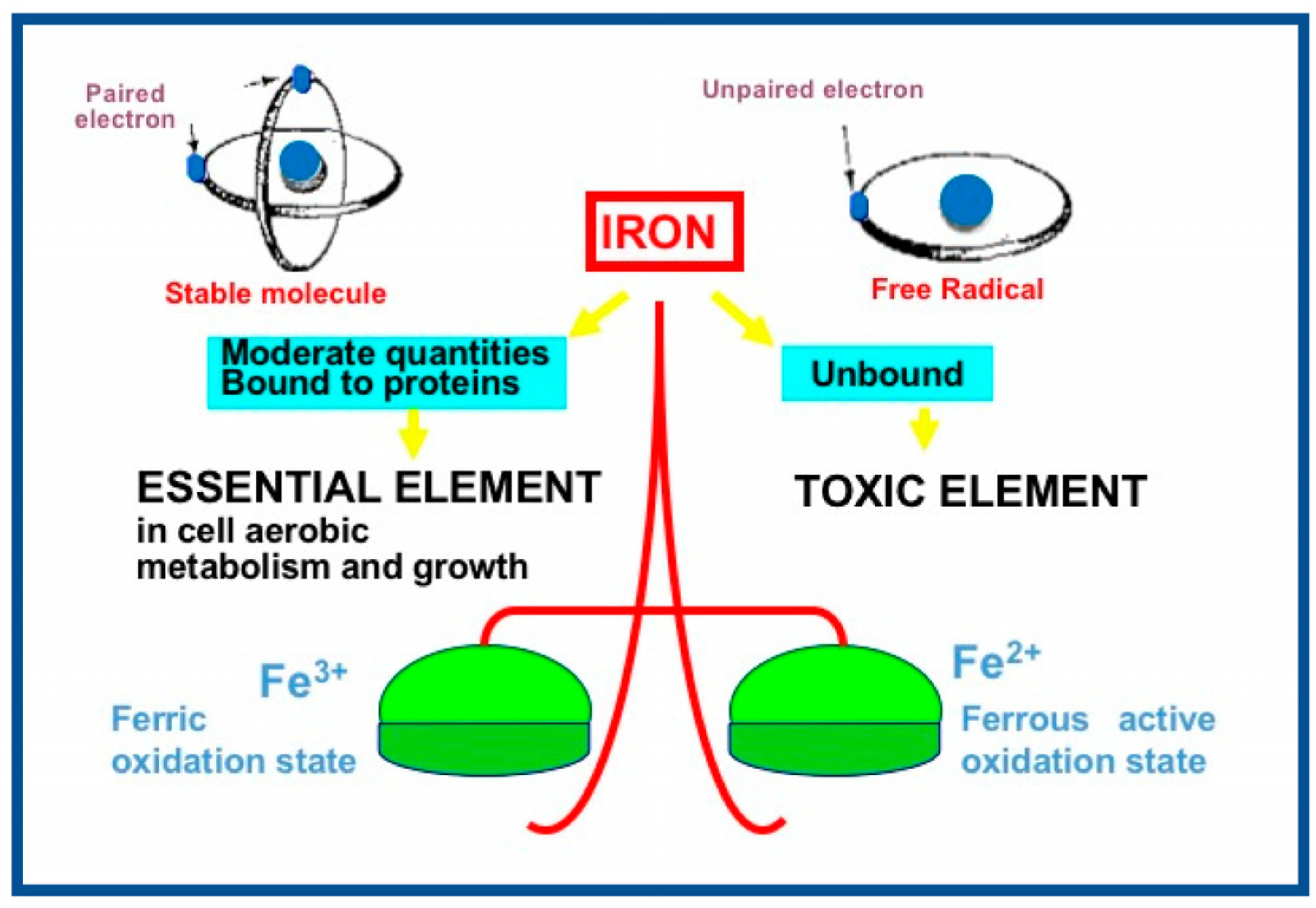
3. Reactive Oxygen and Nitrogen Species
Functions of ROS
4. Antioxidants
4.1. Enzymatic Antioxidants
4.2. Non-Enzymatic Antioxidants
5. Newborn Susceptibility to Oxidants
6. Free Radical Disease in Newborn
6.1. Retinopathy of Prematurity
6.2. Periventricular Leukomalacia
6.3. Bronchopulmonary Dysplasia
6.4. Necrotizing Enterocolitis
6.5. Patent Ductus Arteriosus
7. Discussion
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maltepe, E.; Saugstad, O.D. Oxygen in Health and Disease: Regulation of Oxygen Homeostasis--Clinical Implications. Pediatr. Res. 2009, 65, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Jia, Z.; Trush, M.A. Defining ROS in Biology and Medicine. React. Oxyg. Species Apex NC 2016, 1, 9–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrone, S.; Santacroce, A.; Longini, M.; Proietti, F.; Bazzini, F.; Buonocore, G. The Free Radical Diseases of Prematurity: From Cellular Mechanisms to Bedside. Oxid. Med. Cell. Longev. 2018, 2018, 7483062. [Google Scholar] [CrossRef] [Green Version]
- Weidinger, A.; Kozlov, A.V. Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules 2015, 5, 472–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliwell, B. Reactive Oxygen Species in Living Systems: Source, Biochemistry, and Role in Human Disease. Am. J. Med. 1991, 91, S14–S22. [Google Scholar] [CrossRef] [PubMed]
- Perrone, S.; Laschi, E.; Buonocore, G. Oxidative stress biomarkers in the perinatal period: Diagnostic and prognostic value. Semin. Fetal Neonatal Med. 2020, 25, 101087. [Google Scholar] [CrossRef]
- Studer, A.; Curran, D.P. Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem. Int. Ed. Engl. 2016, 55, 58–102. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Oxidants and Human Disease: Some New Concepts. FASEB J. 1987, 1, 358–364. [Google Scholar] [CrossRef]
- Ozcan, A.; Ogun, M. Biochemistry of Reactive Oxygen and Nitrogen Species. In Basic Principles and Clinical Significance of Oxidative Stress; Gowder, S.J.T., Ed.; InTech: London, UK, 2015; ISBN 978-953-51-2200-5. [Google Scholar]
- Ahmad, M.; Wolberg, A.; Kahwaji, C.I. Biochemistry, Electron Transport Chain. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial Electron Transport Chain, ROS Generation and Uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Navarro, A.; Boveris, A. The Mitochondrial Energy Transduction System and the Aging Process. Am. J. Physiol. Cell Physiol. 2007, 292, C670–C686. [Google Scholar] [CrossRef]
- Illés, E.; Patra, S.G.; Marks, V.; Mizrahi, A.; Meyerstein, D. The FeII(Citrate) Fenton Reaction under Physiological Conditions. J. Inorg. Biochem. 2020, 206, 111018. [Google Scholar] [CrossRef]
- Goldstein, S.; Meyerstein, D.; Czapski, G. The Fenton Reagents. Free Radic. Biol. Med. 1993, 15, 435–445. [Google Scholar] [CrossRef]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron Homeostasis and Oxidative Stress: An Intimate Relationship. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef] [PubMed]
- Kehrer, J.P. The Haber-Weiss Reaction and Mechanisms of Toxicity. Toxicology 2000, 149, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Segal, A.W. How Neutrophils Kill Microbes. Annu. Rev. Immunol. 2005, 23, 197–223. [Google Scholar] [CrossRef] [Green Version]
