

Rare Variants of Dermatofibrosarcoma Protuberans: Clinical, Histologic, and Molecular Features and Diagnostic Pitfalls



{kind=link}
{kind=link}
{kind=link}
{kind=link}
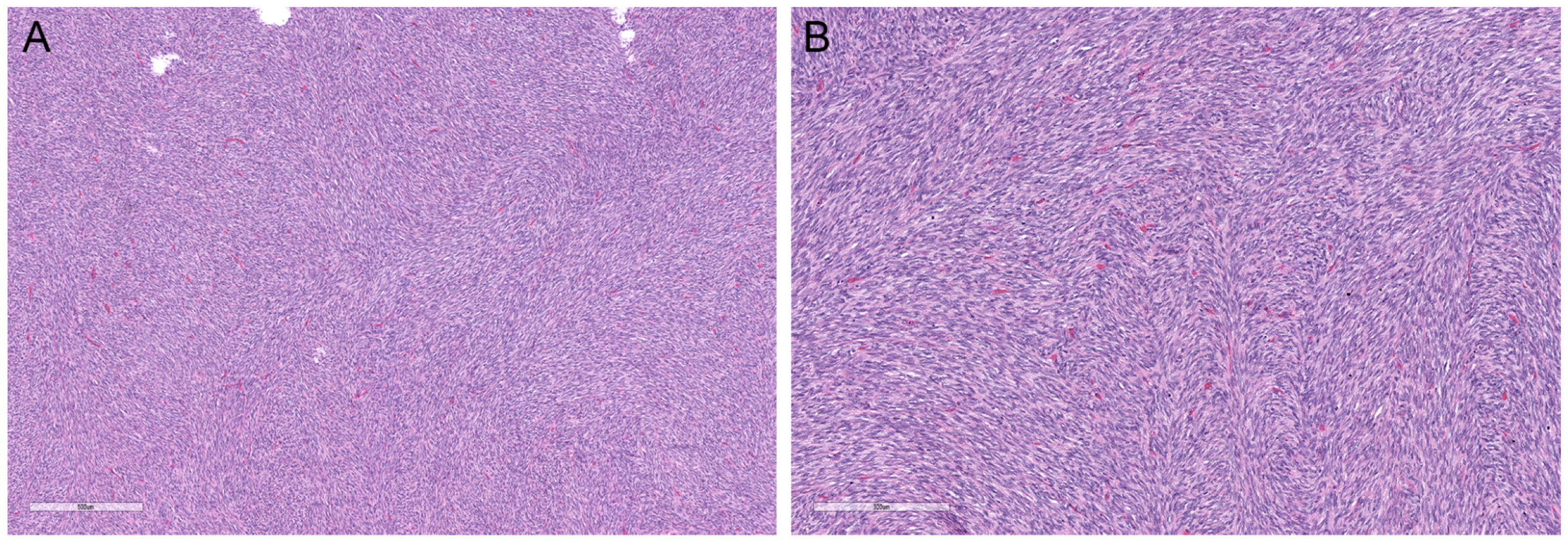{kind=link}
Abstract
:1. Introduction
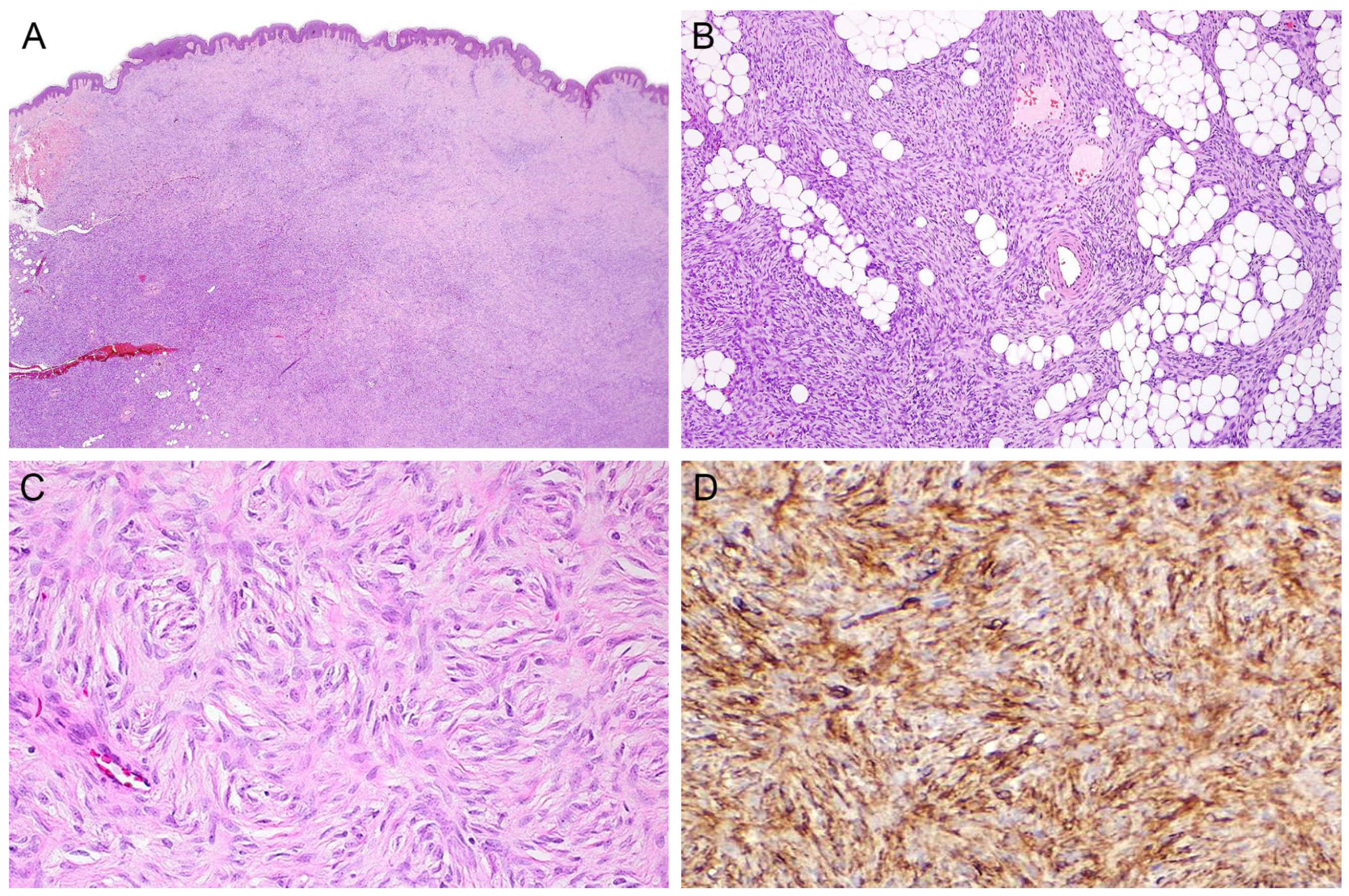
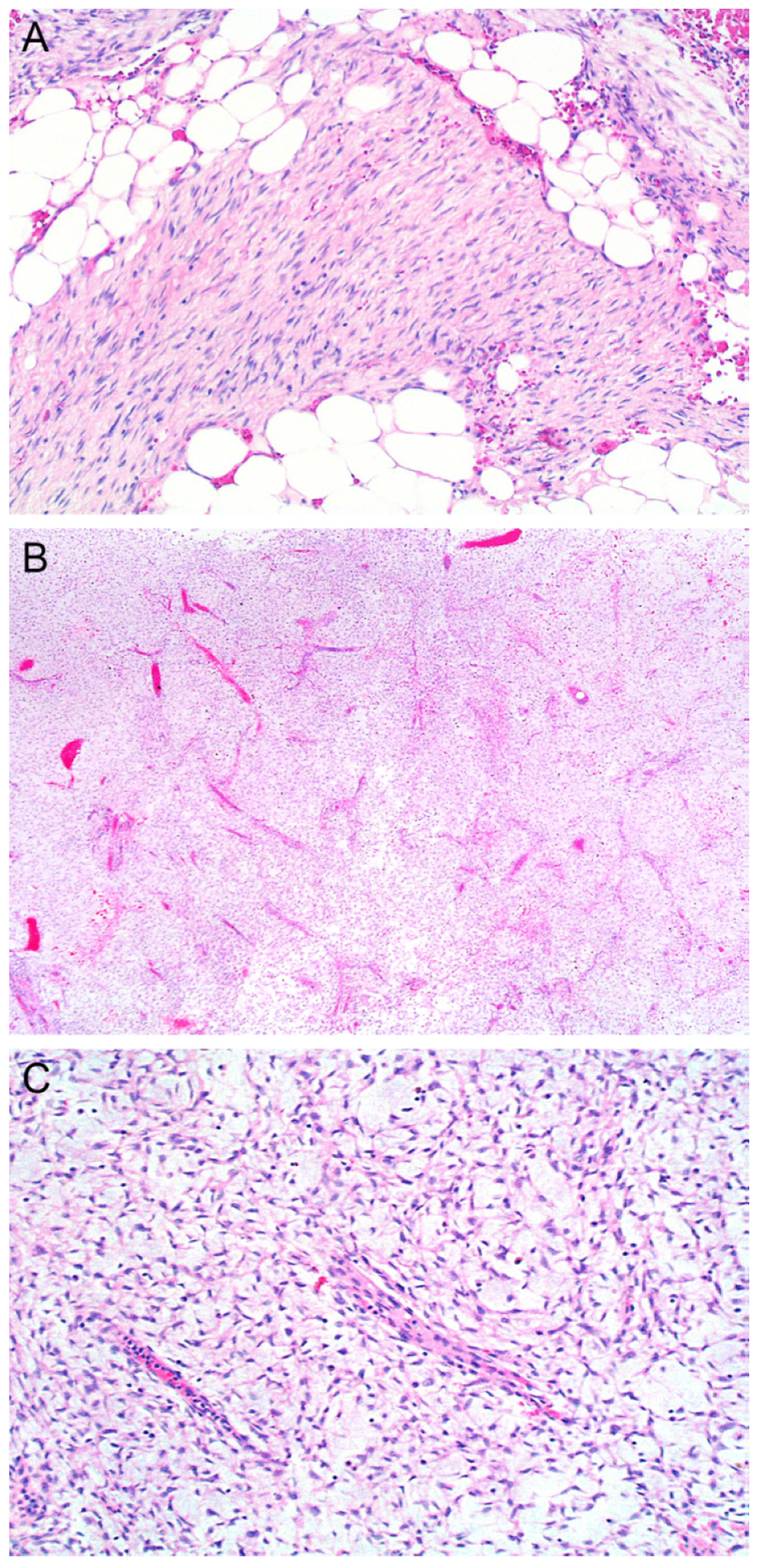
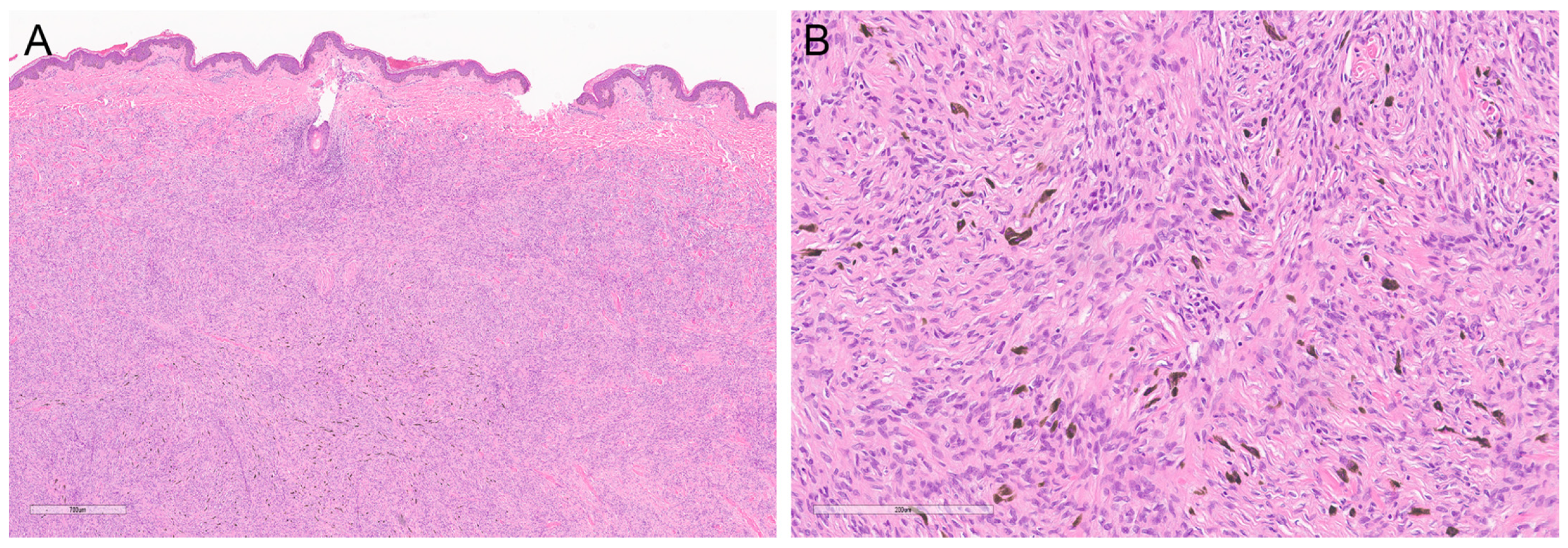
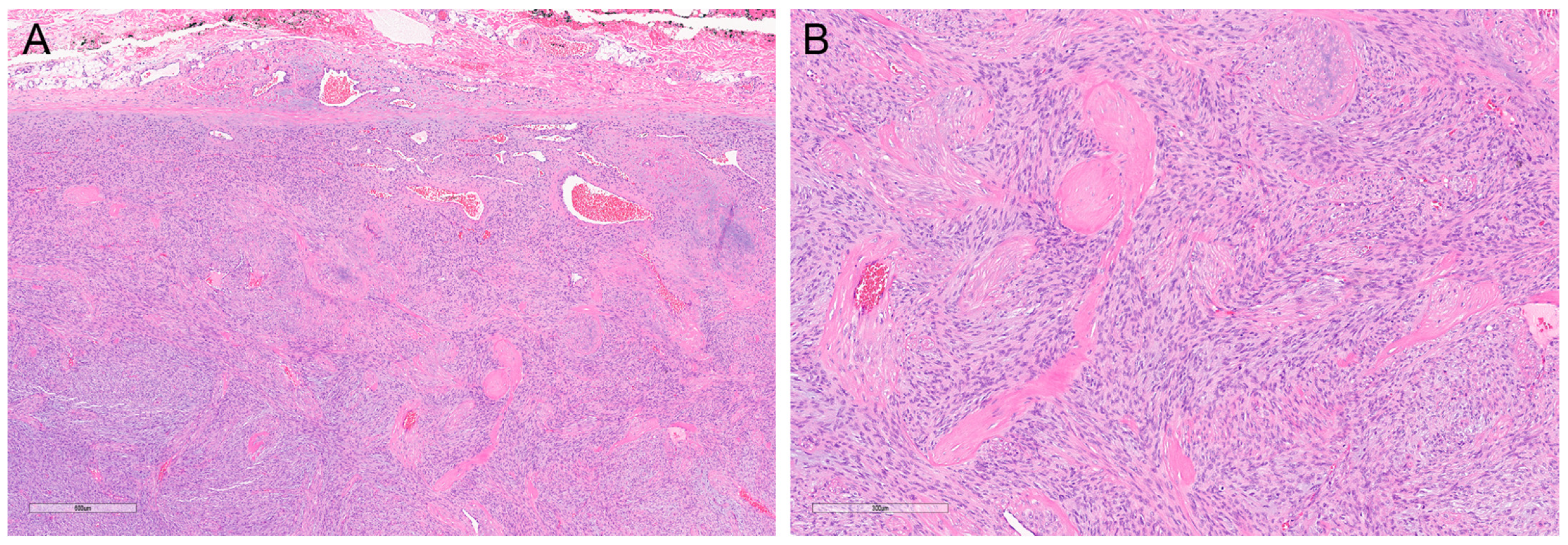
2. Rare Variants of DFSP
2.1. Myxoid
2.2. Pigmented
2.3. Myoid
2.4. Atrophic
2.5. Sclerosing
2.6. Granular Cell
2.7. Fibrosarcomatous
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Elder, D.E.; Massi, D.; Scolyer, R.A.; International Agency for Research on Cancer. WHO Classification of Skin Tumours; International Agency for Research on Cancer: Lyon, France, 2018. [Google Scholar]
- Mentzel, T.; Schärer, L.; Kazakov, D.V.; Michal, M. Myxoid dermatofibrosarcoma protuberans: Clinicopathologic, immunohistochemical, and molecular analysis of eight cases. Am. J. Dermatopathol. 2007, 29, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Socoliuc, C.; Zurac, S.; Andrei, R.; Stăniceanu, F. Multiple Histological Subtypes of Dermatofibrosarcoma Protuberans Occurring in the Same Tumor. Rom. J. Intern. Med. 2015, 53, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.U.; Szabo, S.S.; Hernandez, V.S.; Prieto, V.G.; Abruzzo, L.V.; Lazar, A.J.; López-Terrada, D. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum. Pathol. 2008, 39, 184–193. [Google Scholar] [PubMed]
- Cloutier, J.M.; Allard, G.; Bean, G.R.; Hornick, J.L.; Charville, G.W. PDGFB RNA in situ hybridization for the diagnosis of dermatofibrosarcoma protuberans. Mod. Pathol. 2021, 34, 1521–1529. [Google Scholar] [CrossRef] [PubMed]
- Frierson, H.F.; Cooper, P.H. Myxoid variant of dermatofibrosarcoma protuberans. Am. J. Surg. Pathol. 1983, 7, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Billings, S.D.; Giblen, G.; Fanburg-Smith, J.C. Superficial low-grade fibromyxoid sarcoma (Evans tumor): A clinicopathologic analysis of 19 cases with a unique observation in the pediatric population. Am. J. Surg. Pathol. 2005, 29, 204–210. [Google Scholar] [CrossRef] [PubMed]
- WHO. Soft Tissue and Bone Tumours; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Al-Daraji, W.I.; Miettinen, M. Superficial acral fibromyxoma: A clinicopathological analysis of 32 tumors including 4 in the heel. J. Cutan. Pathol. 2008, 35, 1020–1026. [Google Scholar] [CrossRef]
- Agaimy, A.; Michal, M.; Giedl, J.; Hadravsky, L. Superficial acral fibromyxoma: Clinicopathological, immunohistochemical, and molecular study of 11 cases highlighting frequent Rb1 loss/deletions. Hum. Pathol. 2017, 60, 192–198. [Google Scholar] [CrossRef]
