
Going the Extra Mile: Why Clinical Research in Cystic Fibrosis Must Include Children

 ,
,
Abstract
:1. Introduction to Cystic Fibrosis and the Evolution of Management
2. Why Involve Children in CF Research
2.1. Learning Things We Would Not Otherwise Know
2.2. Preventing End Organ Damage
2.3. Extrapolating from Adults May Be Inappropriate or Misleading
2.4. Rights and Fairness Issues
3. How to Involve Children in Research
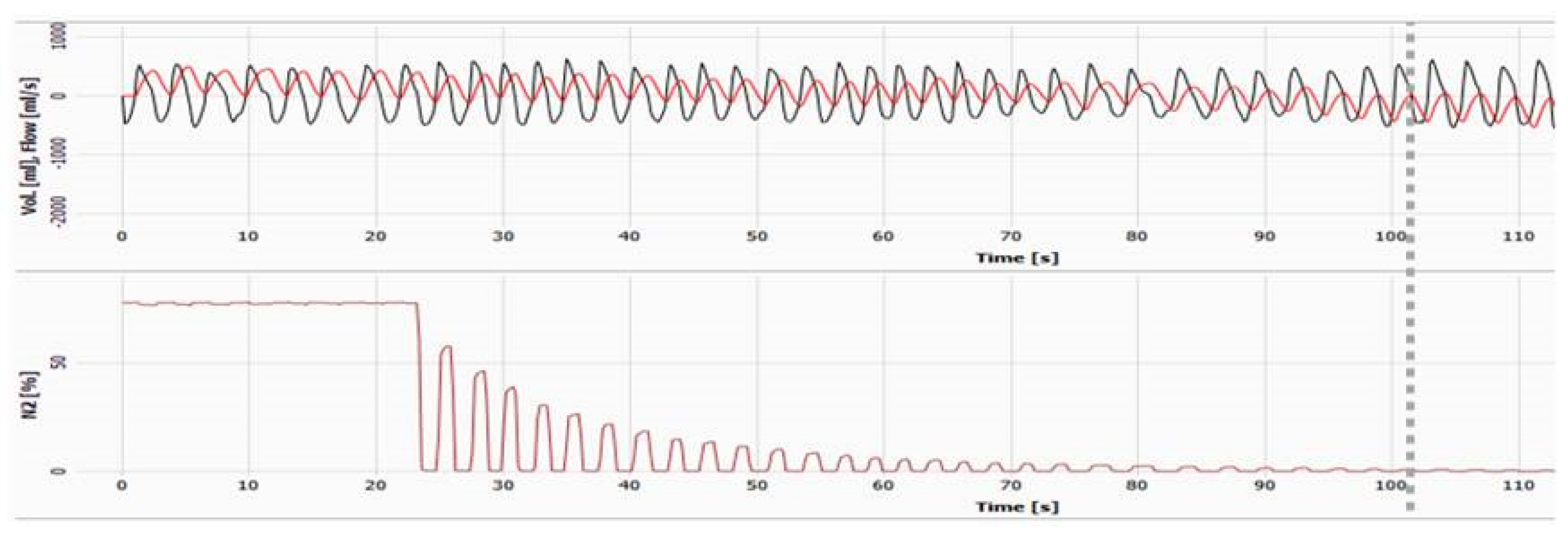
3.1. Choosing Suitable Outcome Measures
3.2. Environment and Team Skills
3.3. Co-Design for Patient-Centred Research
3.4. Approaching Oversubscribed Trials
3.5. Practical Challenges of Recruitment at the Time of Diagnosis
“There is an element of the unknown when deciding to allow your child into a clinical trial. However as hard as it is, medicines and procedures are sadly a very normal part of life with Cystic Fibrosis. We can never know how our child will react to any medicine (new or otherwise) but will have the peace of mind that the trials are heavily regulated and very well managed. Without the trials and the people who have taken part in them over the years, people with CF would not have access to the life changing drugs that are out there today. The existence of these trials gives my daughter the opportunity to live her life as happy and as healthy as she possibly can.”
3.6. Impact of Participation on Schooling and Parental Work
3.7. Blood Tests and Invasive Procedures
3.8. Assent/Consent, Some Practical Tips
“The consent forms are long. They’ve made the children’s one shorter and easier. The language is a bit easier to understand. But they asked my son to sign it as well as write his name. He’s 5. He doesn’t have a signature. He got really frustrated writing his name twice next to each other. I don’t think he’s the only one.”
