
Placental mTOR Signaling and Sexual Dimorphism in Metabolic Health across the Lifespan of Offspring


{kind=link}
Abstract
:1. Introduction
1.1. Fetal Growth Restriction
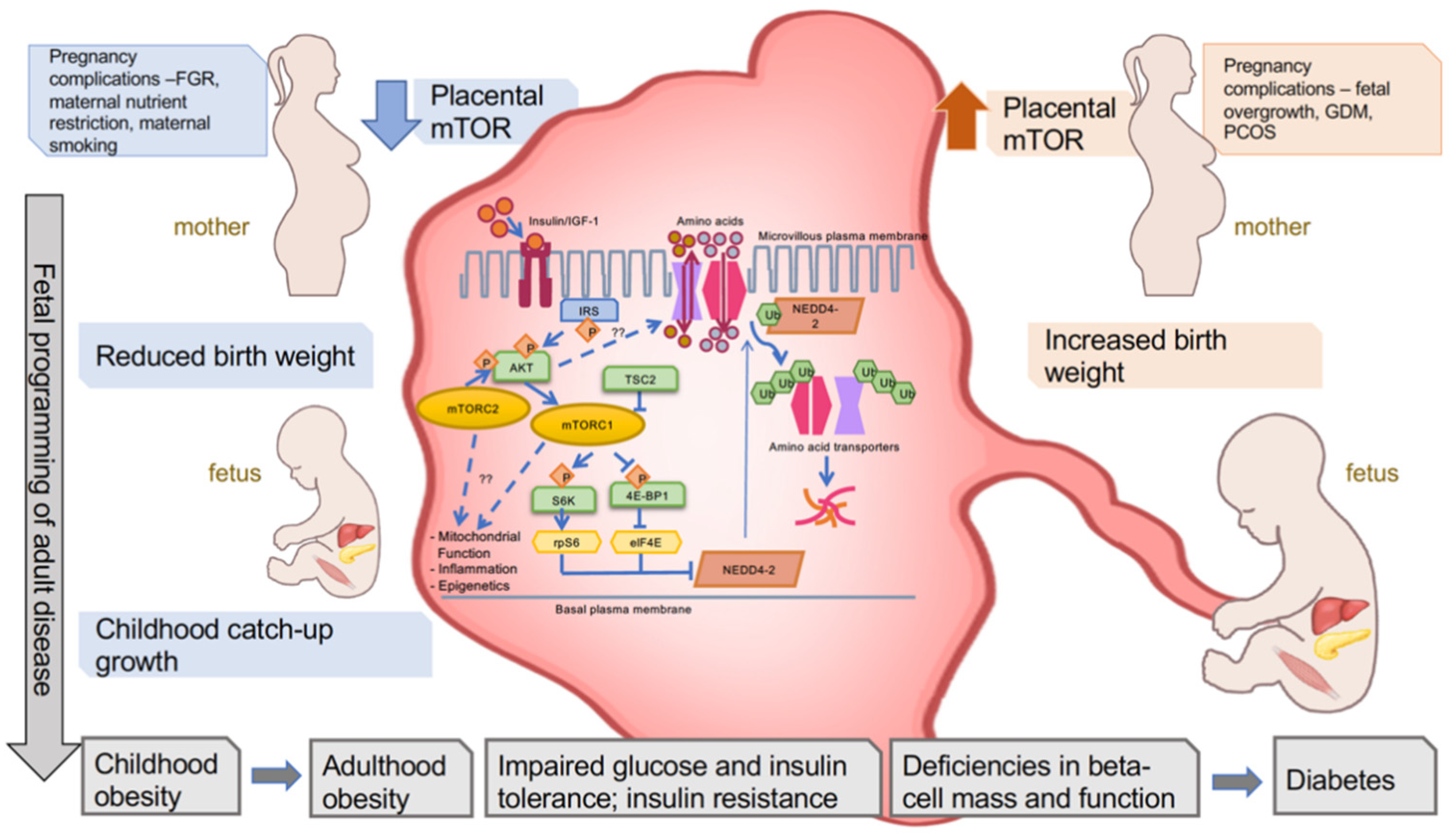
1.1.1. Placental mTOR
1.1.2. Sexual Dimorphism in FGR
1.2. Fetal Overgrowth
1.2.1. Placental mTOR
1.2.2. Sexual Dimorphism in Fetal Overgrowth
1.3. Gestational Diabetes Mellitus
1.3.1. Placental mTOR
1.3.2. Metformin for GDM Treatment
1.3.3. Sexual Dimorphism in GDM
1.4. Polycystic Ovary Syndrome
1.4.1. Placental mTOR
1.4.2. Sexual Dimorphism in PCOS
1.5. Maternal Nutrient Restriction
1.5.1. Placental mTOR
1.5.2. Sexual Dimorphism in MNR
1.6. Preeclampsia
1.6.1. Placental mTOR
1.6.2. Sexual Dimorphism in PE
1.7. Maternal Smoking
1.7.1. Placental mTOR
1.7.2. Sexual Dimorphism in Maternal Smoking
1.8. Relationship between Perturbed Placental mTOR, Birth Weight, and Offspring Obesity
1.8.1. Childhood
1.8.2. Adulthood
1.9. Relationship between Placental mTOR, Birth Weight, and Offspring Insulin Sensitivity
Adulthood
1.10. Relationship between Placental mTOR, Birth Weight, and Beta Cell Mass/Function
Adulthood
2. Biology Underlying Sexual Dimorphism during Fetal Programming
3. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Fall, C.H.D.; Kumaran, K. Metabolic programming in early life in humans. Philos. Trans. R. Soc. 2019, 374, 20180123. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, P.; Morriseau, T.S.; Kereliuk, S.M.; Doucette, C.A.; Wicklow, B.A.; Dolinsky, V.W. Maternal obesity, diabetes during pregnancy and epigenetic mechanisms that influence the developmental origins of cardiometabolic disease in the offspring. Crit. Rev. Clin. Lab. Sci. 2018, 55, 71–101. [Google Scholar] [CrossRef] [PubMed]
- Rosario, F.J.; Gupta, M.B.; Myatt, L.; Powell, T.L.; Glenn, J.P.; Cox, L.; Jansson, T. Mechanistic Target of Rapamycin Complex 1 Promotes the Expression of Genes Encoding Electron Transport Chain Proteins and Stimulates Oxidative Phosphorylation in Primary Human Trophoblast Cells by Regulating Mitochondrial Biogenesis. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Dimasuay, K.G.; Boeuf, P.; Powell, T.L.; Jansson, T. Placental Responses to Changes in the Maternal Environment Determine Fetal Growth. Front. Physiol. 2016, 7, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.D.; Wu, W.K.K.; Wang, H.Y.; Li, X.X. Serine and one-carbon metabolism, a bridge that links mTOR signaling and DNA methylation in cancer. Pharmacol. Res. 2019, 149, 104352. [Google Scholar] [CrossRef] [PubMed]
- Roos, S.; Kanai, Y.; Prasad, P.D.; Powell, T.L.; Jansson, T. Regulation of placental amino acid transporter activity by mammalian target of rapamycin. Am. J. Physiol. Cell Physiol. 2009, 296, C142–C150. [Google Scholar] [CrossRef] [Green Version]
- Rosario, F.J.; Dimasuay, K.G.; Kanai, Y.; Powell, T.L.; Jansson, T. Regulation of amino acid transporter trafficking by mTORC1 in primary human trophoblast cells is mediated by the ubiquitin ligase Nedd4-2. Clin. Sci. 2016, 130, 499–512. [Google Scholar] [CrossRef] [Green Version]