- de Saint Aubain Somerhausen, N.; Rubin, B.P.; Fletcher, C.D. Myxoid solitary fibrous tumor: A study of seven cases with emphasis on differential diagnosis. Mod. Pathol. 1999, 12, 463–471. [Google Scholar]
- Krane, J.F.; Bertoni, F.; Fletcher, C.D. Myxoid synovial sarcoma: An underappreciated morphologic subset. Mod. Pathol. 1999, 12, 456–462. [Google Scholar] [PubMed]
- Bednar, B. Storiform neurofibromas of the skin, pigmented and nonpigmented. Cancer 1957, 10, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Benito, L.M.; Berenguer-Fröhner, B.; Parra-Blanco, V.; Campos-Domínguez, M. Pigmented Dermatofibrosarcoma Protuberans: Description of a pediatric case. Rev. Chil. Pediatr. 2020, 91, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Bernárdez, C.; Molina-Ruiz, A.M.; Requena, L. Bednar Tumor Mimicking Congenital Melanocytic Nevus. Actas Dermo-Sifiliográficas 2016, 107, 523. [Google Scholar] [CrossRef]
- Al-Mendalawi, M.D. Dermatofibrosarcoma protuberans: Two rare variants. Indian J. Dermatol. Venereol. Leprol. 2019, 85, 412. [Google Scholar] [CrossRef]
