New and Emerging Targeted Therapies for Pediatric Acute Myeloid Leukemia (AML)


Abstract
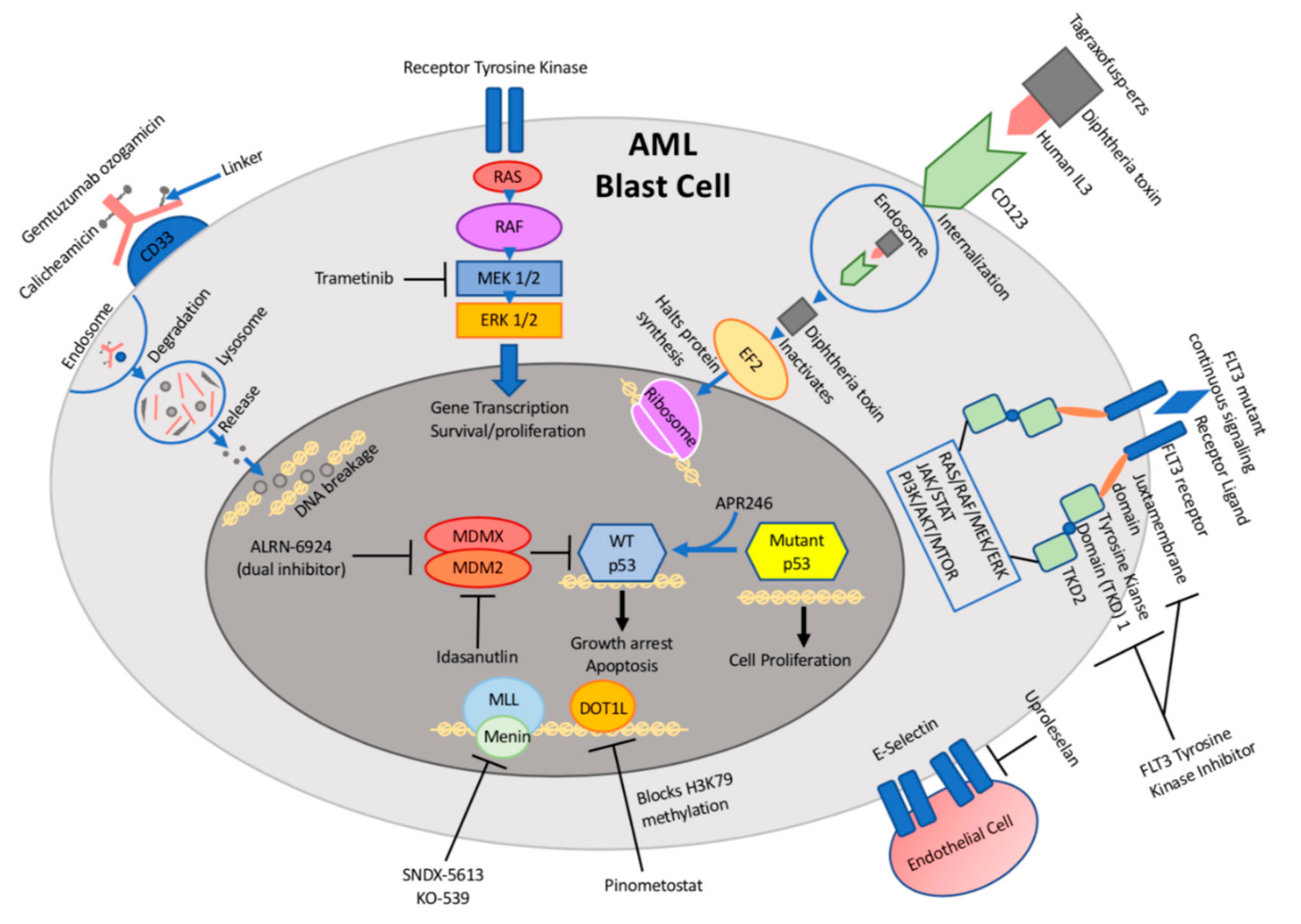
:1. Introduction
2. Antibody Drug Conjugates
2.1. Targeting CD33
2.2. Targeting Mesothelin
2.3. Targeting CD123
3. Small Molecule Inhibitors
3.1. E-Selectin Inhibitors
3.2. Targeting KMT2A-Fusion
3.3. MEK Inhibitors
3.4. MDM2 Antagonists
3.5. Targeting Mutant TP53
4. FLT3 Inhibitors
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gamis, A.S.; Alonzo, T.A.; Perentesis, J.P.; Meshinchi, S.; COG Acute Myeloid Leukemia Committee. Children’s oncology group’s 2013 blueprint for research: Acute myeloid leukemia. Pediatr. Cancer 2013, 60, 964–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamis, A.S.; Alonzo, T.A.; Meshinchi, S.; Sung, L.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Kahwash, S.B.; Heerema-McKenney, A.; Winter, L.; et al. Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: Results from the randomized phase III children’s oncology group trial AAML0531. J. Clin. Oncol. 2014, 32, 3021–3032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasche, M.; Zimmermann, M.; Borschel, L.; Bourquin, J.P.; Dworzak, M.; Klingebiel, T.; Lehrnbecher, T.; Creutzig, U.; Klusmann, J.H.; Reinhardt, D. Successes and challenges in the treatment of pediatric acute myeloid leukemia: A retrospective analysis of the AML-BFM trials from 1987 to 2012. Leukemia 2018, 32, 2167–2177. [Google Scholar] [CrossRef] [Green Version]
- Kuhlen, M.; Klusmann, J.H.; Hoell, J.I. Molecular approaches to treating pediatric leukemias. Front. Pediatr. 2019, 7, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolouri, H.; Farrar, J.E.; Triche, T., Jr.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.A.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018, 24, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohl, S.R.; Bullinger, L.; Rucker, F.G. New targeted agents in acute myeloid leukemia: New hope on the rise. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Walter, R.B. Investigational CD33-targeted therapeutics for acute myeloid leukemia. Expert Opin. Investig. Drugs 2018, 27, 339–348. [Google Scholar] [CrossRef]
- Raza, A.; Jurcic, J.G.; Roboz, G.J.; Maris, M.; Stephenson, J.J.; Wood, B.L.; Feldman, E.J.; Galili, N.; Grove, L.E.; Drachman, J.G.; et al. Complete remissions observed in acute myeloid leukemia following prolonged exposure to lintuzumab: A phase 1 trial. Leuk. Lymphoma 2009, 50, 1336–1344. [Google Scholar] [CrossRef]
- Feldman, E.J.; Brandwein, J.; Stone, R.; Kalaycio, M.; Moore, J.; O’Connor, J.; Wedel, N.; Roboz, G.J.; Miller, C.; Chopra, R.; et al. Phase iii randomized multicenter study of a humanized anti-CD33 monoclonal antibody, lintuzumab, in combination with chemotherapy, versus chemotherapy alone in patients with refractory or first-relapsed acute myeloid leukemia. J. Clin. Oncol. 2005, 23, 4110–4116. [Google Scholar] [CrossRef]
