Neuro-Behavioral Phenotype in 16p11.2 Duplication: A Case Series



Abstract
:1. Introduction
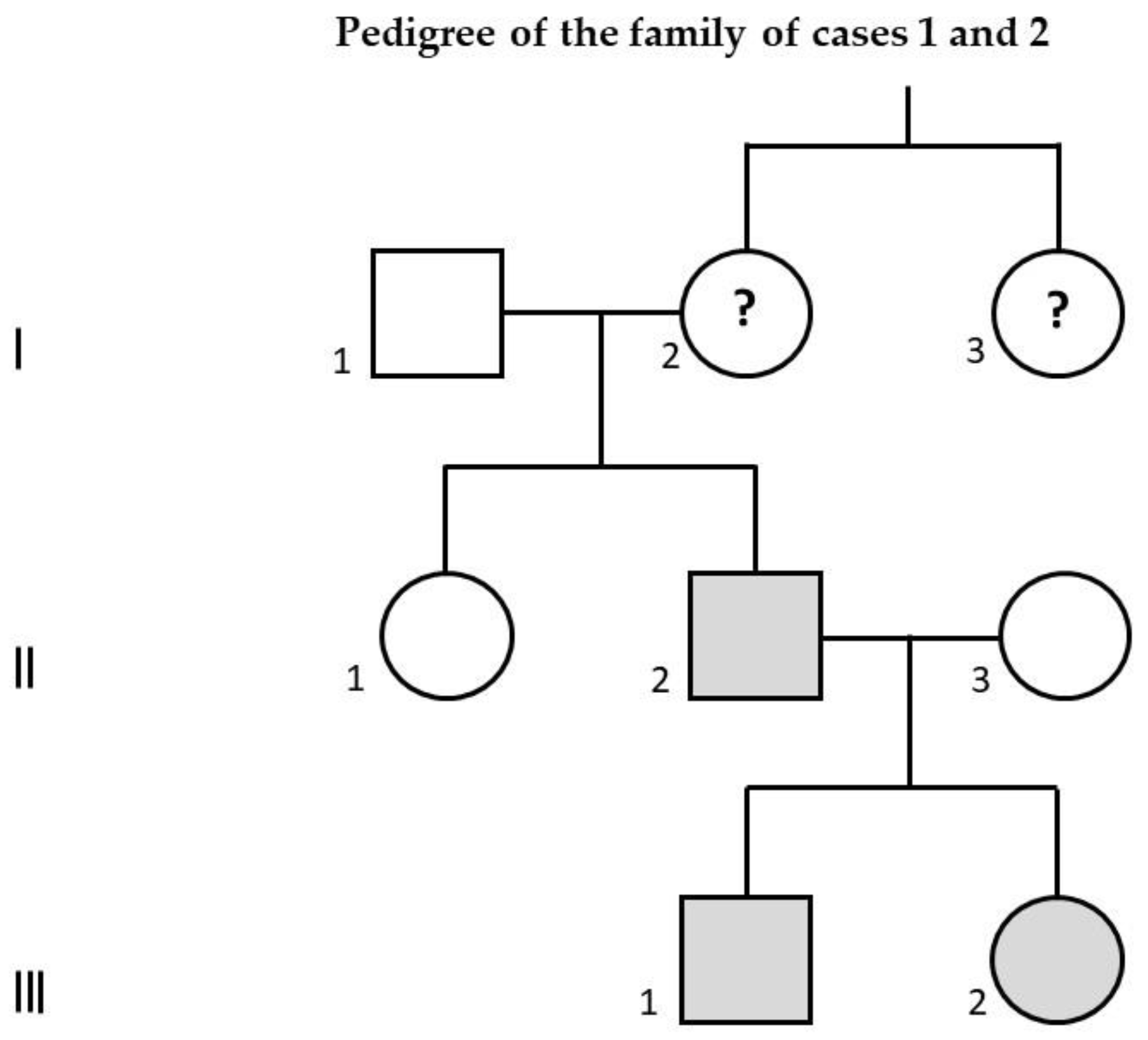
2. Case Presentation
3. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tucker, T.; Giroux, S.; Clément, V.; Langlois, S.; Friedman, J.M.; Rousseau, F. Prevalence of selected genomic deletions and duplications in a French-Canadian population-based sample of newborns. Mol. Genet. Genom. Med. 2013, 1, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Ronemus, M.; Yamrom, B.; Lee, Y.H.; Leotta, A.; Kendall, J.; Marks, S.; Lakshmi, B.; Pai, D.; Ye, K.; et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 2011, 70, 886–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green Snyder, L.; D’Angelo, D.; Chen, Q.; Bernier, R.; Goin-Kochel, R.P.; Wallace, A.S.; Gerdts, J.; Kanne, S.; Berry, L.; Blaskey, L.; et al. Simons VIP consortium. Autism spectrum disorder, developmental and psychiatric features in 16p11.2 duplication. J. Autism Dev. Disord. 2016, 46, 2734–2748. [Google Scholar] [CrossRef] [PubMed]
- Steinman, K.J.; Spence, S.J.; Ramocki, M.B.; Proud, M.B.; Kessler, S.K.; Marco, E.J.; Green Snyder, L.; D’Angelo, D.; Chen, Q.; Chung, W.K.; et al. Simons VIP Consortium. 16p11.2 deletion and duplication: Characterizing neurologic phenotypes in a large clinically ascertained cohort. Am. J. Med. Genet. A 2016, 170, 2943–2955. [Google Scholar] [CrossRef] [PubMed]
- Giaroli, G.; Bass, N.; Strydom, A.; Rantell, K.; McQuillin, A. Does rare matter? Copy number variants at 16p11.2 and the risk of psychosis: A systematic review of literature and meta-analysis. Schizophr. Res. 2014, 159, 340–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angelo, D.; Lebon, S.; Chen, Q.; Martin-Brevet, S.; Snyder, L.G.; Hippolyte, L.; Hanson, E.; Maillard, A.M.; Faucett, W.A.; Macé, A.; et al. Cardiff University Experiences of Children with Copy Number Variants (ECHO) Study; 16p11.2 European Consortium; Simons Variation in Individuals Project (VIP) Consortium. Defining the effect of the 16p11.2 duplication on cognition, behavior, and medical comorbidities. JAMA Psychiatry 2016, 73, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Niarchou, M.; Chawner, S.J.R.A.; Doherty, J.L.; Maillard, A.M.; Jacquemont, S.; Chung, W.K.; Green-Snyder, L.; Bernier, R.A.; Goin-Kochel, R.P.; Hanson, E.; et al. Psychiatric disorders in children with 16p11.2 deletion and duplication. Transl. Psychiatry 2019, 9, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Publishing: Arlington, VA, USA, 2013. [Google Scholar]
- Lord, C.; Rutter, M.; DiLavore, P.C.; Risi, S.; Gotham, K.; Bishop, S. Autism Diagnostic Observation Schedule, 2nd ed.; (ADOS-2) Manual (Part I): Modules 1–4; Western Psychological Services: Torrance, CA, USA, 2012. [Google Scholar]