- Calonje, E.; Fletcher, C.D. Myoid differentiation in dermatofibrosarcoma protuberans and its fibrosarcomatous variant: Clinicopathologic analysis of 5 cases. J. Cutan. Pathol. 1996, 23, 30–36. [Google Scholar] [CrossRef]
- Yadav, S.; Verma, N.; Khurana, N.; Neogi, S. Recurrent Dermatofibrosarcoma Protuberans with Pigmentation and Myoid Differentiation. Sultan Qaboos Univ. Med. J. 2018, 18, e228–e230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellami, R.; Chaabouni, S.; Doghri, R.; Driss, M.; Sassi, S.; Abbes, I.; Karima, M.; Kacem, D.; Ben Ghorbel, R.; Ben Romdhane, K. Myoid differentiation in dermatofibrosarcoma protuberans. Tunis Med. 2012, 90, 185–186. [Google Scholar] [PubMed]
- Lambert, W.C.; Abramovits, W.; Gonzalez-Sevra, A.; Souchon, E.; Schwartz, R.A.; Little, W.P., Jr. Dermatofibrosarcoma non-protuberans: Description and report of five cases of a morpheaform variant of dermatofibrosarcoma. J. Surg. Oncol. 1985, 28, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Yang, Z.; Tu, P.; Li, H. Atrophic pigmented dermatofibrosarcoma protuberans misdiagnosed as hyperpigmentation. Indian J. Dermatol. Venereol. Leprol. 2021, 87, 693–695. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhao, L.; Wang, J. Atrophic dermatofibrosarcoma protuberans: A clinicopathological study of 16 cases. Pathology 2019, 51, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Cascajo, C.; Weyers, W.; Borghi, S. Sclerosing dermatofibrosarcoma protuberans. J. Cutan. Pathol. 1998, 25, 440–444. [Google Scholar] [CrossRef]