- Fathi, A.T.; Erba, H.P.; Lancet, J.E.; Stein, E.M.; Ravandi, F.; Faderl, S.; Walter, R.B.; Advani, A.S.; DeAngelo, D.J.; Kovacsovics, T.J.J.B. A phase 1 trial of vadastuximab talirine combined with hypomethylating agents in patients with CD33-positive AML. Blood 2018, 132, 1125–1133. [Google Scholar] [CrossRef]
- Stein, E.M.; Walter, R.B.; Erba, H.P.; Fathi, A.T.; Advani, A.S.; Lancet, J.E.; Ravandi, F.; Kovacsovics, T.; DeAngelo, D.J.; Bixby, D.J.B. A phase 1 trial of vadastuximab talirine as monotherapy in patients with CD33-positive acute myeloid leukemia. Blood 2018, 131, 387–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garfin, P.M.; Feldman, E.J. Antibody-based treatment of acute myeloid leukemia. Curr. Hematol. Malig. Rep. 2016, 11, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Castaigne, S.; Pautas, C.; Terre, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.N.; Legrand, O.; Thomas, X.; Turlure, P.; Reman, O.; et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet 2012, 379, 1508–1516. [Google Scholar] [CrossRef]
- Burnett, A.K.; Hills, R.K.; Milligan, D.; Kjeldsen, L.; Kell, J.; Russell, N.H.; Yin, J.A.; Hunter, A.; Goldstone, A.H.; Wheatley, K. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: Results of the MRC AML15 trial. J. Clin. Oncol. 2011, 29, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Cooper, T.M.; Franklin, J.; Gerbing, R.B.; Alonzo, T.A.; Hurwitz, C.; Raimondi, S.C.; Hirsch, B.; Smith, F.O.; Mathew, P.; Arceci, R.J.; et al. AAML03P1, a pilot study of the safety of gemtuzumab ozogamicin in combination with chemotherapy for newly diagnosed childhood acute myeloid leukemia: A report from the children’s oncology group. Cancer 2012, 118, 761–769. [Google Scholar] [CrossRef]
- Norsworthy, K.J.; Ko, C.W.; Lee, J.E.; Liu, J.; John, C.S.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. Fda approval summary: Mylotarg for treatment of patients with relapsed or refractory CD33-positive acute myeloid leukemia. Oncologist 2018, 23, 1103–1108. [Google Scholar] [CrossRef] [Green Version]
- Ghafoor, A.; Thomas, A.; Hassan, R. Targeting mesothelin in ovarian cancer. Oncotarget 2018, 9, 36050–36051. [Google Scholar] [CrossRef]
- Tarlock, K.; Kaeding, A.J.; Alonzo, T.A.; Loken, M.R.; Ries, R.E.; Pardo, L.; Gerbing, R.; Farrar, J.E.; Auvil, J.M.G.; Gerhard, D.S. Discovery and validation of cell-surface protein mesothelin (MSLN) as a novel therapeutic target in AML: Results from the COG/NCI target AML initiative. Blood 2016, 128, 2873. [Google Scholar] [CrossRef]
- Golfier, S.; Kopitz, C.; Kahnert, A.; Heisler, I.; Schatz, C.A.; Stelte-Ludwig, B.; Mayer-Bartschmid, A.; Unterschemmann, K.; Bruder, S.; Linden, L.; et al. Anetumab ravtansine: A novel mesothelin-targeting antibody-drug conjugate cures tumors with heterogeneous target expression favored by bystander effect. Mol. Cancer Ther. 2014, 13, 1537–1548. [Google Scholar] [CrossRef] [Green Version]
- Quanz, M.; Hagemann, U.B.; Zitzmann-Kolbe, S.; Stelte-Ludwig, B.; Golfier, S.; Elbi, C.; Mumberg, D.; Ziegelbauer, K.; Schatz, C.A. Anetumab ravtansine inhibits tumor growth and shows additive effect in combination with targeted agents and chemotherapy in mesothelin-expressing human ovarian cancer models. Oncotarget 2018, 9, 34103–34121. [Google Scholar] [CrossRef] [Green Version]