- Demaurex, N.; Schrenzel, J.; Jaconi, M.E.; Lew, D.P.; Krause, K.H. Proton Channels, Plasma Membrane Potential, and Respiratory Burst in Human Neutrophils. Eur. J. Haematol. 1993, 51, 309–312. [Google Scholar] [CrossRef]
- Clark, R.A. Oxidative Inactivation of Pneumolysin by the Myeloperoxidase System and Stimulated Human Neutrophils. J. Immunol. Baltim. Md 1950 1986, 136, 4617–4622. [Google Scholar] [CrossRef]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric Oxide Synthases: Structure, Function and Inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef]
- Brown, O.I.; Bridge, K.I.; Kearney, M.T. Nicotinamide Adenine Dinucleotide Phosphate Oxidases in Glucose Homeostasis and Diabetes-Related Endothelial Cell Dysfunction. Cells 2021, 10, 2315. [Google Scholar] [CrossRef] [PubMed]
- Kovac, S.; Angelova, P.R.; Holmström, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 2015, 1850, 794–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, E.; Steenbergen, C. Mechanisms Underlying Acute Protection from Cardiac Ischemia-Reperfusion Injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef] [Green Version]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial Reactive Oxygen Species: A Double Edged Sword in Ischemia/Reperfusion vs. Preconditioning. Redox Biol. 2014, 2, 702–714. [Google Scholar] [CrossRef] [Green Version]
- Chang, E.; Hornick, K.; Fritz, K.I.; Mishra, O.P.; Delivoria-Papadopoulos, M. Effect of Hyperoxia on Cortical Neuronal Nuclear Function and Programmed Cell Death Mechanisms. Neurochem. Res. 2007, 32, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.-Y.; Sharkis, S.J. A Low Level of Reactive Oxygen Species Selects for Primitive Hematopoietic Stem Cells That May Reside in the Low-Oxygenic Niche. Blood 2007, 110, 3056–3063. [Google Scholar] [CrossRef] [Green Version]
- Le Belle, J.E.; Orozco, N.M.; Paucar, A.A.; Saxe, J.P.; Mottahedeh, J.; Pyle, A.D.; Wu, H.; Kornblum, H.I. Proliferative Neural Stem Cells Have High Endogenous ROS Levels That Regulate Self-Renewal and Neurogenesis in a PI3K/Akt-Dependant Manner. Cell Stem Cell 2011, 8, 59–71. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Roy, J.; Galano, J.-M.; Durand, T.; Le Guennec, J.-Y.; Lee, J.C.-Y. Physiological Role of Reactive Oxygen Species as Promoters of Natural Defenses. FASEB J. 2017, 31, 3729–3745. [Google Scholar] [CrossRef] [Green Version]
- Ishimoto, Y.; Tanaka, T.; Yoshida, Y.; Inagi, R. Physiological and Pathophysiological Role of Reactive Oxygen Species and Reactive Nitrogen Species in the Kidney. Clin. Exp. Pharmacol. Physiol. 2018, 45, 1097–1105. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Su, Y.; Wang, J. ROS-Mediated Platelet Generation: A Microenvironment-Dependent Manner for Megakaryocyte Proliferation, Differentiation, and Maturation. Cell Death Dis. 2013, 4, e722. [Google Scholar] [CrossRef] [Green Version]
- Matés, J.M.; Pérez-Gómez, C.; Núñez de Castro, I. Antioxidant Enzymes and Human Diseases. Clin. Biochem. 1999, 32, 595–603. [Google Scholar] [CrossRef]
- Li, S.; Yan, T.; Yang, J.Q.; Oberley, T.D.; Oberley, L.W. The Role of Cellular Glutathione Peroxidase Redox Regulation in the Suppression of Tumor Cell Growth by Manganese Superoxide Dismutase. Cancer Res. 2000, 60, 3927–3939. [Google Scholar] [PubMed]
