
Microcephaly, Short Stature, Intellectual Disability, Speech Absence and Cataract Are Associated with Novel Bi-Allelic Missense Variant in RTTN Gene: A Seckel Syndrome Case Report

Abstract
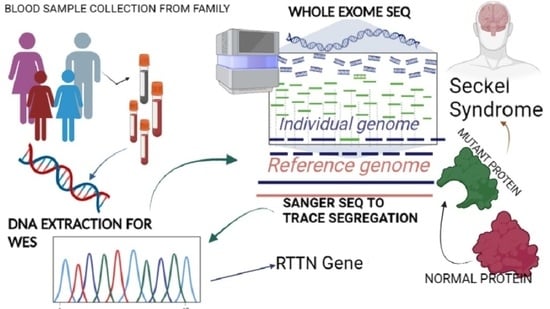
:
1. Introduction
2. Case Presentation
2.1. Clinical Diagnosis
2.2. Genetic Analysis
2.3. Functional Analysis
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MCPH | Microcephaly Primary Hereditary |
| ID | Intellectual Disability |
| DD | Developmental Delay |
| OPD | Outpatient Department |
| WES | Whole-Exome Sequencing |
References
- Pacheco, N.P.; Pettersson, M.; Lindstrand, A.; Grigelioniene, G. Expanding the phenotype of Seckel syndrome associated with biallelic loss-of-function variants in CEP63. Am. J. Med. Genet. Part A 2023. [Google Scholar] [CrossRef] [PubMed]
- Batool, T.; Irshad, S.; Riaz, M.; Baig, S.M.; Nuernberg, P.; Hussain, M.S. Recurrence mutation in RBBP8 gene causing non-syndromic autosomal recessive primary microcephaly; geometric simulation approach for insight into predicted computational models. J. Hum. Genet. 2023, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Majewski, F.; Ranke, M.; Schinzel, A.; Opitz, J.M. Studies of microcephalic primordial dwarfism II: The osteodysplastic type II of primordial dwarfism. Am. J. Med. Genet. 1982, 12, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Carreras-Castañer, X.; Dìaz-Cascajosa, J.; Morales-Ballùs, M.; Català-Mora, J. Bilateral retinal detachment in Hallermann-Streiff syndrome: Case report. J. Français d’Ophtalmologie 2022, 45, e123–e124. [Google Scholar] [CrossRef] [PubMed]
- Saeidi, M.; Shahbandari, M. A Child with Seckel Syndrome and Arterial Stenosis: Case Report and Literature Review. IMCRJ 2020, 13, 159–163. [Google Scholar] [CrossRef]
- TRAIP Promotes DNA Damage Response during Genome Replication and Is Mutated in Primordial Dwarfism | Nature Genetics. Available online: https://www.nature.com/articles/ng.3451 (accessed on 29 April 2023).
- Case of Seckel Syndrome in a 9-Month-Old Girl | Open Access Macedonian Journal of Medical Sciences (OAMJMS). January 2023. Available online: https://oamjms.eu/index.php/mjms/article/view/10988 (accessed on 29 April 2023).
- Khetarpal, P.; Das, S.; Panigrahi, I.; Munshi, A. Primordial dwarfism: Overview of clinical and genetic aspects. Mol. Genet. Genom. 2016, 291, 1–15. [Google Scholar] [CrossRef]
- Zaqout, S.; Kaindl, A.M. Autosomal Recessive Primary Microcephaly: Not Just a Small Brain. Front. Cell Dev. Biol. 2022, 9, 3635. Available online: https://www.frontiersin.org/articles/10.3389/fcell.2021.784700 (accessed on 29 April 2023). [CrossRef]
- Jean, F.; Stuart, A.; Tarailo-Graovac, M. Dissecting the Genetic and Etiological Causes of Primary Microcephaly. Front. Neurol. 2020, 11, 570830. [Google Scholar] [CrossRef]
- Asif, M.; Abdullah, U.; Nürnberg, P.; Tinschert, S.; Hussain, M.S. Congenital Microcephaly: A Debate on Diagnostic Challenges and Etiological Paradigm of the Shift from Isolated/Non-Syndromic to Syndromic Microcephaly. Cells 2023, 12, 642. [Google Scholar] [CrossRef]
- Nasser, H.; Vera, L.; Elmaleh-Bergès, M.; Steindl, K.; Letard, P.; Teissier, N.; Ernault, A.; Guimiot, F.; Afenjar, A.; Moutard, M.L.; et al. CDK5RAP2 primary microcephaly is associated with hypothalamic, retinal and cochlear developmental defects. J. Med. Genet. 2020, 57, 389–399. [Google Scholar] [CrossRef]
- Jacob, A.; Pasquier, J.; Carapito, R.; Auradé, F.; Molitor, A.; Froguel, P.; Fakhro, K.; Halabi, N.; Viot, G.; Bahram, S.; et al. A de novo synonymous variant in EFTUD2 disrupts normal splicing and causes mandibulofacial dysostosis with microcephaly: Case report. BMC Med. Genet. 2020, 21, 182. [Google Scholar] [CrossRef] [PubMed]
- Zaqout, S.; Mannaa, A.; Klein, O.; Krajewski, A.; Klose, J.; Luise-Becker, L.; Elsabagh, A.; Ferih, K.; Kraemer, N.; Ravindran, E.; et al. Proteome changes in autosomal recessive primary microcephaly. Ann. Hum. Genet. 2023, 87, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Siskos, N.; Stylianopoulou, E.; Skavdis, G.; Grigoriou, M.E. Molecular Genetics of Microcephaly Primary Hereditary: An Overview. Brain Sci. 2021, 11, 581. [Google Scholar] [CrossRef]