- Steinbach, D.; Onda, M.; Voigt, A.; Dawczynski, K.; Wittig, S.; Hassan, R.; Gruhn, B.; Pastan, I. Mesothelin, a possible target for immunotherapy, is expressed in primary aml cells. Eur. J. Haematol. 2007, 79, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Kaeding, A.; Tarlock, K.; Barwe, S.; Gopalakrisnapillai, A.; Alonzo, T.A.; Gerbing, R.B.; Loken, M.R.; Ries, R.E.; Pardo, L.; Chou, C. Mesothelin is a novel disease marker and potential therapeutic target in pediatric acute myeloid leukemia. Blood 2017, 130, 2461. [Google Scholar]
- Kaeding, A.; Tarlock, K.; Kolb, E.A.; Meshinchi, S. Immunotherapeutic targeting of mesothelin in acute myeloid leukemia in vitro with anetumab ravtansine and a novel antibody-drug conjugate. Blood 2018, 132, 1448. [Google Scholar] [CrossRef]
- Gopalakrishnapillai, A.; Kaeding, A.; Schatz, C.; Sommer, A.; Meshinchi, S.; Kolb, E.A.; Barwe, S. In vivo evaluation of mesothelin as a therapeutic target in pediatric acute myeloid leukemia. Blood 2019, 134, 1370. [Google Scholar] [CrossRef]
- Kovtun, Y.; Jones, G.E.; Adams, S.; Harvey, L.; Audette, C.A.; Wilhelm, A.; Bai, C.; Rui, L.; Laleau, R.; Liu, F.; et al. A CD123-targeting antibody-drug conjugate, IMGN632, designed to eradicate AML while sparing normal bone marrow cells. Blood Adv. 2018, 2, 848–858. [Google Scholar] [CrossRef] [Green Version]
- Konopleva, M.; Hogge, D.E.; Rizzieri, D.A.; Cirrito, T.P.; Kornblau, S.M.; Borthakur, G.; Bivins, C.; Garcia-Manero, G.; Kadia, T.M.; Ravandi, F. SL-401, a targeted therapy directed to the interleukin-3 receptor present on leukemia blasts and cancer stem cells, is active as a single agent in patients with advanced AML. Blood 2012, 21, 3625. [Google Scholar] [CrossRef]
- Jen, E.Y.; Gao, X.; Li, L.; Zhuang, L.; Simpson, N.E.; Aryal, B.; Wang, R.; Przepiorka, D.; Shen, Y.L.; Leong, R.; et al. FDA approval summary: Tagraxofusp-erzs for treatment of blastic plasmacytoid dendritic cell neoplasm. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef] [Green Version]
- Pemmaraju, N.; Sweet, K.L.; Lane, A.A.; Stein, A.S.; Vasu, S.; Blum, W.; Rizzieri, D.A.; Wang, E.S.; Duvic, M.; Aung, P. Results of pivotal phase 2 trial of SL-401 in patients with blastic plasmacytoid dendritic cell neoplasm (BPDCN). Blood 2018, 132, 765. [Google Scholar] [CrossRef]
- Daver, N.G.; Erba, H.P.; Papadantonakis, N.; DeAngelo, D.J.; Wang, E.S.; Konopleva, M.Y.; Sloss, C.M.; Culm-Merdek, K.; Zweidler-McKay, P.A.; Kantarjian, H.M. A phase I, first-in-human study evaluating the safety and preliminary antileukemia activity of IMGN632, a novel CD123-targeting antibody-drug conjugate, in patients with relapsed/refractory acute myeloid leukemia and other CD123-positive hematologic malignancies. Blood 2018, 132, 27. [Google Scholar]