- Hattori, H. Nodular sclerotic change in dermatofibrosarcoma protuberans: A potential diagnostic problem. Br. J. Dermatol. 2003, 148, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Abdaljaleel, M.Y.; North, J.P. Sclerosing Dermatofibrosarcoma Protuberans Shows Significant Overlap with Sclerotic Fibroma in Both Routine and Immunohistochemical Analysis: A Potential Diagnostic Pitfall. Am. J. Dermatopathol. 2017, 39, 83–88. [Google Scholar] [CrossRef]
- Calonje, J.E.; Brenn, T.; Lazar, A.J.; Billings, S. McKee’s Pathology of the Skin. In With Clinical Correlations, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Banerjee, S.S.; Harris, M.; Eyden, B.P.; Hamid, B.N. Granular cell variant of dermatofibrosarcoma protuberans. Histopathology 1990, 17, 375–378. [Google Scholar] [CrossRef]
- Maire, G.; Pédeutour, F.; Coindre, J.M. COL1A1-PDGFB gene fusion demonstrates a common histogenetic origin for dermatofibrosarcoma protuberans and its granular cell variant. Am. J. Surg. Pathol. 2002, 26, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Saadat, P.; Vadmal, M. Granular Cell Variant of Dermatofibrosarcoma Protuberans (DFSP). J. Cutan. Pathol. 2008, 33, 67. [Google Scholar] [CrossRef]