- Roos, S.; Powell, T.L.; Jansson, T. Placental mTOR links maternal nutrient availability to fetal growth. Biochem. Soc. Trans. 2009, 37, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Roos, S.; Jansson, N.; Palmberg, I.; Säljö, K.; Powell, T.L.; Jansson, T. Mammalian target of rapamycin in the human placenta regulates leucine transport and is down-regulated in restricted fetal growth. J. Physiol. 2007, 582, 449–459. [Google Scholar] [CrossRef]
- Jansson, N.; Rosario, F.J.; Gaccioli, F.; Lager, S.; Jones, H.N.; Roos, S.; Jansson, T.; Powell, T.L. Activation of placental mTOR signaling and amino acid transporters in obese women giving birth to large babies. J. Clin. Endocrinol. Metab. 2013, 98, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Godfrey, K.M.; Reynolds, R.M.; Prescott, S.L.; Nyirenda, M.; Jaddoe, V.W.; Eriksson, J.G.; Broekman, B.F. Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol. 2017, 5, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Bianco, M.E.; Josefson, J.L. Hyperglycemia During Pregnancy and Long-Term Offspring Outcomes. Curr. Diab. Rep. 2019, 19, 1–8. [Google Scholar] [CrossRef]
- Joham, A.E.; Palomba, S.; Hart, R. Polycystic Ovary Syndrome, Obesity, and Pregnancy. Semin. Reprod. Med. 2016, 34, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Moghetti, P.; Tosi, F. Insulin resistance and PCOS: Chicken or egg? J. Endocrinol. Investig. 2021, 44, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Kapral, N.; Miller, S.E.; Scharf, R.J.; Gurka, M.J.; DeBoer, M.D. Associations between birthweight and overweight and obesity in school-age children. Pediatr. Obes. 2018, 13, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Derraik, J.G.B.; Maessen, S.E.; Gibbins, J.D.; Cutfield, W.S.; Lundgren, M.; Ahlsson, F. Large-for-gestational-age phenotypes and obesity risk in adulthood: A study of 195,936 women. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faienza, M.F.; Wang, D.Q.; Frühbeck, G.; Garruti, G.; Portincasa, P. The dangerous link between childhood and adulthood predictors of obesity and metabolic syndrome. Intern. Emerg. Med. 2016, 11, 175–182. [Google Scholar] [CrossRef]
- Akhaphong, B.; Baumann, D.C.; Beetch, M.; Lockridge, A.D.; Jo, S.; Wong, A.; Zemanovic, T.; Mohan, R.; Fondevilla, D.L.; Sia, M.; et al. Placental mTOR complex 1 regulates fetal programming of obesity and insulin resistance in mice. JCI Insight 2021, 6, e149271. [Google Scholar] [CrossRef]
- Barker, D.J.; Gluckman, P.D.; Godfrey, K.M.; Harding, J.E.; Owens, J.A.; Robinson, J.S. Fetal nutrition and cardiovascular disease in adult life. Lancet 1993, 341, 938–941. [Google Scholar] [CrossRef]
- Tsai, K.; Tullis, B.; Jensen, T.; Graff, T.; Reynolds, P.; Arroyo, J. Differential expression of mTOR related molecules in the placenta from gestational diabetes mellitus (GDM), intrauterine growth restriction (IUGR) and preeclampsia patients. Reprod. Biol. 2021, 21, 100503. [Google Scholar] [CrossRef]