- Daver, N.G.; Montesinos, P.; DeAngelo, D.J.; Wang, E.S.; Papadantonakis, N.; Deconinck, E.; Erba, H.P.; Pemmaraju, N.; Lane, A.A.; Rizzieri, D.A. Clinical Profile of IMGN632, a Novel CD123-Targeting Antibody-Drug Conjugate (ADC), in Patients with Relapsed/Refractory (r/r) Acute Myeloid Leukemia (AML) or Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN); American Society of Hematology: Washington, DC, USA, 2019. [Google Scholar]
- Daver, N.G.; Erba, H.P.; Papadantonakis, N.; DeAngelo, D.J.; Wang, E.S.; Konopleva, M.Y.; Sloss, C.M.; Wang, J.; Malcolm, K.E.; Zweidler-McKay, P.A. A phase 1b/2 study of the CD123-targeting antibody-drug conjugate imgn632 as monotherapy or in combination with venetoclax and/or azacitidine for patients with CD123-positive acute myeloid leukemia. Blood 2018, 134, 2601. [Google Scholar] [CrossRef]
- Al-Hussaini, M.; DiPersio, J.F. Small molecule inhibitors in acute myeloid leukemia: From the bench to the clinic. Expert Rev. Hematol. 2014, 7, 439–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashidi, A.; DiPersio, J.F. Targeting the leukemia-stroma interaction in acute myeloid leukemia: Rationale and latest evidence. Ther. Adv. Hematol. 2016, 7, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Winkler, I.G.; Barbier, V.; Pattabiraman, D.R.; Gonda, T.J.; Magnani, J.L.; Levesque, J.-P. Vascular niche e-selectin protects acute myeloid leukaemia stem cells from chemotherapy. Blood 2014, 124, 620. [Google Scholar] [CrossRef]
- Chien, S.; Haq, S.U.; Pawlus, M.; Moon, R.T.; Estey, E.H.; Appelbaum, F.R.; Othus, M.; Magnani, J.L. Adhesion of acute myeloid leukemia blasts to e-selectin in the vascular niche enhances their survival by mechanisms such as wnt activation. Blood 2013, 122, 61. [Google Scholar] [CrossRef]
- DeAngelo, D.J.; Jonas, B.A.; Liesveld, J.L.; Bixby, D.L.; Advani, A.S.; Marlton, P.; O’Dwyer, M.E.; Fogler, W.E.; Magnani, J.L.; Chen, M.M.J.B. High e-selectin ligand expression contributes to chemotherapy-resistance in poor risk relapsed and refractory (r/r) acute myeloid leukemia (AML) patients and can be overcome with the addition of uproleselan. Blood 2019, 134, 2690. [Google Scholar] [CrossRef]
- Chien, S.; Zhao, X.; Brown, M.; Saxena, A.; Patton, J.T.; Magnani, J.L.; Becker, P.S. A novel small molecule e-selectin inhibitor GMI-1271 blocks adhesion of AML blasts to e-selectin and mobilizes cells in nodscid IL2RGC−/− mice engrafted with human AML. Blood 2012, 120, 4092. [Google Scholar] [CrossRef]
- DeAngelo, D.J.; Jonas, B.A.; Liesveld, J.L.; Bixby, D.L.; Advani, A.S.; Marlton, P.; O’Dwyer, M.E.; Fogler, W.E.; Wolfgang, C.D.; Magnani, J.L. Uproleselan (GMI-1271), an e-selectin antagonist, improves the efficacy and safety of chemotherapy in relapsed/refractory (r/r) and newly diagnosed older patients with acute myeloid leukemia: Final, correlative, and subgroup analyses. Blood 2018, 132, 331. [Google Scholar] [CrossRef]
- Leonti, A.R.; Pardo, L.; Alonzo, T.A.; Gerbing, R.B.; Eidenschink Brodersen, L.; Ries, R.E.; Smith, J.L.; Le, Q.; Aplenc, R.; Kolb, E.A. Transcriptome profiling of glycosylation genes defines correlation with e-selectin ligand expression and clinical outcome in AML. Blood 2019, 134, 3772. [Google Scholar] [CrossRef]