- Baldan, F.; Gnan, C.; Franzoni, A.; Ferino, L.; Allegri, L.; Passon, N.; Damante, G. Genomic deletion involving the IMMP2L gene in two cases of autism spectrum disorder. Cytogenet. Genome Res. 2018, 154, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Spielmann, M.; Mundlos, S. Looking beyond the genes: The role of non-coding variants in human disease. Hum. Mol. Genet. 2016, 25, R157–R165. [Google Scholar] [CrossRef] [PubMed]
- Posar, A.; Visconti, P. Autism in 2016: The need for answers. J. Pediatr. (Rio J.) 2017, 93, 111–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Cases: Sex and Age at last Observation | aCGH Features | Family Antecedents | Pre-, Peri-Neonatal Period | Psychomotor Development | Sleep | Neurological Examination | Movement Disorders | Dysmorphisms |
|---|---|---|---|---|---|---|---|---|
| 1. Male, 12 years 10 months | (average spatial resolution ~130 Kb): dup16p11.2 (size: 457 Kb; nucleotides involved: 29,673,954–30,131,105), inherited from the father. | Brother of case 2. Father, carrier of the same 16p11.2 duplication: schizophrenia. Father’s aunt: schizophrenia. | Cesarean section for breech presentation. | Language and motor delay. Age of walking: 17 months. First words: 12 months; first sentences: ~36 months. | Difficulty falling asleep, awakenings, nightmares, pavor nocturnus. | Clumsiness, toe-walking, severe language impairment, echolalia. HC: ~25th percentile. | Stereotypies with upper limbs. | Testicle retained. Weight: ~25th percentile. |
| 2. Female, 11 years | As case 1 (see cell above). | Sister of case 1 (see above). | Normal. | Normal. Age of walking: 12 months. First words: 13 months. | Restless sleep. | Normal. HC: ~50th percentile. | Absent. | Very protruding ears. Weight: ~50th percentile. |
| 3. Male, 12 years 10 months | (average spatial resolution ~100 Kb): dup16p11.2 (size: 590 Kb; nucleotides involved: 28,543–29,133), inherited from the healthy father. | Twin sister: intellectual disability. Mother: sleepwalking. | Twin pregnancy with threatened abortions; at 35–36 weeks of gestation cesarean section for breech presentation. | Language delay (prevailing in the production). Age of walking: 15 months. First words: 36 months. | At first reduced sleep times; later restless sleep and perhaps sleepwalking. | Very poor speech. HC: ~25th percentile. | Absent. | Absent. Weight: ~25th percentile. |
| 4. Male, 15 years 10 months | (average spatial resolution ~100 Kb): (1) dup16p11.2 (size: 525 Kb; nucleotides involved: 29,673,754–30,198,753), including KIF22, PRRT2, and ALDOA genes, inherited from the healthy mother. (2) del7q31.1 (size: 60 Kb), not involving genes, inherited from the healthy mother. | Neoplasms in paternal line. Intellectual disability and psychiatric disorders in two cousins of the mother. | Normal. | Language delay. Age of walking: 13–14 months. First words: 12 months; first sentences: after 36 months. | Difficulty sleeping. Nocturnal awakenings. | Right eye exophoria. HC: ~25th percentile. | Absent. | Retrognathia, thin nose, pectus excavatum, thin fingers, flat pronated feet, thin skin. Weight deficit: <3rd percentile. |