- Dheen, S.T.; Tay, S.S.W.; Boran, J.; Ting, L.W.; Kumar, S.D.; Fu, J.; Ling, E.-A. Recent Studies on Neural Tube Defects in Embryos of Diabetic Pregnancy: An Overview. Curr. Med. Chem. 2009, 16, 2345–2354. [Google Scholar] [CrossRef]
- Buonocore, G.; Groenendaal, F. Anti-Oxidant Strategies. Semin. Fetal. Neonatal Med. 2007, 12, 287–295. [Google Scholar] [CrossRef]
- Silvers, K.M.; Gibson, A.T.; Powers, H.J. High plasma vitamin C concentrations at birth associated with low antioxidant status and poor outcome in premature infants. Arch. Dis. Child. Fetal Neonatal Ed. 1994, 71, F40–F44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, T.M.; Polidori, M.C.; Dabbagh, A.; Evans, P.J.; Halliwell, B.; Morrow, J.D.; Roberts, L.J.; Frei, B. Antioxidant activity of vitamin C in iron-overloaded human plasma. J. Biol. Chem. 1997, 272, 15656–15660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEvoy, C.T.; Schilling, D.; Clay, N.; Jackson, K.; Go, M.D.; Spitale, P.; Bunten, C.; Leiva, M.; Gonzales, D.; Hollister-Smith, J.; et al. Vitamin C supplementation for pregnant smoking women and pulmonary function in their newborn infants: A randomized clinical trial. JAMA 2014, 311, 2074–2082. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C. Chemistry and biology of vitamin E. Mol. Nutr. Food Res. 2005, 49, 7–30. [Google Scholar] [CrossRef]
- Kontush, A.; Finckh, B.; Karten, B.; Kohlschütter, A.; Beisiegel, U. Antioxidant and prooxidant activity of alpha-tocopherol in human plasma and low density lipoprotein. J. Lipid Res. 1996, 37, 1436–1448. [Google Scholar] [CrossRef]
- Bowry, V.W.; Ingold, K.U.; Stocker, R. Vitamin E in human low-density lipoprotein. When and how this antioxidant becomes a pro-oxidant. Biochem. J. 1992, 288 Pt 2, 341–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazawa, T.; Burdeos, G.C.; Itaya, M.; Nakagawa, K.; Miyazawa, T. Vitamin E: Regulatory Redox Interactions. IUBMB Life 2019, 71, 430–441. [Google Scholar] [CrossRef]
- Millán, I.; Piñero-Ramos, J.D.; Lara, I.; Parra-Llorca, A.; Torres-Cuevas, I.; Vento, M. Oxidative Stress in the Newborn Period: Useful Biomarkers in the Clinical Setting. Antioxidants 2018, 7, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitridge, R.; Thompson, M. (Eds.) Mechanisms of Vascular Disease: A Reference Book for Vascular Specialists; University of Adelaide Press: Adelaide, AU, Australia, 2011; ISBN 978-0-9871718-2-5. [Google Scholar]
- Saugstad, O.D. Hypoxanthine as a Measurement of Hypoxia. Pediatr. Res. 1975, 9, 158–161. [Google Scholar] [CrossRef] [Green Version]
- Saugstad, O.D.; Aasen, A.O. Plasma Hypoxanthine Concentrations in Pigs. A Prognostic Aid in Hypoxia. Eur. Surg. Res. Eur. Chir. Forsch. Rech. Chir. Eur. 1980, 12, 123–129. [Google Scholar] [CrossRef]
- Qanungo, S.; Mukherjea, M. Ontogenic Profile of Some Antioxidants and Lipid Peroxidation in Human Placental and Fetal Tissues. Mol. Cell. Biochem. 2000, 215, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Matyas, M.; Zaharie, G. Antioxidants at Newborns. In Antioxidants; Shalaby, E., Ed.; IntechOpen: London, UK, 2019; ISBN 978-1-78923-919-5. [Google Scholar]