- Winters, A.C.; Bernt, K.M. MLL-rearranged leukemias-an update on science and clinical approaches. Front. Pediatr. 2017, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Meyer, C.; Burmeister, T.; Groger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-de-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284. [Google Scholar] [CrossRef]
- De Rooij, J.D.; Masetti, R.; Van Den Heuvel-Eibrink, M.M.; Cayuela, J.-M.; Trka, J.; Reinhardt, D.; Rasche, M.; Sonneveld, E.; Alonzo, T.A.; Fornerod, M.J.B. Recurrent abnormalities can be used for risk group stratification in pediatric AMKL: A retrospective intergroup study. Blood 2016, 127, 3424–3430. [Google Scholar] [CrossRef] [PubMed]
- Bernt, K.M.; Zhu, N.; Sinha, A.U.; Vempati, S.; Faber, J.; Krivtsov, A.V.; Feng, Z.; Punt, N.; Daigle, A.; Bullinger, L.; et al. Mll-rearranged leukemia is dependent on aberrant H3k79 methylation by DOT1L. Cancer Cell 2011, 20, 66–78. [Google Scholar] [CrossRef] [Green Version]
- Krivtsov, A.V.; Eschle, B.K.; Witkin, M.; Gadrey, J.Y.; Uckelmann, H.J.; Kitajima, S.; McGeehan, G.M.; Armstrong, S.A. Vtp50469 is a novel, orally available MENIN-MLL1 inhibitor effective against MLL-rearranged and NPM1-mutant leukemia. Cancer Res. 2018, 78, 4958. [Google Scholar]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-Lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The dot1l inhibitor pinometostat reduces H3k79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018, 131, 2661–2669. [Google Scholar] [CrossRef] [PubMed]
- Shukla, N.; Wetmore, C.; O’Brien, M.M.; Silverman, L.B.; Brown, P.; Cooper, T.M.; Thomson, B.; Blakemore, S.J.; Daigle, S.; Suttle, B. Final report of phase 1 study of the DOT1L inhibitor, pinometostat (EPZ-5676), in children with relapsed or refractory MLL-R acute leukemia. Blood 2016, 128, 2780. [Google Scholar] [CrossRef]
- Borkin, D.; He, S.; Miao, H.; Kempinska, K.; Pollock, J.; Chase, J.; Purohit, T.; Malik, B.; Zhao, T.; Wang, J.; et al. Pharmacologic inhibition of the MENIN-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell 2015, 27, 589–602. [Google Scholar] [CrossRef] [Green Version]
- Cierpicki, T.; Grembecka, J. Challenges and opportunities in targeting the MENIN-MLL interaction. Future Med. Chem. 2014, 6, 447–462. [Google Scholar] [CrossRef] [Green Version]
- Richard, B.L.; Kathryn, E.; Tara, P.; Stephen, W.E.; Yuelong, G.; David, A.C.; Gerard, M.M.; Beverly, A.T.; Malcolm, A.S. Pediatric preclinical testing consortium evaluation of the menin inhibitor, VTP-50469, against xenograft models of MLL-rearranged infant acute lymphoblastic leukemia. In Proceedings of the AACR Annual Meeting 2018, Chicago, IL, USA, 14–18 April 2018. [Google Scholar]
- Burrows, F.; Wu, T.; Kessler, L.; Li, S.; Zhang, J.; Zarrinkar, P.; Li, L.; Cierpicki, T.; Grembecka, J.; Ren, P. Abstract lb-a27: A novel small molecule MENIN-MLL inhibitor for potential treatment of MLL-rearranged leukemias and NPM1/DNMT3A-mutant AML. Available online: https://mct.aacrjournals.org/content/17/1_Supplement/LB-A27 (accessed on 1 February 2020).