| 5. Male, 7 years 3 months | (average spatial resolution ~25 Kb): (1) dup16p11.2 (size: 59 Kb; nucleotides involved: 29,652,999–29,712,097), including SPN and QPRT genes, inherited from the healthy father. (2) del7q31.1 (size: 29 Kb) including intron 5 of IMMP2L gene, inherited from the healthy father. (3) del9p24.3 (size: 66 Kb), not involving genes, inherited from the healthy mother. | Personality disorder: maternal uncle. Alzheimer’s disease: paternal grandmother. | FIVET. Initial biovular twinning with spontaneous interruption of biovularity at the 3rd month of gestation. Hypovalid sucking. | Language delay. Age of walking: 15 months. First words: 12 months; first sentences: ~36 months. | Nocturnal awakenings in the first 2 years of life. | Echolalia. Unintelligible speech. HC: ~75th percentile. | Motor tics. Bruxism. Stereotypies (iterative hops). | Absent. Weight: ~25th percentile. |
| Cases: Sex and Age at last Observation | Intellectual Functioning | Neuropsychological Assessment | Psychiatric and Behavioral Assessment | Epilepsy and Other Paroxysmal Events | EEG Findings | Brain MRI Findings | Medical Comorbidity |
|---|---|---|---|---|---|---|---|
| 1. Male, 12 years 10 months | Moderate intellectual disability. Nonverbal IQ = 40. | Language: phonological alterations. Severe learning disorder in reading, writing, and mathematics. Good visual memory. | Deficits of social communication and social interaction; restricted and repetitive behaviors, interests or activities; sensory abnormalities: autism spectrum disorder (severity level 3: requiring very substantial support, according to DSM-5). Irritability (auto-aggressiveness). Hyperactivity. Attention deficit. ADOS-2: autism; CSS: 8. CBCL: above the clinical threshold for Attention Deficit/Hyperactivity Problems. | Since the age of 7 years, absence seizures; cluster of myoclonic seizures at 9 years and 8 months. No further seizures thereafter. Therapy: lamotrigine. | Multifocal (left parieto-temporal and right fronto-temporal) and diffuse paroxysmal abnormalities at 7 years 9 months. Afterwards, only slow activities prevailing on the right and after on the left hemisphere. | Dilated perivascular spaces in the left retrotrigonal white matter and at the junction between the anterior third and the middle third of the corpus callosum. | None. |
| 2. Female, 11 years | Normal. FSIQ = 99, ICV = 98, IRP = 106, IML = 97, IVE = 94. | Learning disorder in reading, writing, and mathematics. | Hyperactivity. Poor tolerance of frustration. ADOS-2: out of autism spectrum; CSS: 1. CBCL: below the clinical threshold in all areas. | None. | Not performed. | Not performed. | None. |