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bobadilla, J.L.; Macek, M.; Fine, J.P.; Farrell, P.M. Cystic fibrosis: A worldwide analysis of CFTR mutations—Correlation with incidence data and application to screening. Hum. Mutat. 2002, 19, 575–606. Available online: http://www.ncbi.nlm.nih.gov/pubmed/12007216 (accessed on 15 April 2020). [CrossRef] [PubMed]
- Quinton, P.M. Chloride impermeability in cystic fibrosis. Nature 1983, 301, 421–422. [Google Scholar] [CrossRef] [PubMed]
- Di Sant’agnese, P.A.; Darling, R.C.; Perera, G.A.; Shea, E. Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas; clinical significance and relationship to the disease. Pediatrics 1953, 12, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Quinton, P.M. Cystic fibrosis: A disease in electrolyte transport. FASEB J. 1990, 4, 2709–2710. Available online: https://onlinelibrary.wiley.com/doi/full/10.1096/fasebj.4.10.2197151 (accessed on 14 June 2022). [CrossRef]
- Cystic Fibrosis Genotype-Phenotype Consortium. Correlation between Genotype and Phenotype in Patients with Cystic Fibrosis. N. Engl. J. Med. 1993, 329, 1308–1313. [Google Scholar] [CrossRef]
- Pezzulo, A.A.; Tang, X.X.; Hoegger, M.J.; Abou Alaiwa, M.H.; Ramachandran, S.; Moninger, T.O.; Karp, P.H.; Wohlford-Lenane, C.L.; Haagsman, H.P.; van Eijk, M. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012, 487, 109–113. Available online: http://www.nature.com/doifinder/10.1038/nature11130 (accessed on 13 November 2017). [CrossRef]
- Roesch, E.A.; Nichols, D.P.; Chmiel, J.F. Inflammation in cystic fibrosis: An update. Pediatr. Pulmonol. 2018, 53, S30–S50. Available online: https://pubmed.ncbi.nlm.nih.gov/29999593/ (accessed on 29 June 2022). [CrossRef] [Green Version]
- Olivier, A.K.; Yi, Y.; Sun, X.; Sui, H.; Liang, B.; Hu, S.; Xie, W.; Fisher, J.T.; Keiser, N.W.; Lei, D. Abnormal endocrine pancreas function at birth in cystic fibrosis ferrets. J. Clin. Investig. 2012, 122, 3755–3768. Available online: http://www.jci.org/articles/view/60610 (accessed on 13 November 2017). [CrossRef] [Green Version]
- Benninger, L.; Trillo, C.A.; Lascano, J. Tp032 Epidemiology, Biomarkers, and Therapy in Cf And Non-Cf Bronchiectasis/Thematic Poster Session CFTR Modulator Use in Post-Lung Transplant Recipients. Available online: https://www.atsjournals.org/doi/abs/10.1164/ajrccm-conference.2021.203.1_MeetingAbstracts.A2027 (accessed on 13 June 2022).
- Gibson, L.E.; Cooke, R.E. A test for concentration of electrolytes in sweat in cystic fibrosis of the pancreas utilizing pilocarpine by iontophoresis. Pediatrics 1959, 23, 545–549. Available online: http://www.ncbi.nlm.nih.gov/pubmed/13633369 (accessed on 15 April 2020). [CrossRef]
- Andersen, D.H.; Hodges, R.G. Celiac syndrome; genetics of cystic fibrosis of the pancreas, with a consideration of etiology. Am. J. Dis. Child. 1946, 72, 62–80. [Google Scholar] [CrossRef]
- Kerem, B.S.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.C. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerem, E.; Reisman, J.; Corey, M.; Canny, G.J.; Levison, H. Prediction of mortality in patients with cystic fibrosis. N. Engl. J. Med. 1992, 326, 1187–1191. Available online: http://www.ncbi.nlm.nih.gov/pubmed/1285737 (accessed on 15 April 2020). [CrossRef] [PubMed]
- Bell, S.C.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.-R.; Tullis, E.; Castaños, C.; Castellani, C.; et al. The future of cystic fibrosis care: A global perspective. Lancet Respir. Med. 2020, 8, 65–124. Available online: https://linkinghub.elsevier.com/retrieve/pii/S2213260019303376 (accessed on 3 September 2020). [CrossRef] [Green Version]
- Corriveau, S.; Sykes, J.; Stephenson, A.L. Cystic fibrosis survival: The changing epidemiology. Curr. Opin. Pulm. Med. 2018, 24, 574–578. Available online: https://pubmed.ncbi.nlm.nih.gov/30281026/ (accessed on 13 June 2022). [CrossRef]
- McCormick, J.; Green, M.W.; Mehta, G.; Culross, F.; Mehta, A. Demographics of the, U.K. cystic fibrosis population: Implications for neonatal screening. Eur. J. Hum. Genet. 2002, 10, 583–590. Available online: https://pubmed.ncbi.nlm.nih.gov/12357328/ (accessed on 13 June 2022). [CrossRef]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Dřevínek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [Green Version]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. Available online: http://www.nejm.org/doi/10.1056/NEJMoa1709846 (accessed on 13 November 2017). [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- McNamara, J.J.; McColley, S.A.; Marigowda, G.; Liu, F.; Tian, S.; Owen, C.A.; Stiles, D.; Li, C.; Waltz, D.; Wang, L.T.; et al. Safety, pharmacokinetics, and pharmacodynamics of lumacaftor and ivacaftor combination therapy in children aged 2–5 years with cystic fibrosis homozygous for F508del-CFTR: An open-label phase 3 study. Lancet Respir. Med. 2019, 7, 325–335. [Google Scholar] [CrossRef]