- Dafflon, C.; Craig, V.J.; Mereau, H.; Grasel, J.; Schacher Engstler, B.; Hoffman, G.; Nigsch, F.; Gaulis, S.; Barys, L.; Ito, M.; et al. Complementary activities of DOT1L and MENIN inhibitors in MLL-rearranged leukemia. Leukemia 2017, 31, 1269–1277. [Google Scholar] [CrossRef]
- Steelman, L.S.; Franklin, R.A.; Abrams, S.L.; Chappell, W.; Kempf, C.R.; Basecke, J.; Stivala, F.; Donia, M.; Fagone, P.; Nicoletti, F.; et al. Roles of the RAS/RAF/MEK/ERK pathway in leukemia therapy. Leukemia 2011, 25, 1080–1094. [Google Scholar] [CrossRef] [Green Version]
- Knight, T.; Irving, J.A. RAS/Raf/MEK/ERK pathway activation in childhood acute lymphoblastic leukemia and its therapeutic targeting. Front. Oncol. 2014, 4, 160. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.S.; Shapiro, P.S.; Ahn, N.G. Signal transduction through MAP kinase cascades. Adv. Cancer Res. 1998, 74, 49–139. [Google Scholar] [PubMed]
- Towatari, M.; Iida, H.; Tanimoto, M.; Iwata, H.; Hamaguchi, M.; Saito, H. Constitutive activation of mitogen-activated protein kinase pathway in acute leukemia cells. Leukemia 1997, 11, 479–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goemans, B.F.; Zwaan, C.M.; Miller, M.; Zimmermann, M.; Harlow, A.; Meshinchi, S.; Loonen, A.H.; Hahlen, K.; Reinhardt, D.; Creutzig, U.; et al. Mutations in kit and RAS are frequent events in pediatric core-binding factor acute myeloid leukemia. Leukemia 2005, 19, 1536–1542. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.B.; Smalley, K.S.; Sosman, J.A. Molecular pathways: Targeting NRAS in melanoma and acute myelogenous leukemia. Clin. Cancer Res. 2014, 20, 4186–4192. [Google Scholar] [CrossRef] [Green Version]
- Burgess, M.R.; Hwang, E.; Firestone, A.J.; Huang, T.; Xu, J.; Zuber, J.; Bohin, N.; Wen, T.; Kogan, S.C.; Haigis, K.M.; et al. Preclinical efficacy of MEK inhibition in NRAS-mutant AML. Blood 2014, 124, 3947–3955. [Google Scholar] [CrossRef]
- Kerstjens, M.; Pinhancos, S.S.; Castro, P.G.; Schneider, P.; Wander, P.; Pieters, R.; Stam, R.W. Trametinib inhibits RAS-mutant MLL-rearranged acute lymphoblastic leukemia at specific niche sites and reduces erk phosphorylation in vivo. Haematologica 2018, 103, 147–150. [Google Scholar] [CrossRef] [Green Version]
- Pikman, Y.; Tasian, S.K.; Sulis, M.L.; Cooper, T.M.; Pauly, M.; Maloney, K.W.; Burke, M.J.; Brown, P.; Gossai, N.; Cole, P. Matched targeted therapy for pediatric patients with relapsed, refractory or high-risk leukemias: A report from the leap consortium. J. Clin. Oncol. 2018. [Google Scholar] [CrossRef]
- Borthakur, G.; Popplewell, L.; Boyiadzis, M.; Foran, J.; Platzbecker, U.; Vey, N.; Walter, R.B.; Olin, R.; Raza, A.; Giagounidis, A.; et al. Activity of the oral mitogen-activated protein kinase kinase inhibitor trametinib in ras-mutant relapsed or refractory myeloid malignancies. Cancer 2016, 122, 1871–1879. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, K.; Li, S.; Adams, P.D.; Deshpande, A.J. The role of tp53 in acute myeloid leukemia: Challenges and opportunities. Genes Chromosomes Cancer 2019, 58, 875–888. [Google Scholar] [CrossRef] [Green Version]
- Faderl, S.; Kantarjian, H.M.; Estey, E.; Manshouri, T.; Chan, C.Y.; Rahman Elsaied, A.; Kornblau, S.M.; Cortes, J.; Thomas, D.A.; Pierce, S.; et al. The prognostic significance of p16(INK4A)/p14(ARF) locus deletion and MDM-2 protein expression in adult acute myelogenous leukemia. Cancer 2000, 89, 1976–1982. [Google Scholar] [CrossRef]
- Iwakuma, T.; Lozano, G. MDM2, an introduction. Mol. Cancer Res. 2003, 1, 993–1000. [Google Scholar] [PubMed]
- Oliner, J.D.; Pietenpol, J.A.; Thiagalingam, S.; Gyuris, J.; Kinzler, K.W.; Vogelstein, B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 1993, 362, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, M.F.; Morano, W.F.; Lee, J.; Gleeson, E.; Babcock, B.D.; Michl, J.; Sarafraz-Yazdi, E.; Pincus, M.R.; Bowne, W.B. Emerging role of MDM2 as target for anti-cancer therapy: A review. Ann. Clin. Lab. Sci. 2016, 46, 627–634. [Google Scholar] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, K.; Konopleva, M.; Samudio, I.J.; Shikami, M.; Cabreira-Hansen, M.; McQueen, T.; Ruvolo, V.; Tsao, T.; Zeng, Z.; Vassilev, L.T.; et al. MDM2 antagonists induce p53-dependent apoptosis in aml: Implications for leukemia therapy. Blood 2005, 106, 3150–3159. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, E.; Halilovic, E.; Cooke, V.G.; Nonami, A.; Ren, T.; Sanda, T.; Simkin, I.; Yuan, J.; Antonakos, B.; Barys, L.; et al. Inhibition of wild-type p53-expressing aml by the novel small molecule HDM2 inhibitor CGM097. Mol. Cancer Ther. 2015, 14, 2249–2259. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, G.; Pappayannidis, C.; Yee, K.; Vey, N.; Drummond, M.; Kelly, K.; Dickinson, M.; Lee, J.; Seiter, K.; Yoon, S.J.H. Phase 1b results of idasanutlin+ cytarabine (ARA-C) in acute myeloid leukemia (AML) patients (Pts). Available online: https://library.ehaweb.org/eha/2016/21st/135260/cristina.pappayannidis.phase.1b.results.of.idasanutlin.2B.cytarabine.28ara-c29.in.html (accessed on 1 February 2020).