- Fahlbusch, F.B.; Hartner, A.; Menendez-Castro, C.; Nögel, S.C.; Marek, I.; Beckmann, M.W.; Schleussner, E.; Ruebner, M.; Huebner, H.; Dörr, H.G.; et al. The placental mTOR-pathway: Correlation with early growth trajectories following intrauterine growth restriction? J. Dev. Orig. Health Dis. 2015, 6, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Thayer, Z.M.; Feranil, A.B.; Kuzawa, C.W. Maternal cortisol disproportionately impacts fetal growth in male offspring: Evidence from the Philippines. Am. J. Hum. Biol. 2012, 24, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, B.; Morgan, E.; Alejandro, E.U. Nutrient sensor signaling pathways and cellular stress in fetal growth restriction. J. Mol. Endocrinol. 2019, 62, R155–R165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebire, N.J.; Jolly, M.; Harris, J.P.; Wadsworth, J.; Joffe, M.; Beard, R.W.; Regan, L.; Robinson, S. Maternal obesity and pregnancy outcome: A study of 287,213 pregnancies in London. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 1175–1182. [Google Scholar] [CrossRef] [Green Version]
- Rosario, F.J.; Kanai, Y.; Powell, T.L.; Jansson, T. Increased placental nutrient transport in a novel mouse model of maternal obesity with fetal overgrowth. Obesity 2015, 23, 1663–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosario, F.J.; Powell, T.L.; Jansson, T. Activation of placental insulin and mTOR signaling in a mouse model of maternal obesity associated with fetal overgrowth. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R87–R93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Renzo, G.C.; Rosati, A.; Sarti, R.D.; Cruciani, L.; Cutuli, A.M. Does fetal sex affect pregnancy outcome? Gend. Med. 2007, 4, 19–30. [Google Scholar] [CrossRef]
- Alur, P. Sex Differences in Nutrition, Growth, and Metabolism in Preterm Infants. Front. Pediatr. 2019, 7, 22. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, F.; Parker, M.; Cerda, S.; Pearson, C.; Fu, L.; Gillman, M.W.; Zuckerman, B.; Wang, X. Placental weight mediates the effects of prenatal factors on fetal growth: The extent differs by preterm status. Obesity 2013, 21, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Gallou-Kabani, C.; Gabory, A.; Tost, J.; Karimi, M.; Mayeur, S.; Lesage, J.; Boudadi, E.; Gross, M.S.; Taurelle, J.; Vigé, A.; et al. Sex- and diet-specific changes of imprinted gene expression and DNA methylation in mouse placenta under a high-fat diet. PLoS ONE 2010, 5, e14398. [Google Scholar] [CrossRef]
- Sati, L.; Soygur, B.; Celik-Ozenci, C. Expression of Mammalian Target of Rapamycin and Downstream Targets in Normal and Gestational Diabetic Human Term Placenta. Reprod. Sci. 2016, 23, 324–332. [Google Scholar] [CrossRef]
- Shang, M.; Wen, Z. Increased placental IGF-1/mTOR activity in macrosomia born to women with gestational diabetes. Diabetes Res. Clin. Pract. 2018, 146, 211–219. [Google Scholar] [CrossRef]