- Da Silva Sanchez, A.; Paunovska, K.; Cristian, A.; Dahlman, J. Treating Cystic Fibrosis with mRNA and, C.R.ISPR. Hum. Gene Ther. 2020, 31, 940–955. Available online: https://pubmed.ncbi.nlm.nih.gov/32799680/ (accessed on 29 June 2022). [CrossRef]
- Sly, P.D.; Brennan, S.; Gangell, C.; de Klerk, N.; Murray, C.; Mott, L.; Stick, S.M.; Robinson, P.J.; Robertson, C.F.; Ranganathan, S.C.; et al. Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. Am. J. Respir. Crit. Care Med. 2009, 180, 146–152. Available online: http://www.atsjournals.org/doi/abs/10.1164/rccm.200901-0069OC (accessed on 13 November 2017). [CrossRef] [PubMed]
- Linnane, B.M.; Hall, G.L.; Nolan, G.; Brennan, S.; Stick, S.M.; Sly, P.D.; Ranganathan, S.C. Lung function in infants with cystic fibrosis diagnosed by newborn screening. Am. J. Respir. Crit. Care Med. 2008, 178, 1238–1244. Available online: http://www.atsjournals.org/doi/abs/10.1164/rccm.200804-551OC (accessed on 13 November 2017). [CrossRef] [PubMed]
- Ranganathan, S.C.; Dezateux, C.; Bush, A.; Carr, S.B.; Castle, R.A.; Madge, S.; Stocks, J. Airway function in infants newly diagnosed with cystic fibrosis. Lancet 2001, 358, 1964–1965. Available online: http://www.thelancet.com/article/S0140673601069707/fulltext (accessed on 16 June 2022). [CrossRef]
- Ramsey, K.A.; Ranganathan, S.; Park, J.; Skoric, B.; Adams, A.M.; Simpson, S.J.; Hall, G.L. Early respiratory infection is associated with reduced spirometry in children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 1111–1116. Available online: https://pubmed.ncbi.nlm.nih.gov/25321321/ (accessed on 29 June 2022). [CrossRef] [PubMed] [Green Version]
- Lange, P.; Celli, B.; Agustí, A.; Boje Jensen, G.; Divo, M.; Faner, R.; Vestbo, J. Lung-Function Trajectories Leading to Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2015, 373, 111–122. Available online: https://pubmed.ncbi.nlm.nih.gov/26154786/ (accessed on 29 June 2022). [CrossRef] [Green Version]
- Schlüter, D.K.; Ostrenga, J.S.; Carr, S.B.; Fink, A.K.; Faro, A.; Szczesniak, R.D.; Taylor-Robinson, D. Lung Function in Children with Cystic Fibrosis in the, U.S.A. and, U.K.: A Comparative Longitudinal Analysis of National Registry Data. Thorax 2022, 77, 136–142. [Google Scholar] [CrossRef]
- Ratjen, F.; Davis, S.D.; Stanojevic, S.; Kronmal, R.A.; Hinckley Stukovsky, K.D.; Jorgensen, N.; Richmond, M. Inhaled hypertonic saline in preschool children with cystic fibrosis (SHIP): A multicentre, randomised, double-blind, placebo-controlled trial. Lancet Respir. Med. 2019, 7, 802–809. Available online: https://pubmed.ncbi.nlm.nih.gov/31178421/ (accessed on 29 June 2022). [CrossRef]
- Rosenfeld, M.; Wainwright, C.E.; Higgins, M.; Wang, L.T.; McKee, C.; Campbell, D.; Robinson, P. Ivacaftor treatment of cystic fibrosis in children aged 12 to <24 months and with a CFTR gating mutation (ARRIVAL): A phase 3 single-arm study. Lancet Respir. Med. 2018, 6, 545–553. Available online: http://www.ncbi.nlm.nih.gov/pubmed/29886024 (accessed on 15 April 2020). [CrossRef]
- Davies, J.C.; Cunningham, S.; Harris, W.T.; Lapey, A.; Regelmann, W.E.; Sawicki, G.S.; Southern, K.W.; Robertson, S.; Green, Y.; Cooke, J.; et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2–5 years with cystic fibrosis and a CFTR gating mutation (KIWI): An open-label, single-arm study. Lancet Respir. Med. 2016, 4, 107–115. Available online: http://linkinghub.elsevier.com/retrieve/pii/S2213260015005457 (accessed on 12 August 2017). [CrossRef]
- Hutchinson, I.; McNally, P. Appearance of Pancreatic Sufficiency and Discontinuation of Pancreatic Enzyme Replacement Therapy in Children with Cystic Fibrosis on Ivacaftor. Ann. Am. Thorac. Soc. 2021, 18, 182–183. Available online: https://pubmed.ncbi.nlm.nih.gov/32931706/ (accessed on 29 June 2022). [CrossRef]