- Dangl, M.; Chien, Y.; Lehmann, C.; Friess, T. Synergistic anticancer activity of clinical stage, non-genotoxic apoptosis inducing agents Rg7388 (MDM2 antagonist) and ABT-199 (GDC-0199, BCL2 inhibitor) in p53 wild-type aml tumor models. Cancer Res. 2014, 74, 5505. [Google Scholar]
- Daver, N.G.; Pollyea, D.A.; Garcia, J.S.; Jonas, B.A.; Yee, K.W.; Fenaux, P.; Assouline, S.; Vey, N.; Olin, R.; Roboz, G.J. Safety, efficacy, pharmacokinetic (pk) and biomarker analyses of BCL2 inhibitor venetoclax (Ven) plus MDM2 inhibitor Idasanutlin (IDASA) in patients (PTS) with relapsed or refractory (r/r) AML: A phase Ib, non-randomized, open-label study. Blood 2018. [Google Scholar] [CrossRef]
- Khurana, A.; Shafer, D.A. MDM2 antagonists as a novel treatment option for acute myeloid leukemia: Perspectives on the therapeutic potential of idasanutlin (RG7388). Onco. Targets Ther. 2019, 12, 2903–2910. [Google Scholar] [CrossRef] [Green Version]
- Carol, H.; Reynolds, C.P.; Kang, M.H.; Keir, S.T.; Maris, J.M.; Gorlick, R.; Kolb, E.A.; Billups, C.A.; Geier, B.; Kurmasheva, R.T.; et al. Initial testing of the MDM2 inhibitor RG7112 by the pediatric preclinical testing program. Pediatr. Cancer 2013, 60, 633–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. Tp53 variations in human cancers: New lessons from the IARC TP53 database and genomics data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Grimwade, D.; Ivey, A.; Huntly, B.J. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood 2016, 127, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Kadia, T.M.; Jain, P.; Ravandi, F.; Garcia-Manero, G.; Andreef, M.; Takahashi, K.; Borthakur, G.; Jabbour, E.; Konopleva, M.; Daver, N.G.; et al. Tp53 mutations in newly diagnosed acute myeloid leukemia: Clinicomolecular characteristics, response to therapy, and outcomes. Cancer 2016, 122, 3484–3491. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Maslah, N.; Salomao, N.; Drevon, L.; Verger, E.; Partouche, N.; Ly, P.; Aubin, P.; Naoui, N.; Schlageter, M.H.; Bally, C.; et al. Synergistic effects of PRIMA-1MET (APR-246) and azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2019. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.J.; Zhang, Q.; Zhang, M.; Ceder, S.; Abrahmsen, L.; Wiman, K.G. Targeting of mutant p53 and the cellular redox balance by APR-246 as a strategy for efficient cancer therapy. Front. Oncol. 2016, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.F.; McGraw, K. Phase 2 Results of APR-246 and Azacitidine (Aza) in Patients with Tp53 Mutant Myelodysplastic Syndromes (Mds) and Oligoblastic Acute Myeloid Leukemia (Aml); American Society of Hematology: Washington, DC, USA, 2019. [Google Scholar]
- Ravandi, F.; Cortes, J.E.; Jones, D.; Faderl, S.; Garcia-Manero, G.; Konopleva, M.Y.; O’Brien, S.; Estrov, Z.; Borthakur, G.; Thomas, D.J.J.o.c.o. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 1856. [Google Scholar] [CrossRef]
- Rollig, C.; Serve, H.; Huttmann, A.; Noppeney, R.; Muller-Tidow, C.; Krug, U.; Baldus, C.D.; Brandts, C.H.; Kunzmann, V.; Einsele, H.; et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): A multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015, 16, 1691–1699. [Google Scholar] [CrossRef]
- Pollard, J.A.; Alonzo, T.A.; Brown, P.A.; Gerbing, R.B.; Fox, E.; Choi, J.K.; Fisher, B.T.; Hirsch, B.A.; Kahwash, S.; Levine, J.E. Sorafenib in Combination with Standard Chemotherapy for Children with High Allelic Ratio Flt3/Itd+ Aml Improves Event-Free Survival and Reduces Relapse Risk: A Report From the Children’s Oncology Group Protocol Aaml1031; American Society of Hematology: Washington, DC, USA, 2019. [Google Scholar]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Dohner, K.; Marcucci, G.; et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Zwaan, C.M.; Söderhäll, S.; Brethon, B.; Luciani, M.; Rizzari, C.; Stam, R.W.; Besse, E.; Dutreix, C.; Fagioli, F.; Ho, P.A.J.B.j.o.h. A phase 1/2, open-label, dose-escalation study of midostaurin in children with relapsed or refractory acute leukaemia. Br. J. Haematol. 2019, 185, 623. [Google Scholar] [CrossRef] [PubMed]