- Martino, J.; Sebert, S.; Segura, M.T.; García-Valdés, L.; Florido, J.; Padilla, M.C.; Marcos, A.; Rueda, R.; McArdle, H.J.; Budge, H.; et al. Maternal Body Weight and Gestational Diabetes Differentially Influence Placental and Pregnancy Outcomes. J. Clin. Endocrinol. Metab. 2016, 101, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capobianco, E.; Fornes, D.; Linenberg, I.; Powell, T.L.; Jansson, T.; Jawerbaum, A. A novel rat model of gestational diabetes induced by intrauterine programming is associated with alterations in placental signaling and fetal overgrowth. Mol. Cell Endocrinol. 2016, 422, 221–232. [Google Scholar] [CrossRef]
- Tarry-Adkins, J.L.; Aiken, C.E.; Ozanne, S.E. Neonatal, infant, and childhood growth following metformin versus insulin treatment for gestational diabetes: A systematic review and meta-analysis. PLoS Med. 2019, 16, e1002848. [Google Scholar] [CrossRef] [Green Version]
- Grace, M.R.; Dotters-Katz, S.K.; Zhou, C.; Manuck, T.; Boggess, K.; Bae-Jump, V. Effect of a High-Fat Diet and Metformin on Placental mTOR Signaling in Mice. AJP Rep. 2019, 9, e138–e143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregg, B.; Elghazi, L.; Alejandro, E.U.; Smith, M.R.; Blandino-Rosano, M.; El-Gabri, D.; Cras-Méneur, C.; Bernal-Mizrachi, E. Exposure of mouse embryonic pancreas to metformin enhances the number of pancreatic progenitors. Diabetologia 2014, 57, 2566–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Wang, Z.; Mo, M.; Muyiduli, X.; Wang, S.; Li, M.; Jiang, S.; Wu, Y.; Shao, B.; Shen, Y.; et al. The association of gestational diabetes mellitus with fetal birth weight. J. Diabetes Complicat. 2018, 32, 635–642. [Google Scholar] [CrossRef]
- Kc, K.; Shakya, S.; Zhang, H. Gestational diabetes mellitus and macrosomia: A literature review. Ann. Nutr. Metab. 2015, 66 (Suppl. S2), 14–20. [Google Scholar] [CrossRef]
- Strøm-Roum, E.M.; Haavaldsen, C.; Tanbo, T.G.; Eskild, A. Placental weight relative to birthweight in pregnancies with maternal diabetes mellitus. Acta Obstet. Gynecol. Scand. 2013, 92, 783–789. [Google Scholar] [CrossRef]
- Oken, E.; Morton-Eggleston, E.; Rifas-Shiman, S.L.; Switkowski, K.M.; Hivert, M.F.; Fleisch, A.F.; Mantzoros, C.; Gillman, M.W. Sex-Specific Associations of Maternal Gestational Glycemia with Hormones in Umbilical Cord Blood at Delivery. Am. J. Perinatol. 2016, 33, 1273–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Moullec, N.; Fianu, A.; Maillard, O.; Chazelle, E.; Naty, N.; Schneebeli, C.; Gérardin, P.; Huiart, L.; Charles, M.A.; Favier, F. Sexual dimorphism in the association between gestational diabetes mellitus and overweight in offspring at 5–7 years: The OBEGEST cohort study. PLoS ONE 2018, 13, e0195531. [Google Scholar] [CrossRef] [PubMed]
- Verburg, P.E.; Tucker, G.; Scheil, W.; Erwich, J.J.; Dekker, G.A.; Roberts, C.T. Sexual Dimorphism in Adverse Pregnancy Outcomes—A Retrospective Australian Population Study 1981–2011. PLoS ONE 2016, 11, e0158807. [Google Scholar] [CrossRef] [PubMed]
- Alejandro, E.U.; Mamerto, T.P.; Chung, G.; Villavieja, A.; Gaus, N.L.; Morgan, E.; Pineda-Cortel, M.R.B. Gestational Diabetes Mellitus: A Harbinger of the Vicious Cycle of Diabetes. Int. J. Mol. Sci. 2020, 21, 5003. [Google Scholar] [CrossRef]