- Bellin, M.D.; Laguna, T.; Leschyshyn, J.; Regelmann, W.; Dunitz, J.; Billings, J.; Moran, A. Insulin secretion improves in cystic fibrosis following ivacaftor correction of, C.F.TR: A small pilot study. Pediatr. Diabetes 2013, 14, 417–421. Available online: http://doi.wiley.com/10.1111/pedi.12026 (accessed on 13 November 2017). [CrossRef] [PubMed] [Green Version]
- Kelly, A.; De Leon, D.D.; Sheikh, S.; Camburn, D.; Kubrak, C.; Peleckis, A.J.; Rubenstein, R.C. Islet Hormone and Incretin Secretion in Cystic Fibrosis after Four Months of Ivacaftor Therapy. Am. J. Respir. Crit. Care Med. 2019, 1199, 342–351. Available online: https://pubmed.ncbi.nlm.nih.gov/30130412/ (accessed on 29 June 2022). [CrossRef] [PubMed]
- Bessonova, L.; Volkova, N.; Higgins, M.; Bengtsson, L.; Tian, S.; Simard, C.; Bilton, D. Data from the, U.S. and, U.K. cystic fibrosis registries support disease modification by CFTR modulation with ivacaftor. Thorax 2018, 73, 731–740. Available online: https://pubmed.ncbi.nlm.nih.gov/29748252/ (accessed on 29 June 2022). [CrossRef] [PubMed] [Green Version]
- Volkova, N.; Moy, K.; Evans, J.; Campbell, D.; Tian, S.; Simard, C.; Bilton, D. Disease progression in patients with cystic fibrosis treated with ivacaftor: Data from national, U.S. and, U.K. registries. J. Cyst. Fibros. 2020, 19, 68–79. Available online: http://www.ncbi.nlm.nih.gov/pubmed/31196670 (accessed on 15 April 2020). [CrossRef]
- Balfour-Lynn, I.M.; King, J.A. CFTR modulator therapies—Effect on life expectancy in people with cystic fibrosis. Paediatr. Respir. Rev. 2022, 42, 3–8. Available online: https://pubmed.ncbi.nlm.nih.gov/32565113/ (accessed on 29 June 2022). [CrossRef]
- Bui, S.; Masson, A.; Enaud, R.; Roditis, L.; Dournes, G.; Galode, F.; Mittaine, M. Long-Term Outcomes in Real Life of Lumacaftor-Ivacaftor Treatment in Adolescents With Cystic Fibrosis. Front. Pediatr. 2021, 9, 744705. Available online: https://pubmed.ncbi.nlm.nih.gov/34869102/ (accessed on 29 June 2022). [CrossRef]
- Stalvey, M.S.; Pace, J.; Niknian, M.; Higgins, M.N.; Tarn, V.; Davis, J.; Rowe, S.M. Growth in Prepubertal Children With Cystic Fibrosis Treated With Ivacaftor. Pediatrics 2017, 139, e20162522. Available online: https://pubmed.ncbi.nlm.nih.gov/28143919/ (accessed on 29 June 2022). [CrossRef] [Green Version]
- Sun, X.; Yi, Y.; Yan, Z.; Rosen, B.H.; Liang, B.; Winter, M.C.; Engelhardt, J.F. In utero and postnatal, V.X.-770 administration rescues multiorgan disease in a ferret model of cystic fibrosis. Sci. Transl. Med. 2019, 11, eaau7531. Available online: https://pubmed.ncbi.nlm.nih.gov/30918114/ (accessed on 29 June 2022). [CrossRef] [Green Version]
- Fortner, C.N.; Seguin, J.M.; Kay, D.M. Normal pancreatic function and false-negative, C.F. newborn screen in a child born to a mother taking CFTR modulator therapy during pregnancy. J. Cyst. Fibros. 2021, 20, 835–836. Available online: https://pubmed.ncbi.nlm.nih.gov/33846105/ (accessed on 29 June 2022). [CrossRef]
- Kelly, H.W.; Sternberg, A.L.; Lescher, R.; Fuhlbrigge, A.L.; Williams, P.; Zeiger, R.S.; Strunk, R.C. Effect of inhaled glucocorticoids in childhood on adult height. N. Engl. J. Med. 2012, 367, 904–912. Available online: https://pubmed.ncbi.nlm.nih.gov/22938716/ (accessed on 29 June 2022). [CrossRef] [Green Version]
- Smits, A.; Annaert, P.; Cavallaro, G.; De Cock, P.A.J.G.; de Wildt, S.N.; Kindblom, J.M.; Allegaert, K. Current knowledge, challenges and innovations in developmental pharmacology: A combined conect4children Expert Group and European Society for Developmental, Perinatal and Paediatric Pharmacology White Paper. Br. J. Clin. Pharmacol. 2021, 29, 30. Available online: https://onlinelibrary.wiley.com/doi/full/10.1111/bcp.14958 (accessed on 29 June 2022). [CrossRef]
- PharmD, R.D.B.; PharmD, M.C.N. Aerosolized dornase alpha (rhDNase) in cystic fibrosis. J. Clin. Pharm. Ther. 1995, 20, 313–315. Available online: https://pubmed.ncbi.nlm.nih.gov/8847368/ (accessed on 29 June 2022). [CrossRef] [PubMed]
- Kirwan, L.; Fletcher, G.; Harrington, M.; Jeleniewska, P.; Zhou, S.; Casserly, B.; Jackson, A.D. Longitudinal trends in real-world outcomes after initiation of ivacaftor: A cohort study from the cystic fibrosis registry of Ireland. Ann. Am. Thorac. Soc. 2019, 16, 209–216. Available online: https://www.atsjournals.org/doi/full/10.1513/AnnalsATS.201802-149OC (accessed on 28 March 2022). [CrossRef] [PubMed]
- Nations, U. Chapter IV. Human Rights. 1989. Available online: https://www.unicef.org/child-rights-convention/convention-text# (accessed on 13 July 2022).