- Cooper, T.M.; Cassar, J.; Eckroth, E.; Malvar, J.; Sposto, R.; Gaynon, P.; Chang, B.H.; Gore, L.; August, K.; Pollard, J.A.; et al. A phase I study of quizartinib combined with chemotherapy in relapsed childhood leukemia: A therapeutic advances in childhood leukemia & lymphoma (TACL) study. Clin. Cancer Res. 2016, 22, 4014–4022. [Google Scholar] [PubMed] [Green Version]
- Perl, A.E.; Altman, J.K.; Cortes, J.; Smith, C.; Litzow, M.; Baer, M.R.; Claxton, D.; Erba, H.P.; Gill, S.; Goldberg, S.; et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: A multicentre, first-in-human, open-label, phase 1-2 study. Lancet Oncol. 2017, 18, 1061–1075. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Therapy | Type | Target | Clinical Trial | Patient Population |
|---|---|---|---|---|
| Gemtuzumab ozogamicin + chemotherapy | Antibody drug conjugate | CD33 | Phase III COG AAML1831 GO included in standard of care treatment backbone—in development | Newly diagnosed AML in children |
| Anetumab ravtensine | Antibody drug conjugate | Mesothelin | Phase I (COG AAML2011)—in development | ≥2nd relapse AML in children with mesothelin+ AML |
| IMGN632 | Antibody drug conjugate | CD123 | Phase I (NCT03386513) Phase I/II—in development | R/R AML Adults with CD123+ AML and other hematologic malignancies R/R AML in children |
| Uproleselan (GMI-1271) | Small molecule inhibitor | E-selectin | Randomized phase III (NCT03616470) | R/R AML in adults |
| Pinometostat + Azacitidine | Small molecular inhibitor | DOT1L | Phase I/II (NCT03701295) | Newly diagnosed or R/R AML with KMT2A rearrangement in adults |
| SNDX-5613 (VTP-50469) | Small molecule inhibitor | KMT2A rearrangement or NPM1 mutation | Phase I/II (NCT04065399) | Phase I: R/R acute leukemia Phase II in adults:
|
| KO = 539 | Small Molecule Inhibitor | KMT2A rearrangement | Phase I/II (NCT04067336) | R/R AML in adults |
| Trametinib | Small molecule inhibitor | RAS-pathway mutations | COG Phase II ADVL1521 (NCT03190915) | R/R juvenile myelomonocytic leukemia (JMML) in children |
| Idasanutlin+Cytarabine | Small molecule inhibitor | MDM2 antagonist | Randomized Phase III (NCT02545283) | R/R AML with WT and mutated TP53 in adults |
| ALRN-6924 (dual MDM2/MDMX inhibitor) | Small molecule inhibitor | MDM2 antagonist | Phase I (NCT03654716) | R/R AML, ALL, MPAL, or other undifferentiated acute leukemia (Cohort C) with WT TP53 in children |
| APR-246 | Small molecule inhibitor | TP53 | Randomized Phase III (NCT03745716) | TP53-mutated MDS in adults |
| Sorafenib + Palbociclib | Tyrosine kinase inhibitor | FLT3 | Phase I (NCT03132454) | R/R AML and ALL in adolescents and adults |
| Midostaurin + chemotherapy | Tyrosine kinase inhibitor | FLT3 | Phase II (NCT03591510) | Newly diangosed FLT3-mutated AML in children |
| Quizartinib + chemotherapy | Tyrosine kinase inhibitor | FLT3 | Phase I/II (NCT03793478) | R/R FLT3-mutated AML in children |
| Gilteritinib + chemotherapy | Tyrosine kinase inhibitor | FLT3 | Phase III (COG AAML1831)—in development | Newly diagnosed FLT3-mutated AML in children |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Glasser, C.L. New and Emerging Targeted Therapies for Pediatric Acute Myeloid Leukemia (AML). Children 2020, 7, 12. https://doi.org/10.3390/children7020012
Chen J, Glasser CL. New and Emerging Targeted Therapies for Pediatric Acute Myeloid Leukemia (AML). Children. 2020; 7(2):12. https://doi.org/10.3390/children7020012
Chicago/Turabian StyleChen, Jing, and Chana L. Glasser. 2020. "New and Emerging Targeted Therapies for Pediatric Acute Myeloid Leukemia (AML)" Children 7, no. 2: 12. https://doi.org/10.3390/children7020012
APA StyleChen, J., & Glasser, C. L. (2020). New and Emerging Targeted Therapies for Pediatric Acute Myeloid Leukemia (AML). Children, 7(2), 12. https://doi.org/10.3390/children7020012