- Sir-Petermann, T.; Hitchsfeld, C.; Maliqueo, M.; Codner, E.; Echiburú, B.; Gazitúa, R.; Recabarren, S.; Cassorla, F. Birth weight in offspring of mothers with polycystic ovarian syndrome. Hum. Reprod. 2005, 20, 2122–2126. [Google Scholar] [CrossRef] [Green Version]
- Maliqueo, M.; Sundström Poromaa, I.; Vanky, E.; Fornes, R.; Benrick, A.; Åkerud, H.; Stridsklev, S.; Labrie, F.; Jansson, T.; Stener-Victorin, E. Placental STAT3 signaling is activated in women with polycystic ovary syndrome. Hum. Reprod. 2015, 30, 692–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, K.; Roberts, V.H.J.; Gaffney, J.; Takahashi, D.L.; Morgan, T.; Lo, J.O.; Stouffer, R.L.; Frias, A.E. Maternal High-Fat Diet Consumption and Chronic Hyperandrogenemia Are Associated With Placental Dysfunction in Female Rhesus Macaques. Endocrinology 2019, 160, 1937–1949. [Google Scholar] [CrossRef] [PubMed]
- Bishop, C.V.; Mishler, E.C.; Takahashi, D.L.; Reiter, T.E.; Bond, K.R.; True, C.A.; Slayden, O.D.; Stouffer, R.L. Chronic hyperandrogenemia in the presence and absence of a western-style diet impairs ovarian and uterine structure/function in young adult rhesus monkeys. Hum. Reprod. 2018, 33, 128–139. [Google Scholar] [CrossRef]
- Stein, Z.; Susser, M. The Dutch famine, 1944–1945, and the reproductive process. I. Effects on six indices at birth. Pediatr. Res. 1975, 9, 70–76. [Google Scholar] [CrossRef] [Green Version]
- Kavitha, J.V.; Rosario, F.J.; Nijland, M.J.; McDonald, T.J.; Wu, G.; Kanai, Y.; Powell, T.L.; Nathanielsz, P.W.; Jansson, T. Down-regulation of placental mTOR, insulin/IGF-I signaling, and nutrient transporters in response to maternal nutrient restriction in the baboon. FASEB J. 2014, 28, 1294–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosario, F.J.; Jansson, N.; Kanai, Y.; Prasad, P.D.; Powell, T.L.; Jansson, T. Maternal protein restriction in the rat inhibits placental insulin, mTOR, and STAT3 signaling and down-regulates placental amino acid transporters. Endocrinology 2011, 152, 1119–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roseboom, T.J.; Painter, R.C.; de Rooij, S.R.; van Abeelen, A.F.; Veenendaal, M.V.; Osmond, C.; Barker, D.J. Effects of famine on placental size and efficiency. Placenta 2011, 32, 395–399. [Google Scholar] [CrossRef]
- Alejandro, E.U.; Gregg, B.; Wallen, T.; Kumusoglu, D.; Meister, D.; Chen, A.; Merrins, M.J.; Satin, L.S.; Liu, M.; Arvan, P.; et al. Maternal diet-induced microRNAs and mTOR underlie β cell dysfunction in offspring. J. Clin. Investig. 2014, 124, 4395–4410. [Google Scholar] [CrossRef] [Green Version]