- Regulations Requiring Manufacturers to Assess the Safety and Effectiveness of New Drugs and Biological Products in Pediatric Patients—FDA. Final Rule—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/10338880/ (accessed on 29 June 2022).
- Regulation (EC) No 1901/2006 of the European Parliament and of the Council of 12 December 2006 on Medicinal Products for Paediatric Use and Amending Regulation (EEC) No 1768/92, Directive 2001/20/EC, Directive 2001/83/EC and Regulation (EC) No 726/2004 (Text with, E.E.A Relevance). Available online: https://www.legislation.gov.uk/eur/2006/1901/contents (accessed on 29 June 2022).
- Research Charter for Infants’, Children’s and Young People’s Child Health|RCPCH. Available online: https://www.rcpch.ac.uk/resources/research-charter-infants-childrens-young-peoples-child-health (accessed on 29 June 2022).
- CF STORM. Available online: https://www.cfstorm.org.uk/ (accessed on 13 June 2022).
- Dobra, R.; Elborn, S.; Madge, S.; Allen, L.; Saunders, C.; Goundry, S.; Davies, J. P361 What influences participation in clinical trials by people with cystic fibrosis? A national delphi study. J. Cyst. Fibros. 2020, 19, S157. [Google Scholar] [CrossRef]
- Callahan, P.; Pinto, S.J.; Kurland, G.; Cain, J.G.; Motoyama, E.K.; Weiner, D.J. Dexmedetomidine for infant pulmonary function testing. Pediatr. Pulmonol. 2014, 50, 150–154. Available online: https://europepmc.org/article/MED/25187360 (accessed on 29 June 2022). [CrossRef]
- Chen, M.L.; Chen, Q.; Xu, F.; Zhang, J.X.; Su, X.Y.; Tu, X.Z. Safety and efficacy of chloral hydrate for conscious sedation of infants in the pediatric cardiovascular intensive care unit. Medcine 2017, 96, e5842. [Google Scholar] [CrossRef]
- Davies, J.C.; Wainwright, C.E.; Sawicki, G.S.; Higgins, M.N.; Campbell, D.; Harris, C.; Rosenfeld, M. Ivacaftor in Infants Aged 4 to <12 Months with Cystic Fibrosis and a Gating Mutation. Results of a Two-Part Phase 3 Clinical Trial. Am. J. Respir. Crit. Care Med. 2021, 203, 585–593. Available online: https://europepmc.org/articles/PMC7924576 (accessed on 29 June 2022). [CrossRef]
- Harris, J.K.; Wagner, B.D.; Zemanick, E.T.; Robertson, C.E.; Stevens, M.J.; Heltshe, S.L.; Sagel, S.D. Changes in airway microbiome and inflammation with ivacaftor treatment in patients with cystic fibrosis and the G551D mutation. Ann. Am. Thorac. Soc. 2020, 17, 212–220. [Google Scholar] [CrossRef]
- Rosenthal, M. Annual assessment spirometry, plethysmography, and gas transfer in cystic fibrosis: Do they predict death or transplantation. Pediatr. Pulmonol. 2008, 43, 945–952. Available online: https://onlinelibrary.wiley.com/doi/full/10.1002/ppul.20879 (accessed on 29 June 2022). [CrossRef]
- Aurora, P.; Stocks, J.; Oliver, C.; Saunders, C.; Castle, R.; Chaziparasidis, G.; Bush, A. Quality control for spirometry in preschool children with and without lung disease. Am. J. Respir. Crit. Care Med. 2004, 169, 1152–1159. Available online: https://pubmed.ncbi.nlm.nih.gov/15028561/ (accessed on 29 June 2022). [CrossRef]
- Nguyen, T.T.D.; Thia, L.P.; Hoo, A.F.; Bush, A.; Aurora, P.; Wade, A.; Chudleigh, J.; Lum, S.; Stocks, J.; London Cystic Fibrosis Collaboration (LCFC). Evolution of lung function during the first year of life in newborn screened cystic fibrosis infants. Thorax 2014, 69, 910–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aurora, P.; Bush, A.; Gustafsson, P.; Oliver, C.; Wallis, C.; Price, J.; Stocks, J. Multiple-breath washout as a marker of lung disease in preschool children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2005, 171, 249–256. Available online: https://pubmed.ncbi.nlm.nih.gov/15516530/ (accessed on 29 June 2022). [CrossRef] [PubMed]