- ìGveric-Ahmetasevic, S.; Sunjic, S.B.; Skala, H.; Andrisic, L.; Stroser, M.; Zarkovic, K.; Skrablin, S.; Tatzber, F.; Cipak, A.; Jaganjac, M.; et al. Oxidative Stress in Small-for-Gestational Age (SGA) Term Newborns and Their Mothers. Free Radic. Res. 2009, 43, 376–384. [Google Scholar] [CrossRef]
- Baydas, G.; Karatas, F.; Gursu, M.F.; Bozkurt, H.A.; Ilhan, N.; Yasar, A.; Canatan, H. Antioxidant Vitamin Levels in Term and Preterm Infants and Their Relation to Maternal Vitamin Status. Arch. Med. Res. 2002, 33, 276–280. [Google Scholar] [CrossRef]
- Conti, M.G.; Angelidou, A.; Diray-Arce, J.; Smolen, K.K.; Lasky-Su, J.; De Curtis, M.; Levy, O. Immunometabolic Approaches to Prevent, Detect, and Treat Neonatal Sepsis. Pediatr. Res. 2020, 87, 399–405. [Google Scholar] [CrossRef]
- Saugstad, O.D. Oxidative Stress in the Newborn--a 30-Year Perspective. Biol. Neonate 2005, 88, 228–236. [Google Scholar] [CrossRef]
- Saugstad, O.D. The Oxygen Radical Disease in Neonatology. Indian J. Pediatr. 1989, 56, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Pierce, E.A.; Foley, E.D.; Smith, L.E. Regulation of Vascular Endothelial Growth Factor by Oxygen in a Model of Retinopathy of Prematurity. Arch. Ophthalmol. Chic. Ill 1960 1996, 114, 1219–1228. [Google Scholar] [CrossRef]
- Carnesecchi, S.; Deffert, C.; Pagano, A.; Garrido-Urbani, S.; Métrailler-Ruchonnet, I.; Schäppi, M.; Donati, Y.; Matthay, M.A.; Krause, K.-H.; Barazzone Argiroffo, C. NADPH Oxidase-1 Plays a Crucial Role in Hyperoxia-Induced Acute Lung Injury in Mice. Am. J. Respir. Crit. Care Med. 2009, 180, 972–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graziosi, A.; Perrotta, M.; Russo, D.; Gasparroni, G.; D’Egidio, C.; Marinelli, B.; Di Marzio, G.; Falconio, G.; Mastropasqua, L.; Li Volti, G.; et al. Oxidative Stress Markers and the Retinopathy of Prematurity. J. Clin. Med. 2020, 9, 2711. [Google Scholar] [CrossRef]
- Volpe, J.J. Neurobiology of Periventricular Leukomalacia in the Premature Infant. Pediatr. Res. 2001, 50, 553–562. [Google Scholar] [CrossRef] [Green Version]
- Coviello, C.; Perrone, S.; Buonocore, G.; Negro, S.; Longini, M.; Dani, C.; de Vries, L.S.; Groenendaal, F.; Vijlbrief, D.C.; Benders, M.J.N.L.; et al. Isoprostanes as Biomarker for White Matter Injury in Extremely Preterm Infants. Front. Pediatr. 2021, 8, 618622. [Google Scholar] [CrossRef]
- Saugstad, O.D. Bronchopulmonary Dysplasia-Oxidative Stress and Antioxidants. Semin. Neonatol. 2003, 8, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Manti, S.; Galdo, F.; Parisi, G.F.; Napolitano, M.; Decimo, F.; Leonardi, S.; Miraglia Del Giudice, M. Long-term effects of bronchopulmonary dysplasia on lung function: A pilot study in preschool children’s cohort. J. Asthma 2021, 58, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Hartnett, M.E. Pathophysiology and Mechanisms of Severe Retinopathy of Prematurity. Ophthalmology 2015, 122, 200–210. [Google Scholar] [CrossRef] [Green Version]
- Couroucli, X.I. Oxidative Stress in the Retina: Implications for Retinopathy of Prematurity. Curr. Opin. Toxicol. 2018, 7, 102–109. [Google Scholar] [CrossRef]