- Alejandro, E.U.; Jo, S.; Akhaphong, B.; Llacer, P.R.; Gianchandani, M.; Gregg, B.; Parlee, S.D.; MacDougald, O.A.; Bernal-Mizrachi, E. Maternal low-protein diet on the last week of pregnancy contributes to insulin resistance and β-cell dysfunction in the mouse offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 319, R485–R496. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Y.; Qu, H.; Xu, F.; Hu, H.; Zhang, Q.; Ye, Y. Reduced ELABELA expression attenuates trophoblast invasion through the PI3K/AKT/mTOR pathway in early onset preeclampsia. Placenta 2019, 87, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yue, C.; Xu, J.; Zhan, Y.; Zhao, H.; Li, Y.; Ye, Y. Downregulation of receptor tyrosine kinase-like orphan receptor 1 in preeclampsia placenta inhibits human trophoblast cell proliferation, migration, and invasion by PI3K/AKT/mTOR pathway accommodation. Placenta 2019, 82, 17–24. [Google Scholar] [CrossRef]
- Aiko, Y.; Askew, D.J.; Aramaki, S.; Myoga, M.; Tomonaga, C.; Hachisuga, T.; Suga, R.; Kawamoto, T.; Tsuji, M.; Shibata, E. Differential levels of amino acid transporters System L and ASCT2, and the mTOR protein in placenta of preeclampsia and IUGR. BMC Pregnancy Childbirth 2014, 14, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, X.; Demianczuk, N.N.; Saunders, L.D.; Wang, F.L.; Fraser, W.D. Impact of preeclampsia and gestational hypertension on birth weight by gestational age. Am. J. Epidemiol. 2002, 155, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Akhaphong, B.; Lockridge, A.; Jo, S.; Mohan, R.; Wilcox, J.A.; Wing, C.R.; Regal, J.F.; Alejandro, E.U. Reduced uterine perfusion pressure causes loss of pancreatic β-cell area but normal function in fetal rat offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R1220–R1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, M.J.; Clifton, V.L.; Wright, I.M. Neonates born to mothers with preeclampsia exhibit sex-specific alterations in microvascular function. Pediatr. Res. 2009, 65, 292–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Intapad, S.; Warrington, J.P.; Spradley, F.T.; Palei, A.C.; Drummond, H.A.; Ryan, M.J.; Granger, J.P.; Alexander, B.T. Reduced uterine perfusion pressure induces hypertension in the pregnant mouse. Am. J. Physiol. Regul. Integr. Comp. Physiol 2014, 307, R1353–R1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heltemes, A.; Gingery, A.; Soldner, E.L.; Bozadjieva, N.; Jahr, K.N.; Johnson, B.K.; Gilbert, J.S. Chronic placental ischemia alters amniotic fluid milieu and results in impaired glucose tolerance, insulin resistance and hyperleptinemia in young rats. Exp. Biol. Med. 2010, 235, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Natale, B.V.; Mehta, P.; Vu, P.; Schweitzer, C.; Gustin, K.; Kotadia, R.; Natale, D.R.C. Reduced Uteroplacental Perfusion Pressure (RUPP) causes altered trophoblast differentiation and pericyte reduction in the mouse placenta labyrinth. Sci. Rep. 2018, 8, 1–21. [Google Scholar] [CrossRef]
- Wang, N.; Tikellis, G.; Sun, C.; Pezic, A.; Wang, L.; Wells, J.C.; Cochrane, J.; Ponsonby, A.L.; Dwyer, T. The effect of maternal prenatal smoking and alcohol consumption on the placenta-to-birth weight ratio. Placenta 2014, 35, 437–441. [Google Scholar] [CrossRef] [Green Version]