- Robinson, P.D.; Latzin, P.; Ramsey, K.A.; Stanojevic, S.; Aurora, P.; Davis, S.D.; Ratjen, F. Preschool multiple-breath washout testing an official American thoracic society technical statement. Am. J. Respir. Crit. Care Med. 2018, 197, e1–e19. [Google Scholar] [CrossRef]
- Kent, L.; Reix, P.; Innes, J.A.; Zielen, S.; Le Bourgeois, M.; Braggion, C.; European Cystic Fibrosis Society Clinical Trial Network (ECFS-CTN) Standardisation Committee. Lung clearance index: Evidence for use in clinical trials in cystic fibrosis. J. Cyst. Fibros. 2014, 13, 123–138. Available online: https://pure.ulster.ac.uk/en/publications/lung-clearance-index-evidence-for-use-in-clinical-trials-in-cysti-3 (accessed on 29 June 2022). [CrossRef] [Green Version]
- Davies, J.; Sheridan, H.; Bell, N.; Cunningham, S.; Davis, S.D.; Elborn, J.S.; Ratjen, F. Assessment of clinical response to ivacaftor with lung clearance index in cystic fibrosis patients with a G551D-CFTR mutation and preserved spirometry: A randomised controlled trial. Lancet Respir. Med. 2013, 1, 630–638. Available online: https://pubmed.ncbi.nlm.nih.gov/24461666/ (accessed on 29 June 2022). [CrossRef]
- Ratjen, F.; Hug, C.; Marigowda, G.; Tian, S.; Huang, X.; Stanojevic, S.; Hjelte, L. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR: A randomised, placebo-controlled phase 3 trial. Lancet Respir. Med. 2017, 5, 557–567. Available online: https://pubmed.ncbi.nlm.nih.gov/28606620/ (accessed on 29 June 2022). [CrossRef]
- Pillarisetti, N.; Williamson, E.; Linnane, B.; Skoric, B.; Robertson, C.F.; Robinson, P.; Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST CF). Infection, Inflammation, and Lung Function Decline in Infants with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 75–81. Available online: http://www.ncbi.nlm.nih.gov/pubmed/21493738 (accessed on 13 November 2017). [CrossRef]
- Thia, L.P.; Calder, A.; Stocks, J.; Bush, A.; Owens, C.M.; Wallis, C.; London Cystic Fibrosis Collaboration. Is chest, C.T. useful in newborn screened infants with cystic fibrosis at 1 year of age? Thorax 2014, 69, 320–327. [Google Scholar] [CrossRef] [Green Version]
- Rosenow, T.; Oudraad, M.C.J.; Murray, C.P.; Turkovic, L.; Kuo, W.; de Bruijne, M.; Stick, S.M. PRAGMA-CF. A Quantitative Structural Lung Disease Computed Tomography Outcome in Young Children with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 191, 1158–1165. Available online: https://www.atsjournals.org/doi/full/10.1164/rccm.201501-0061OC (accessed on 1 July 2022). [CrossRef] [Green Version]
- Tiddens, H.A.W.M. Chest computed tomography scans should be considered as a routine investigation in cystic fibrosis. Paediatr. Respir. Rev. 2006, 7, 202–208. Available online: https://pubmed.ncbi.nlm.nih.gov/16938643/ (accessed on 1 July 2022). [CrossRef]
- Cooper, P.; MacLean, J. High-resolution computed tomography (HRCT) should not be considered as a routine assessment method in cystic fibrosis lung disease. Paediatr. Respir. Rev. 2006, 7, 197–201. Available online: https://pubmed.ncbi.nlm.nih.gov/16938642/ (accessed on 1 July 2022). [CrossRef] [PubMed]
- Dournes, G.; Walkup, L.L.; Benlala, I.; Willmering, M.M.; Macey, J.; Bui, S.; Woods, J.C. The Clinical Use of Lung, M.R.I in Cystic Fibrosis: What, Now, How? Chest 2021, 159, 2205. Available online: https://pubmed.ncbi.nlm.nih.gov/33345950/ (accessed on 1 July 2022). [CrossRef] [PubMed]
- Involve Patients|NIHR. Available online: https://www.nihr.ac.uk/health-and-care-professionals/engagement-and-participation-in-research/involve-patients.htm (accessed on 29 August 2020).