- Chou, E.-J.; Tang, T.K. Human Microcephaly Protein RTTN Is Required for Proper Mitotic Progression and Correct Spindle Position. Cells 2021, 10, 1441. [Google Scholar] [CrossRef] [PubMed]
- Grandone, A.; Torella, A.; Santoro, C.; Giugliano, T.; Del Vecchio Blanco, F.; Mutarelli, M.; Cirillo, G.; Piluso, G.; Capristo, C.; Festa, A.; et al. Expanding the phenotype of RTTN variations: A new family with primary microcephaly, severe growth failure, brain malformations and dermatitis. Clin. Genet. 2016, 90, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Zakaria, M.; Fatima, A.; Klar, J.; Wikström, J.; Abdullah, U.; Ali, Z.; Akram, T.; Tariq, M.; Ahmad, H.; Schuster, J.; et al. Primary microcephaly, primordial dwarfism, and brachydactyly in adult cases with biallelic skipping of RTTN exon 42. Hum. Mutat. 2019, 40, 899–903. [Google Scholar] [CrossRef]
- STIL Balancing Primary Microcephaly and Cancer | Cell Death & Disease. Available online: https://www.nature.com/articles/s41419-017-0101-9 (accessed on 23 April 2023).
- Sapir, T.; Sela-Donenfeld, D.; Karlinski, M.; Reiner, O. Brain Organization and Human Diseases. Cells 2022, 11, 1642. [Google Scholar] [CrossRef]
- Farcy, S.; Albert, A.; Gressens, P.; Baffet, A.D.; el Ghouzzi, V. Cortical Organoids to Model Microcephaly. Cells 2022, 11, 2135. [Google Scholar] [CrossRef]
- Zaidi, D.; Chinnappa, K.; Francis, F. Primary Cilia Influence Progenitor Function during Cortical Development. Cells 2022, 11, 2895. [Google Scholar] [CrossRef]
- Naseer, M.I.; Abdulkareem, A.A.; Muthaffar, O.Y.; Chaudhary, A.G. Exome sequencing reveled a compound heterozygous mutation in RTTN gene causing developmental delay and primary microcephaly. Saudi J. Biol. Sci. 2021, 28, 2824–2829. [Google Scholar] [CrossRef]
- Loss of CPAP in Developing Mouse Brain and Its Functional Implication for Human Primary Microcephaly | Journal of Cell Science | The Company of Biologists. Available online: https://journals.biologists.com/jcs/article/133/12/jcs243592/224078/Loss-of-CPAP-in-developing-mouse-brain-and-its (accessed on 28 April 2023).
- Sabbagh, Q.; Tharreau, M.; Cenni, C.; Sanchez, E.; Ruiz-Pallares, N.; Alkar, F.; Amouroux, C.; David, S.; Prodhomme, O.; Leboucq, N.; et al. Association of Meier-Gorlin and microcephalic osteodysplastic primordial dwarfism type II clinical features in an individual with CDK5RAP2 primary microcephaly. Eur. J. Med. Genet. 2023, 66, 104733. [Google Scholar] [CrossRef] [PubMed]
- Secondary Childhood Glaucoma—A Rare Association in Seckel Syndrome—Manju R Pillai, Srilekha Pallamparthy, Subathra Gnanavelu. 2023. Available online: https://journals.sagepub.com/doi/abs/10.1177/11206721211060949 (accessed on 30 April 2023).
- Krzyżanowska-Berkowska, P.; Szumny, D.; Młyńczak, T.; Kisza, K.; Oficjalska, J. Bilateral retinal detachment in Seckel syndrome. Can. J. Ophthalmol. 2014, 49, e130–e131. [Google Scholar] [CrossRef] [PubMed]
- Inaloo, S.; Khorshidi, S.; Jalli, R.; Haghbin, S. Seckel Syndrome and Vasculopathy: A Case Report. J. Pediatr. Neurol. 2016, 14, 122–125. [Google Scholar] [CrossRef]
- Llorens-Agost, M.; Luessing, J.; van Beneden, A.; Eykelenboom, J.; O’Reilly, D.; Bicknell, L.S.; Reynolds, J.J.; van Koegelenberg, M.; Hurles, M.E.; Brady, A.F.; et al. Analysis of novel missense ATR mutations reveals new splicing defects underlying Seckel syndrome. Hum. Mutat. 2018, 39, 1847–1853. [Google Scholar] [CrossRef] [Green Version]
- Halperin, D.; Agam, N.; Hallak, M.; Feinstein, M.; Drabkin, M.; Yogev, Y.; Wormser, O.; Shavit, E.; Gradstein, L.; Shelef, I.; et al. A syndrome of severe intellectual disability, hypotonia, failure to thrive, dysmorphism, and thinning of corpus callosum maps to chromosome 7q21.13-q21.3. Clin. Genet. 2022, 102, 123–129. [Google Scholar] [CrossRef]
- Heterogeneous Clinical Phenotypes and Cerebral Malformations Reflected by Rotatin Cellular Dynamics|Brain|Oxford Academic. Available online: https://academic.oup.com/brain/article/142/4/867/5382247 (accessed on 30 April 2023).