- Moore, B.F.; Starling, A.P.; Magzamen, S.; Harrod, C.S.; Allshouse, W.B.; Adgate, J.L.; Ringham, B.M.; Glueck, D.H.; Dabelea, D. Fetal exposure to maternal active and secondhand smoking with offspring early-life growth in the Healthy Start study. Int. J. Obes. 2019, 43, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.M. Smoking and pregnancy: Epigenetics and developmental origins of the metabolic syndrome. Birth Defects Res. 2019, 111, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Mejia, C.; Lewis, J.; Jordan, C.; Mejia, J.; Ogden, C.; Monson, T.; Winden, D.; Watson, M.; Reynolds, P.R.; Arroyo, J.A. Decreased activation of placental mTOR family members is associated with the induction of intrauterine growth restriction by secondhand smoke in the mouse. Cell Tissue Res. 2017, 367, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Crowther, C.A.; Hiller, J.E.; Moss, J.R.; McPhee, A.J.; Jeffries, W.S.; Robinson, J.S. Effect of treatment of gestational diabetes mellitus on pregnancy outcomes. N. Engl. J. Med. 2005, 352, 2477–2486. [Google Scholar] [CrossRef] [Green Version]
- Gillman, M.W.; Oakey, H.; Baghurst, P.A.; Volkmer, R.E.; Robinson, J.S.; Crowther, C.A. Effect of treatment of gestational diabetes mellitus on obesity in the next generation. Diabetes Care 2010, 33, 964–968. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Koivuaho, E.; Piltonen, T.T.; Gissler, M.; Lavebratt, C. Association of maternal polycystic ovary syndrome or anovulatory infertility with obesity and diabetes in offspring: A population-based cohort study. Hum. Reprod. 2021, 36, 2345–2357. [Google Scholar] [CrossRef]
- Lisboa, P.C.; de Oliveira, E.; de Moura, E.G. Obesity and endocrine dysfunction programmed by maternal smoking in pregnancy and lactation. Front. Physiol. 2012, 3, 437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergen, H.T. Exposure to smoke during development: Fetal programming of adult disease. Tob. Induc. Dis. 2006, 3, 5–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.J.; Kelly, R.B. Effect of prenatal or perinatal nicotine exposure on neonatal thyroid status and offspring growth in rats. Life Sci. 2005, 76, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, G.P.; Stein, Z.A.; Susser, M.W. Obesity in young men after famine exposure in utero and early infancy. N. Engl. J. Med. 1976, 295, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, S.M.; Ekbom, A. Smoking during pregnancy and diabetes mellitus in a British longitudinal birth cohort. BMJ 2002, 324, 26–27. [Google Scholar] [CrossRef] [Green Version]
- Doherty, D.A.; Newnham, J.P.; Bower, C.; Hart, R. Implications of polycystic ovary syndrome for pregnancy and for the health of offspring. Obstet. Gynecol. 2015, 125, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Gao, J.M.; Zhang, C.M.; Zhao, H.C.; Zhao, Y.; Li, R.; Yu, Y.; Qiao, J. Assessment of growth and metabolism characteristics in offspring of dehydroepiandrosterone-induced polycystic ovary syndrome adults. Reproduction 2016, 152, 705–714. [Google Scholar] [CrossRef] [Green Version]