- Mentzel, T.; Beham, A.; Katenkamp, D.; Dei Tos, A.P.; Fletcher, C.D. Fibrosarcomatous (“high-grade”) dermatofibrosarcoma protuberans: Clinicopathologic and immunohistochemical study of a series of 41 cases with emphasis on prognostic significance. Am. J. Surg. Pathol. 1998, 22, 576–587. [Google Scholar] [CrossRef]
- Abbott, J.J.; Erickson-Johnson, M.; Wang, X.; Nascimento, A.G.; Oliveira, A.M. Gains of COL1A1-PDGFB genomic copies occur in fibrosarcomatous transformation of dermatofibrosarcoma protuberans. Mod. Pathol. 2006, 19, 1512–1518. [Google Scholar] [CrossRef] [Green Version]
- Abbott, J.J.; Oliveira, A.M.; Nascimento, A.G. The prognostic significance of fibrosarcomatous transformation in dermatofibrosarcoma protuberans. Am. J. Surg. Pathol. 2006, 30, 436–443. [Google Scholar] [CrossRef]
- Erdem, O.; Wyatt, A.J.; Lin, E.; Wang, X.; Prieto, V.G. Dermatofibrosarcoma protuberans treated with wide local excision and followed at a cancer hospital: Prognostic significance of clinicopathologic variables. Am. J. Dermatopathol. 2012, 34, 24–34. [Google Scholar] [CrossRef]