- Sapieha, P.; Hamel, D.; Shao, Z.; Rivera, J.C.; Zaniolo, K.; Joyal, J.S.; Chemtob, S. Proliferative Retinopathies: Angiogenesis That Blinds. Int. J. Biochem. Cell Biol. 2010, 42, 5–12. [Google Scholar] [CrossRef]
- Rivera, J.C.; Dabouz, R.; Noueihed, B.; Omri, S.; Tahiri, H.; Chemtob, S. Ischemic Retinopathies: Oxidative Stress and Inflammation. Oxid. Med. Cell. Longev. 2017, 2017, 3940241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banjac, L.; Banjac, G.; Kotur-Stevuljević, J.; Spasojević-Kalimanovska, V.; Gojković, T.; Bogavac-Stanojević, N.; Jelić-Ivanović, Z.; Banjac, G. Pro-oxidants and antioxidants in retinopathy of prematurity. Acta Clin. Croat. 2018, 57, 458–463. [Google Scholar] [CrossRef] [Green Version]
- Folkerth, R.D. Periventricular Leukomalacia: Overview and Recent Findings. Pediatr. Dev. Pathol. 2006, 9, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Coviello, C.; Perrone, S.; Buonocore, G.; Negro, S.; Longini, M.; Groenendaal, F.; Vijlbrief, D.C.; Dani, C.; Benders, M.J.N.L.; Tataranno, M.L. Oxidative Stress Biomarkers and Early Brain Activity in Extremely Preterm Infants: A Prospective Cohort Study. Children 2022, 9, 1376. [Google Scholar] [CrossRef]
- Bancalari, E.; Jain, D. Bronchopulmonary Dysplasia: 50 Years after the Original Description. Neonatology 2019, 115, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Boscarino, G.; Conti, M.G.; De Luca, F.; Di Chiara, M.; Deli, G.; Bianchi, M.; Favata, P.; Cardilli, V.; Di Nardo, G.; Parisi, P.; et al. Intravenous Lipid Emulsions Affect Respiratory Outcome in Preterm Newborn: A Case-Control Study. Nutrients 2021, 13, 1243. [Google Scholar] [CrossRef]
- Haagsman, H.P. Interactions of Surfactant Protein A with Pathogens. Biochim. Biophys. Acta 1998, 1408, 264–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, A.; Fu, J.; Yang, H.; Zhu, Y.; Pan, Y.; Xu, S.; Xue, X. Hyperoxia Stimulates the Transdifferentiation of Type II Alveolar Epithelial Cells in Newborn Rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L861–L872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrone, S.; Laschi, E.; Grande, E.; Buonocore, G. Bronchopulmonary Dysplasia and Oxidative Stress in the Newborn. In Oxidative Stress in Lung Diseases; Chakraborti, S., Chakraborti, T., Das, S.K., Chattopadhyay, D., Eds.; Springer Nature Singapore Pte Ltd.: Singapore, 2019; Volume 16, pp. 309–324. ISBN 978-981-13-8412-7/978-981-13-8413-4. [Google Scholar] [CrossRef]
- Terrin, G.; Scipione, A.; De Curtis, M. Update in Pathogenesis and Prospective in Treatment of Necrotizing Enterocolitis. BioMed Res. Int. 2014, 2014, 543765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baregamian, N.; Song, J.; Papaconstantinou, J.; Hawkins, H.K.; Evers, B.M.; Chung, D.H. Intestinal Mitochondrial Apoptotic Signaling Is Activated during Oxidative Stress. Pediatr. Surg. Int. 2011, 27, 871–877. [Google Scholar] [CrossRef] [Green Version]
- Perrone, S.; Tataranno, M.L.; Negro, S.; Cornacchione, S.; Longini, M.; Proietti, F.; Soubasi, V.; Benders, M.J.; Van Bel, F.; Buonocore, G. May Oxidative Stress Biomarkers in Cord Blood Predict the Occurrence of Necrotizing Enterocolitis in Preterm Infants? J. Matern.-Fetal Neonatal Med. 2012, 25 (Suppl. 1), 128–131. [Google Scholar] [CrossRef] [PubMed]