- de Rooij, S.R.; Painter, R.C.; Phillips, D.I.; Osmond, C.; Michels, R.P.; Godsland, I.F.; Bossuyt, P.M.; Bleker, O.P.; Roseboom, T.J. Impaired insulin secretion after prenatal exposure to the Dutch famine. Diabetes Care 2006, 29, 1897–1901. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wan, H.; Chen, C.; Chen, Y.; Xia, F.; Han, B.; Li, Q.; Wang, N.; Lu, Y. Association between famine exposure in early life with insulin resistance and beta cell dysfunction in adulthood. Nutr. Diabetes 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Somm, E.; Schwitzgebel, V.M.; Vauthay, D.M.; Aubert, M.L.; Hüppi, P.S. Prenatal nicotine exposure and the programming of metabolic and cardiovascular disorders. Mol. Cell Endocrinol. 2009, 304, 69–77. [Google Scholar] [CrossRef]
- Dearden, L.; Bouret, S.G.; Ozanne, S.E. Sex and gender differences in developmental programming of metabolism. Mol. Metab. 2018, 15, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Santos, R.S.; Criollo, A.; Nelson, M.D.; Palmer, B.F.; Clegg, D.J. The effects of oestrogens and their receptors on cardiometabolic health. Nat. Rev. Endocrinol. 2017, 13, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Clegg, D.J.; Riedy, C.A.; Smith, K.A.; Benoit, S.C.; Woods, S.C. Differential sensitivity to central leptin and insulin in male and female rats. Diabetes 2003, 52, 682–687. [Google Scholar] [CrossRef] [Green Version]
- Galetzka, D.; Weis, E.; Tralau, T.; Seidmann, L.; Haaf, T. Sex-specific windows for high mRNA expression of DNA methyltransferases 1 and 3A and methyl-CpG-binding domain proteins 2 and 4 in human fetal gonads. Mol. Reprod. Dev. 2007, 74, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Howerton, C.L.; Morgan, C.P.; Fischer, D.B.; Bale, T.L. O-GlcNAc transferase (OGT) as a placental biomarker of maternal stress and reprogramming of CNS gene transcription in development. Proc. Natl Acad. Sci. USA 2013, 110, 5169–5174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellone, S.; Rapa, A.; Petri, A.; Zavallone, A.; Strigini, L.; Chiorboli, E.; Ciardi, L.; Aguzzi, A.; Bona, G. Leptin levels as function of age, gender, auxological and hormonal parameters in 202 healthy neonates at birth and during the first month of life. J. Endocrinol. Investig. 2004, 27, 18–23. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beetch, M.; Alejandro, E.U. Placental mTOR Signaling and Sexual Dimorphism in Metabolic Health across the Lifespan of Offspring. Children 2021, 8, 970. https://doi.org/10.3390/children8110970
Beetch M, Alejandro EU. Placental mTOR Signaling and Sexual Dimorphism in Metabolic Health across the Lifespan of Offspring. Children. 2021; 8(11):970. https://doi.org/10.3390/children8110970
Chicago/Turabian StyleBeetch, Megan, and Emilyn U. Alejandro. 2021. "Placental mTOR Signaling and Sexual Dimorphism in Metabolic Health across the Lifespan of Offspring" Children 8, no. 11: 970. https://doi.org/10.3390/children8110970
APA StyleBeetch, M., & Alejandro, E. U. (2021). Placental mTOR Signaling and Sexual Dimorphism in Metabolic Health across the Lifespan of Offspring. Children, 8(11), 970. https://doi.org/10.3390/children8110970