- Swaby, M.G.; Evans, H.L.; Fletcher, C.D.; Prieto, V.G.; Patel, K.U.; Lev, D.C.; Lòpez-Terrada, D.; Lazar, A.J.; Wang, W.L. Dermatofibrosarcoma protuberans with unusual sarcomatous transformation: A series of 4 cases with molecular confirmation. Am. J. Dermatopathol. 2011, 33, 354–360. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trinidad, C.M.; Wangsiricharoen, S.; Prieto, V.G.; Aung, P.P. Rare Variants of Dermatofibrosarcoma Protuberans: Clinical, Histologic, and Molecular Features and Diagnostic Pitfalls. Dermatopathology 2023, 10, 54-62. https://doi.org/10.3390/dermatopathology10010008
Trinidad CM, Wangsiricharoen S, Prieto VG, Aung PP. Rare Variants of Dermatofibrosarcoma Protuberans: Clinical, Histologic, and Molecular Features and Diagnostic Pitfalls. Dermatopathology. 2023; 10(1):54-62. https://doi.org/10.3390/dermatopathology10010008
Chicago/Turabian StyleTrinidad, Celestine M., Sintawat Wangsiricharoen, Victor G. Prieto, and Phyu P. Aung. 2023. "Rare Variants of Dermatofibrosarcoma Protuberans: Clinical, Histologic, and Molecular Features and Diagnostic Pitfalls" Dermatopathology 10, no. 1: 54-62. https://doi.org/10.3390/dermatopathology10010008
APA StyleTrinidad, C. M., Wangsiricharoen, S., Prieto, V. G., & Aung, P. P. (2023). Rare Variants of Dermatofibrosarcoma Protuberans: Clinical, Histologic, and Molecular Features and Diagnostic Pitfalls. Dermatopathology, 10(1), 54-62. https://doi.org/10.3390/dermatopathology10010008