- Erdeve, Ö.; Okulu, E.; Singh, Y.; Sindelar, R.; Oncel, M.Y.; Terrin, G.; Boscarino, G.; Bülbül, A.; Sallmon, H.; Atasay, B.; et al. An Update on Patent Ductus Arteriosus and What Is Coming Next. Turk. Arch. Pediatr. 2022, 57, 118–131. [Google Scholar] [CrossRef]
- Lembo, C.; Buonocore, G.; Perrone, S. Oxidative Stress in Preterm Newborns. Antioxidants 2021, 10, 1672. [Google Scholar] [CrossRef]
- Chen, J.-X.; O’Mara, P.W.; Poole, S.D.; Brown, N.; Ehinger, N.J.; Slaughter, J.C.; Paria, B.C.; Aschner, J.L.; Reese, J. Isoprostanes as Physiological Mediators of Transition to Newborn Life: Novel Mechanisms Regulating Patency of the Term and Preterm Ductus Arteriosus. Pediatr. Res. 2012, 72, 122–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamrick, S.E.G.; Hansmann, G. Patent Ductus Arteriosus of the Preterm Infant. Pediatrics 2010, 125, 1020–1030. [Google Scholar] [CrossRef] [Green Version]
- Terrin, G.; Di Chiara, M.; Boscarino, G.; Metrangolo, V.; Faccioli, F.; Onestà, E.; Giancotti, A.; Di Donato, V.; Cardilli, V.; De Curtis, M. Morbidity Associated with Patent Ductus Arteriosus in Preterm Newborns: A Retrospective Case-Control Study. Ital. J. Pediatr. 2021, 47, 9. [Google Scholar] [CrossRef]
- Longini, M.; Perrone, S.; Vezzosi, P.; Proietti, F.; Marzocchi, B.; Buonocore, G.; Fanos, V.; Antonucci, R.; Brunoldi, E. Isoprostane levels in urine of preterm newborns treated with ibuprofen for patent ductus arteriosus closure. Pediatr. Nephrol. 2011, 26, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.D. Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiac Care. Emergency Cardiac Care Committee and Subcommittees, American Heart Association. Part I. Introduction. JAMA 1992, 268, 2171–2183. [Google Scholar]
- Campbell, K. Intensive Oxygen Therapy as a Possible Cause of Retrolental Fibroplasia; a Clinical Approach. Med. J. Aust. 1951, 2, 48–50. [Google Scholar] [CrossRef]
- Usher, R.H. Clinical Investigation of the Respiratory Distress Syndrome of Prematurity. Interim Report. N. Y. State J. Med. 1961, 61, 1677–1696. [Google Scholar]
- STOP-ROP Multicenter Study Group. Supplemental Therapeutic Oxygen for Prethreshold Retinopathy of Prematurity (STOP-ROP), a Randomized, Controlled Trial. I: Primary Outcomes. Pediatrics 2000, 105, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Askie, L.M.; Henderson-Smart, D.J.; Irwig, L.; Simpson, J.M. Oxygen-Saturation Targets and Outcomes in Extremely Preterm Infants. N. Engl. J. Med. 2003, 349, 959–967. [Google Scholar] [CrossRef] [Green Version]
- Askie, L.M.; Brocklehurst, P.; Darlow, B.A.; Finer, N.; Schmidt, B.; Tarnow-Mordi, W. NeOProM Collaborative Group NeOProM: Neonatal Oxygenation Prospective Meta-Analysis Collaboration Study Protocol. BMC Pediatr. 2011, 11, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweet, D.G.; Carnielli, V.; Greisen, G.; Hallman, M.; Ozek, E.; Te Pas, A.; Plavka, R.; Roehr, C.C.; Saugstad, O.D.; Simeoni, U.; et al. European Consensus Guidelines on the Management of Respiratory Distress Syndrome—2019 Update. Neonatology 2019, 115, 432–450. [Google Scholar] [CrossRef] [Green Version]
- Maternal, W.H.O.; Motherhood, N.H. Basic Newborn Resuscitation: A Practical Guide; WHO/RHT/MSM/98.1; World Health Organization: Geneva, Switzerland, 1998.