- Functional Characterization of Biallelic RTTN Variants Identified in an Infant with Microcephaly, Simplified Gyral Pattern, Pontocerebellar Hypoplasia, and Seizures|Pediatric Research. Available online: https://www.nature.com/articles/s41390-018-0083-z (accessed on 30 April 2023).
- Zollo, M.; Ahmed, M.; Ferrucci, V.; Salpietro, V.; Asadzadeh, F.; Carotenuto, M.; Maroofian, R.; Al-Amri, A.; Singh, R.; Scognamiglio, I.; et al. PRUNE is crucial for normal brain development and mutated in microcephaly with neurodevelopmental impairment. Brain 2017, 140, 940–952. [Google Scholar] [CrossRef] [Green Version]
- Gai, M.; Bianchi, F.T.; Vagnoni, C.; Vernì, F.; Bonaccorsi, S.; Pasquero, S.; Berto, G.E.; Sgrò, F.; Chiotto, A.M.A.; Annaratone, L.; et al. ASPM and CITK regulate spindle orientation by affecting the dynamics of astral microtubules. EMBO Rep. 2017, 18, 1870. [Google Scholar] [CrossRef] [Green Version]
- Duerinckx, S.; Abramowicz, M. The genetics of congenitally small brains. Semin. Cell Dev. Biol. 2018, 76, 76–85. [Google Scholar] [CrossRef]
- Watanabe, K.; Takao, D.; Ito, K.K.; Takahashi, M.; Kitagawa, D. The Cep57-pericentrin module organizes PCM expansion and centriole engagement. Nat. Commun. 2019, 10, 931. [Google Scholar] [CrossRef] [Green Version]
- Akalın, A.; Şimşek-Kiper, P.Ö.; Taşkıran, E.Z.; Karaosmanoğlu, B.; Utine, G.E.; Boduroğlu, K. A novel biallelic CRIPT variant in a patient with short stature, microcephaly, and distinctive facial features. Am. J. Med. Genet. Part A 2023, 191, 1119–1127. [Google Scholar] [CrossRef]
- Case Report: Compound Heterozygous NUP85 Variants Cause Autosomal Recessive Primary Microcephaly—PMC. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9947397/ (accessed on 28 May 2023).
- PPP1R35 Is a Novel Centrosomal Protein That Regulates Centriole Length in Concert with the Microcephaly Protein RTTN|eLife. Available online: https://elifesciences.org/articles/37846 (accessed on 30 April 2023).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Age | Gender | Onset Age | Primary Condition | Associated Conditions | Clinical Diagnosis |
|---|---|---|---|---|---|---|
| Proband V-2 | 10 years | F | Congenital | MCPH Short | ID, Seizures, Cataract | Seckel Syndrome Suspected |
| V-3 | 8 years | F | Congenital | MCPH Short | ID, Seizures, Cataract | Seckel Syndrome Suspected |
| Gene | Chromosome | Ref | Alt | Transcript (Exon) | Nucleotide Change | AA Change | Variation Type (Effect) |
|---|---|---|---|---|---|---|---|
| RTTN | 18 | C | A | NM_173630.4 (2) | c.57G > T | pGlu19Asp | Missense |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mudassir, B.U.; Agha, Z. Microcephaly, Short Stature, Intellectual Disability, Speech Absence and Cataract Are Associated with Novel Bi-Allelic Missense Variant in RTTN Gene: A Seckel Syndrome Case Report. Children 2023, 10, 1027. https://doi.org/10.3390/children10061027
Mudassir BU, Agha Z. Microcephaly, Short Stature, Intellectual Disability, Speech Absence and Cataract Are Associated with Novel Bi-Allelic Missense Variant in RTTN Gene: A Seckel Syndrome Case Report. Children. 2023; 10(6):1027. https://doi.org/10.3390/children10061027
Chicago/Turabian StyleMudassir, Behjat Ul, and Zehra Agha. 2023. "Microcephaly, Short Stature, Intellectual Disability, Speech Absence and Cataract Are Associated with Novel Bi-Allelic Missense Variant in RTTN Gene: A Seckel Syndrome Case Report" Children 10, no. 6: 1027. https://doi.org/10.3390/children10061027
APA StyleMudassir, B. U., & Agha, Z. (2023). Microcephaly, Short Stature, Intellectual Disability, Speech Absence and Cataract Are Associated with Novel Bi-Allelic Missense Variant in RTTN Gene: A Seckel Syndrome Case Report. Children, 10(6), 1027. https://doi.org/10.3390/children10061027