- Briel, M.; Elger, B.; von Elm, E.; Satalkar, P. Insufficient recruitment and premature discontinuation of clinical trials in Switzerland: Qualitative study with trialists and other stakeholders. Swiss Med. Wkly. 2017, 147, w14556. Available online: http://doi.emh.ch/smw.2017.14556 (accessed on 6 February 2019). [CrossRef] [PubMed]
- Sacristán, J.A.; Aguarón, A.; Avendaño-Solá, C.; Garrido, P.; Carrión, J.; Gutiérrez, A.; Flores, A. Patient involvement in clinical research: Why, when, and how. Patient Prefer. Adherence 2016, 10, 631–640. Available online: http://www.ncbi.nlm.nih.gov/pubmed/27175063 (accessed on 2 March 2017). [CrossRef] [Green Version]
- Domecq, J.P.; Prutsky, G.; Elraiyah, T.; Wang, Z.; Nabhan, M.; Shippee, N.; Murad, M.H. Patient engagement in research: A systematic review. BMC Health Serv. Res. 2014, 14, 89. Available online: http://www.biomedcentral.com/1472-6963/14/89 (accessed on 26 June 2019). [CrossRef] [Green Version]
- Dobra, R.; Elborn, J.S.; Madge, S.; Allen, L.; Boeri, M.; Kee, F.; Davies, J.C. Guiding the rational design of patient-centred drug trials in Cystic Fibrosis: A Delphi study. J. Cyst. Fibros. 2021, 20, 986–993. Available online: https://pubmed.ncbi.nlm.nih.gov/33895096/ (accessed on 30 May 2022). [CrossRef]
- Harris, J.; Haltbakk, J.; Dunning, T.; Austrheim, G.; Kirkevold, M.; Johnson, M.; Graue, M. How patient and community involvement in diabetes research influences health outcomes: A realist review. Health Expect. 2019, 22, 907–920. [Google Scholar] [CrossRef] [Green Version]
- Cystic Fibrosis Trust—Announcing the New Clinical Trials Accelerator Platform! Available online: https://www.cysticfibrosis.org.uk/news/announcing-the-new-clinical-trials-accelerator-platform (accessed on 14 March 2018).
- Introduction | European Cystic Fibrosis Society (ECFS). Available online: https://www.ecfs.eu/ctn (accessed on 14 March 2018).
- Nguyen, T.T.; Jayadeva, V.; Cizza, G.; Brown, R.J.; Nandagopal, R.; Rodriguez, L.M.; Rother, K.I. Challenging Recruitment of Youth With Type 2 Diabetes Into Clinical Trials. J Adolesc. Health 2014, 54, 247–254. Available online: http://www.ncbi.nlm.nih.gov/pubmed/24161585 (accessed on 6 February 2019). [CrossRef] [Green Version]
- Wendler, D.; Jenkins, T. Children’s and Their Parents’ Views on Facing Research Risks for the Benefit of Others. Arch. Pediatr. Adolesc. Med. 2008, 162, 9. Available online: http://www.ncbi.nlm.nih.gov/pubmed/18180406 (accessed on 6 February 2019). [CrossRef] [Green Version]
- Spinetta, J.J.; Masera, G.; Jankovic, M.; Oppenheim, D.; Martins, A.G.; Ben Arush, M.W.; Eden, T. Valid informed consent and participative decision-making in children with cancer and their parents: A. report of the, S.I.OP working committee on psychosocial issues in pediatric oncology. Med. Pediatr. Oncol. 2003, 40, 244–246. Available online: http://www.ncbi.nlm.nih.gov/pubmed/12555253 (accessed on 6 February 2019). [CrossRef]
- Rowbotham, N.J.; Smith, S.; Leighton, P.A.; Rayner, O.C.; Gathercole, K.; Elliott, Z.C.; Smyth, A.R. The top 10 research priorities in cystic fibrosis developed by a partnership between people with, C.F. and healthcare providers. Thorax 2018, 73, 388–390. Available online: http://www.ncbi.nlm.nih.gov/pubmed/28778919 (accessed on 15 April 2020). [CrossRef] [PubMed] [Green Version]
- Dobra, R.; Davies, G.; Pike, K.; Strassle, C.; Allen, L.; Brendell, R.; Davies, J.C. Optimising equity of access: How should we allocate slots to the most competitive trials in Cystic Fibrosis (CF)? J. Cyst. Fibros. 2021, 20, 978–985. Available online: https://pubmed.ncbi.nlm.nih.gov/33875366/ (accessed on 11 May 2022). [CrossRef] [PubMed]
- Kilgore, K.; Pulungan, Z.; Teigland, C.; Parente, A. The Impact of Demographic And Socio-Economic Factors on Medication Adherence. Value Health 2016, 19, A289. Available online: http://www.valueinhealthjournal.com/article/S1098301516008196/fulltext (accessed on 16 January 2021). [CrossRef]