- Kattwinkel, J.; Niermeyer, S.; Nadkarni, V.; Tibballs, J.; Phillips, B.; Zideman, D.; Van Reempts, P.; Osmond, M. Resuscitation of the Newly Born Infant: An Advisory Statement from the Pediatric Working Group of the International Liaison Committee on Resuscitation. Resuscitation 1999, 40, 71–88. [Google Scholar] [CrossRef]
- Niermeyer, S.; Kattwinkel, J.; Van Reempts, P.; Nadkarni, V.; Phillips, B.; Zideman, D.; Azzopardi, D.; Berg, R.; Boyle, D.; Boyle, R.; et al. International Guidelines for Neonatal Resuscitation: An Excerpt from the Guidelines 2000 for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care: International Consensus on Science. Contributors and Reviewers for the Neonatal Resuscitation Guidelines. Pediatrics 2000, 106, e29. [Google Scholar] [CrossRef] [Green Version]
- Saugstad, O.D.; Rootwelt, T.; Aalen, O. Resuscitation of Asphyxiated Newborn Infants with Room Air or Oxygen: An International Controlled Trial: The Resair 2 Study. Pediatrics 1998, 102, e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramji, S.; Rasaily, R.; Mishra, P.K.; Narang, A.; Jayam, S.; Kapoor, A.N.; Kambo, I.; Mathur, A.; Saxena, B.N. Resuscitation of Asphyxiated Newborns with Room Air or 100% Oxygen at Birth: A Multicentric Clinical Trial. Indian Pediatr. 2003, 40, 510–517. [Google Scholar] [PubMed]
- Vento, M.; Asensi, M.; Sastre, J.; García-Sala, F.; Viña, J. Six Years of Experience with the Use of Room Air for the Resuscitation of Asphyxiated Newly Born Term Infants. Biol. Neonate 2001, 79, 261–267. [Google Scholar] [CrossRef]
- Vento, M.; Asensi, M.; Sastre, J.; Lloret, A.; García-Sala, F.; Viña, J. Oxidative Stress in Asphyxiated Term Infants Resuscitated with 100% Oxygen. J. Pediatr. 2003, 142, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Saugstad, O.D.; Ramji, S.; Vento, M. Resuscitation of Depressed Newborn Infants with Ambient Air or Pure Oxygen: A Meta-Analysis. Biol. Neonate 2005, 87, 27–34. [Google Scholar] [CrossRef]
- Welsford, M.; Nishiyama, C.; Shortt, C.; Isayama, T.; Dawson, J.A.; Weiner, G.; Roehr, C.C.; Wyckoff, M.H.; Rabi, Y. International Liaison Committee on Resuscitation Neonatal Life Support Task Force Room Air for Initiating Term Newborn Resuscitation: A Systematic Review With Meta-Analysis. Pediatrics 2019, 143, e20181825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, K.; Lee, H.C.; Escobedo, M.B.; Hoover, A.V.; Kamath-Rayne, B.D.; Kapadia, V.S.; Magid, D.J.; Niermeyer, S.; Schmölzer, G.M.; Szyld, E.; et al. Part 5: Neonatal Resuscitation: 2020 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation 2020, 142, S524–S550. [Google Scholar] [CrossRef]
- Tiwari, S.; Tiwari, S.; Nangia, S.; Saili, A. Oxygen Saturation Profile in Healthy Term Neonates in the Immediate Post Natal Period. Int. J. Clin. Pediatr. 2013, 2, 19–23. [Google Scholar] [CrossRef] [Green Version]
- Rabi, Y.; Yee, W.; Chen, S.Y.; Singhal, N. Oxygen Saturation Trends Immediately after Birth. J. Pediatr. 2006, 148, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Pulvirenti, G.; Parisi, G.F.; Giallongo, A.; Papale, M.; Manti, S.; Savasta, S.; Licari, A.; Marseglia, G.L.; Leonardi, S. Lower Airway Microbiota. Front. Pediatr. 2019, 7, 393. [Google Scholar] [CrossRef] [Green Version]
- Perrone, S.; Giordano, M.; De Bernardo, G.; Lugani, P.; Sarnacchiaro, P.; Stazzoni, G.; Buonocore, G.; Esposito, S.; Tataranno, M.L. Management of oxygen saturation monitoring in preterm newborns in the NICU: The Italian picture. Ital. J. Pediatr. 2021, 47, 104. [Google Scholar] [CrossRef]