| 3. Male, 12 years 10 months | Moderate intellectual disability. Nonverbal IQ = 44. | Learning: severe impairment of reading, writing and, above all, mathematics. | Oppositional defiant disorder. Hyperactivity. Attention deficit. Disinhibition. Poor frustration tolerance. Hetero-aggressiveness. Depressive and anxious symptoms. Difficulty relating to peers. Therapy: risperidone. ADOS-2: out of autism spectrum; CSS: 2. CBCL: above the clinical threshold for Affective Problems, Anxiety Problems, Attention Deficit/Hyperactivity Problems, and Oppositional Defiant Problems. | At 7 years 9 months, one generalized tonic vibratory seizure. After, some prolonged paroxysmal events with falls, pain at lower limbs, inability to walk, and preserved consciousness (ictal EEG: normal): periodic paralysis? | At 5 years 8 months, right temporal paroxysmal abnormalities during wake and sleep. Afterwards, normal. | Mild dilatation of lateral ventricles. | Laryngospasm. Polyallergic individual. |
| 4. Male, 15 years 10 months | Mild intellectual disability. FSIQ = 68, ICV = 70, IRP = 69, IML = 79, IVE = 91. | Learning problems (reading, writing, and mathematics). Impairment of verbal memory. | Oppositional defiant behavior. Depression. Anxiety. Phobia for insects. Attention deficit. Problematic relationships with peers. Introversion. Impulsiveness. Hetero-aggressiveness. Poor tolerance of frustration. Persecutory ideas. Important dependence on adult reference figures. ADOS-2: out of autism spectrum; CSS: 2. CBCL: above the clinical threshold for Affective Problems, Anxiety Problems, and Oppositional Defiant Problems. | None. | Normal in wake and sleep. | Reduced thickness of the pituitary gland and dilatation of some perivascular spaces in the retrotrigonal area. | None. |
| 5. Male, 7 years 3 months | Mild intellectual disability. Nonverbal IQ = 70. | Language: phonological alterations. Unable to draw. Recognizes letters and numbers. Temporo-spatial disorientation. Deficit of fine motor skills. Visual memory above normal. | Deficits of social communication and social interaction; restricted and repetitive behaviors, interests or activities; sensory abnormalities: autism spectrum disorder (severity level 2: requiring substantial support, according to DSM-5). ADOS-2: autism; CSS: 6. CBCL: above the clinical threshold for Attention Deficit/Hyperactivity Problems. | None. | During wakefulness, slow spike-waves in the left posterior regions at 4 years 5 months; normal at 7 years 3 months. | Normal. | Frequent upper respiratory tract infections in the first 2 years of life. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Posar, A.; Visconti, P. Neuro-Behavioral Phenotype in 16p11.2 Duplication: A Case Series. Children 2020, 7, 190. https://doi.org/10.3390/children7100190
Posar A, Visconti P. Neuro-Behavioral Phenotype in 16p11.2 Duplication: A Case Series. Children. 2020; 7(10):190. https://doi.org/10.3390/children7100190
Chicago/Turabian StylePosar, Annio, and Paola Visconti. 2020. "Neuro-Behavioral Phenotype in 16p11.2 Duplication: A Case Series" Children 7, no. 10: 190. https://doi.org/10.3390/children7100190
APA StylePosar, A., & Visconti, P. (2020). Neuro-Behavioral Phenotype in 16p11.2 Duplication: A Case Series. Children, 7(10), 190. https://doi.org/10.3390/children7100190