- Wickwire, E.M.; Jobe, S.L.; Oldstone, L.M.; Scharf, S.M.; Johnson, A.M.; Albrecht, J.S. Lower socioeconomic status and co-morbid conditions are associated with reduced continuous positive airway pressure adherence among older adult medicare beneficiaries with obstructive sleep apnea. Sleep 2020, 43, zsaa122. Available online: https://academic.oup.com/sleep/article/43/12/zsaa122/5861663 (accessed on 1 July 2022). [CrossRef] [PubMed]
- Mills, E.J.; Seely, D.; Rachlis, B.; Griffith, L.; Wu, P.; Wilson, K.; Wright, J.R. Barriers to participation in clinical trials of cancer: A meta-analysis and systematic review of patient-reported factors. Lancet Oncol. 2006, 7, 141–148. Available online: http://www.ncbi.nlm.nih.gov/pubmed/16455478 (accessed on 11 December 2018). [CrossRef]
- Harrison, J.D.; Solomon, M.J.; Young, J.M.; Meagher, A.; Hruby, G.; Salkeld, G.; Clarke, S. Surgical and oncology trials for rectal cancer: Who will participate? Surgery 2007, 142, 94–101.e20. Available online: http://www.ncbi.nlm.nih.gov/pubmed/17630005 (accessed on 6 February 2019). [CrossRef]
- CF START—A Cystic Fibrosis Randomised Registry Trial. Available online: https://cfstart.org.uk/ (accessed on 13 June 2022).
- Weitzman, M. School absence rates as outcome measures in studies of children with chronic illness. J. Chronic. Dis. 1986, 39, 799–808. [Google Scholar] [CrossRef]
- Emerson, N.D.; Distelberg, B.; Morrell, H.E.R.; Williams-Reade, J.; Tapanes, D.; Montgomery, S. Quality of Life and School Absenteeism in Children With Chronic Illness. J. Sch. Nurs. 2016, 32, 258–266. Available online: https://pubmed.ncbi.nlm.nih.gov/26572160/ (accessed on 1 July 2022). [CrossRef] [Green Version]
- ECFS. The Early Cystic Fibrosis Years, 1st ed.; De Boeck, K., Southern, K., Eds.; ECFS: New York, NY, USA, 2018; pp. 260–271. [Google Scholar]
- McIntosh, N. Guidelines for the ethical conduct of medical research involving children. Arch. Dis. Child. 2000, 82, 177–182. [Google Scholar]
- Chiaruttini, G.; Felisi, M.; Bonifazi, D. Challenges in Paediatric Clinical Trials: How to Make It Feasible. In The Management of Clinical Trials; Books on Demand: Norderstedt, Germany, 2018. [Google Scholar]


{kind=link}
| Modulator | Mutation Profile | License Age in UK |
|---|---|---|
| Single agent ivacaftor | Gating mutations | ≥4 months |
| Lumacaftor/ivacaftor | Homozygous F508del | ≥2 years |
| Tezacaftor/ivacaftor | Homozygous F508del or F508del/residual function | ≥6 years |
| Elexecaftor/tezacaftor/ivacaftor (ETI) | 1 or 2 F508del mutations | ≥6 years |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobra, R.; Bentley, S.; Edmondson, C.; Ovens, M.; Saunders, C.; Short, C.; Wilson, G.; Davies, J.C.; Bush, A. Going the Extra Mile: Why Clinical Research in Cystic Fibrosis Must Include Children. Children 2022, 9, 1080. https://doi.org/10.3390/children9071080
Dobra R, Bentley S, Edmondson C, Ovens M, Saunders C, Short C, Wilson G, Davies JC, Bush A. Going the Extra Mile: Why Clinical Research in Cystic Fibrosis Must Include Children. Children. 2022; 9(7):1080. https://doi.org/10.3390/children9071080
Chicago/Turabian StyleDobra, Rebecca, Siân Bentley, Claire Edmondson, Maxine Ovens, Clare Saunders, Christopher Short, Gemma Wilson, Jane C. Davies, and Andrew Bush. 2022. "Going the Extra Mile: Why Clinical Research in Cystic Fibrosis Must Include Children" Children 9, no. 7: 1080. https://doi.org/10.3390/children9071080
APA StyleDobra, R., Bentley, S., Edmondson, C., Ovens, M., Saunders, C., Short, C., Wilson, G., Davies, J. C., & Bush, A. (2022). Going the Extra Mile: Why Clinical Research in Cystic Fibrosis Must Include Children. Children, 9(7), 1080. https://doi.org/10.3390/children9071080