- Foglia, E.E.; Carper, B.; Gantz, M.; DeMauro, S.B.; Lakshminrusimha, S.; Walsh, M.; Schmidt, B.; Caplan, M.S.; Laptook, A.R.; Keszler, M.; et al. Association between Policy Changes for Oxygen Saturation Alarm Settings and Neonatal Morbidity and Mortality in Infants Born Very Preterm. J. Pediatr. 2019, 209, 17–22.e2. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Disease | Reference | Population | OS Biomarker | Results |
|---|---|---|---|---|
| ROP | Pierce 1996 [56] | Neonatal mice exposed to hyperoxia | VEGF | The expression of VEGF in the peripheral retina was down-regulated by hyperoxia in conjunction with the arrest of growth and the loss of some of the developing vasculature. |
| Banjac 2018 [51] | Preterm newborns | TOS, TAS, MDA and PON1 | TOS and MDA were significantly higher in infants with ROP as compared to infants without ROP | |
| WMI | Coviello 2021 [54] | Preterm newborns | Cord blood and plasma IPs | Cord blood IPs were not correlated with white matter injury score, whereas higher plasma IPs and lower gestational age predicted higher white matter injury score |
| Coviello 2022 [55] | Preterm newborns | IPs | Higher plasma IPs levels are associated with decreased functional brain activity. | |
| BPD | Carnesecchi 2009 [57] | Wild-type and NOX1- and NOX2-deficient mice | ROS | ROS production was reduced in lung from NOX1-deficient mice. |
| Hou 2015 [58] | Newborn rats | AEC I (aquaporin 5, T1α) and AEC II markers (SP-C, SP-B) | Authors found an increase in AEC II-to-AEC I transdifferentiation in a hyperoxia-induced, BPD-like model at both the tissue and cellular levels. | |
| NEC | Baregamian 2011 [59] | Rat and human fetal intestinal epithelial cells | ROS | OS leads to increased intracellular ROS production by mitochondria and activation of mitochondrial apoptotic signaling pathways in both human fetal and rat intestinal epithelial cells. |
| Perrone 2012 [60] | Preterm newborns | NPBI, AOPP and TH | AOPP, TH and NPBI cord blood levels were significantly higher in babies with NEC | |
| PDA | Longini 2010 [61] | Preterm newborns (<33 weeks of gestational age) | urinary IPs | Ibuprofen therapy for PDA closure reduces the risk of OS, inducing a decrease of urinary IPs. |
| Chen 2012 [62] | Newborn mouse | IPs | IPs levels are increased shortly after birth in response to increased oxygen tension and this may serve as a novel physiological signal to stimulate postnatal PDA closure. Authors found that IPs have both vasoconstrictive and vasodilatory effects. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perrone, S.; Manti, S.; Petrolini, C.; Dell’Orto, V.G.; Boscarino, G.; Ceccotti, C.; Bertini, M.; Buonocore, G.; Esposito, S.M.R.; Gitto, E. Oxygen for the Newborn: Friend or Foe? Children 2023, 10, 579. https://doi.org/10.3390/children10030579
Perrone S, Manti S, Petrolini C, Dell’Orto VG, Boscarino G, Ceccotti C, Bertini M, Buonocore G, Esposito SMR, Gitto E. Oxygen for the Newborn: Friend or Foe? Children. 2023; 10(3):579. https://doi.org/10.3390/children10030579
Chicago/Turabian StylePerrone, Serafina, Sara Manti, Chiara Petrolini, Valentina Giovanna Dell’Orto, Giovanni Boscarino, Chiara Ceccotti, Mattia Bertini, Giuseppe Buonocore, Susanna Maria Roberta Esposito, and Eloisa Gitto. 2023. "Oxygen for the Newborn: Friend or Foe?" Children 10, no. 3: 579. https://doi.org/10.3390/children10030579
APA StylePerrone, S., Manti, S., Petrolini, C., Dell’Orto, V. G., Boscarino, G., Ceccotti, C., Bertini, M., Buonocore, G., Esposito, S. M. R., & Gitto, E. (2023). Oxygen for the Newborn: Friend or Foe? Children, 10(3), 579. https://doi.org/10.3390/children10030579