
Conceptual Evolution and Current Approach to Spitz Tumors


Abstract
:Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Ackerman, L.V. Malignant melanoma of the skin. Clinical and histologic analysis of 75 cases. Am. J. Clin. Pathol. 1948, 18, 602–624. [Google Scholar] [CrossRef] [PubMed]
- Pack, G.T.; Anglem, T.J. Tumors of soft somatic tissue in infancy and childhood. J. Pediatr. 1939, 15, 372–400. [Google Scholar] [CrossRef]
- Pack, G.T. Prepubertal melanoma of the skin. Surg. Gynecol. Obstet. 1948, 86, 374–375. [Google Scholar] [PubMed]
- Spitz, S. Melanoma of childhood. Am. J. Pathol. 1948, 24, 591–609. [Google Scholar]
- Allen, A.C. A reorientation of the histogenesis and clinical significance of cutaneous nevi and melanomas. Cancer 1949, 2, 28–56. [Google Scholar] [CrossRef]
- Allen, A.C.; Spitz, S. Malignant melanoma: A clinicopathological analysis of criteria for diagnosis and prognosis. Cancer 1953, 6, 1–45. [Google Scholar] [CrossRef]
- Kernen, J.A.; Ackerman, L.V. Spindle cell nevi and epithelioid cell nevi (so-called juvenile melanomas) in children and adults: A clinicopathologic study of 27 cases. Cancer 1960, 13, 612–625. [Google Scholar] [CrossRef]
- Australian Committee of Pathologists. Moles and malignant melanoma: Terminology and classification. Med. J. Aust. 1967, 1, 123–124. [Google Scholar] [CrossRef]
- Weedon, D.; Little, J.H. Spindle and/or epithelioid cell nevi in children and in adults. Cancer 1977, 40, 217–225. [Google Scholar] [CrossRef]
- Paniago-Pereira, C.; Maize, J.C.; Ackerman, A.B. Nevus of large spindle and/or epithelioid cells (Spitz’s nevus). Arch. Dermatol. 1978, 114, 1811–1823. [Google Scholar] [CrossRef]
- Okun, M.R. Melanoma resembling spindle and epithelioid cell nevus. Arch. Dermatol. 1979, 115, 1416–1420. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, A.B.; Miyauchi, Y.; Yakeuchi, A. Melanomas that simulate Spitz’s nevi histopathologically (and viceversa): An exercise in differentiation based on dependable criteria. Dermatopathol. Pract. Concept. 1999, 5, 9–13. [Google Scholar]
- Farmer, E.R.; Gonin, R.; Hanna, M.P. Discordance in the histopathologic diagnosis of melanoma and melanocytic neoplasms between expert pathologists. Hum. Pathol. 1996, 27, 528–531. [Google Scholar] [CrossRef]
- Smith, K.J.; Barrett, T.L.; Skelton, H.G., 3rd; Lupton, G.P.; Graham, J.H. Spindle cell and epithelioid cell nevi with atypia and metastasis (malignant Spitz nevus). Am. J. Surg. Pathol. 1989, 13, 931–939. [Google Scholar] [CrossRef]
- Barnhill, R.L.; Flotte, T.J.; Fleischli, M.; Perez-Atayde, A. Cutaneous melanoma and atypical Spitz tumors in children. Cancer 1995, 76, 1833–1845. [Google Scholar] [CrossRef]
- Barnhill, R.L.; Argenyi, Z.B.; From, L.; Glass, L.; Maize, J.C.; Mihm, M.C.; Rabkin, M.S.; Ronan, S.G.; White, W.L.; Piepkorn, M. Atypical Spitz nevi/tumor: Lack of consensus for diagnosis, discrimination from melanoma, and prediction of outcome. Hum. Pathol. 1999, 30, 513–520. [Google Scholar] [CrossRef]
- Mones, J.M.; Ackerman, A.B. Melanomas in prepubescent children. Review comprehensively, critique historically, criteria diagnostically, and course biologically. Am. J. Dermatopathol. 2003, 25, 223–238. [Google Scholar] [CrossRef]
- Mones, J.M.; Ackerman, A.B. “Atypical” Spitz’s nevus, “Malignant” Spitz’s nevus, and “Metastasizing” Spitz’s nevus: A critique in historical perspective of three concepts flawed fatally. Am. J. Dermatopathol. 2004, 26, 310–333. [Google Scholar] [CrossRef]
- Shimek, C.M.; Golitz, L.E. The golden anniversary of the Spitz nevus. Arch. Dermatol. 1999, 135, 333–335. [Google Scholar] [CrossRef]
- Shapiro, P.E. Spitz nevi (Letter). J. Am. Acad. Dermatol. 1993, 28, 667–668. [Google Scholar] [CrossRef]
- Piepkorn, M. On the nature of histologic observations: The case of the Spitz nevus. J. Am. Acad. Dermatol. 1995, 32, 248–254. [Google Scholar] [CrossRef]
- Elder, D.E.; Murphy, G.F. Melanocytic tumors of the skin. In Atlas of Tumor Pathology; 3rd Series; Fascicle 2; AFIP: Washington, DC, USA, 1991; p. 40. [Google Scholar]
- Binder, S.W.; Asnong, C.; Paul, E.; Cochran, A.J. The histology and differential diagnosis of Spitz nevus. Sem. Diagn. Pathol. 1993, 10, 36–46. [Google Scholar]
- Cochran, A.J.; Bailly, C.; Paul, E.; Remotti, F. Melanocytic Tumors. A Guide to Diagnosis; Lippincott-Raven Publishers: Philadelphia, PA, USA; New York, NY, USA, 1997; p. 125. [Google Scholar]
- Barnhill, R.L.; Mihm, M.C., Jr. Histopathology of malignant melanoma and its precursor lesions. In Cutaneous Melanoma, 3rd ed.; Balch, C.M., Houghton, A.N., Sober, A.J., Soong, S.-J., Eds.; Quality Medical: St. Louis, MO, USA, 1998; pp. 105–107. [Google Scholar]
- Barnhill, R.L.; Busam, K.L. Spitz nevus and variants. In Pathology of Melanocytic Nevi and Malignant Melanoma, 2nd ed.; Barnhill, R.L., Piepkorn, M., Busam, K.L., Eds.; Springer: New York, NY, USA, 2004; pp. 148–319. [Google Scholar]
- McWhorter, H.E.; Woolner, L.B. Pigmented nevi, juvenile melanomas, and malignant melanomas in children. Cancer 1954, 7, 564–585. [Google Scholar] [CrossRef]
- Piepkorn, M. The Spitz nevus is melanoma. Am. J. Dermatopathol. 2005, 27, 367–369. [Google Scholar] [CrossRef] [PubMed]
- Urso, C. A new perspective for Spitz tumors? Am. J. Dermatopathol. 2005, 27, 364–366. [Google Scholar] [CrossRef]
- Urso, C. Melanocytic lesions, Spitz tumors, and Don Ferrante’s logic. Am. J. Dermatopathol. 2007, 29, 491–494. [Google Scholar] [CrossRef]
- Massi, D.; De Giorgi, V.; Mandalà, M. The complex management of atypical Spitz tumours. Pathology 2016, 48, 132–141. [Google Scholar] [CrossRef]
- Wiesner, T.; He, J.; Yelensky, R.; Esteve-Puig, R.; Botton, T.; Yeh, I.; Lipson, D.; Otto, G.; Brennan, K.; Murali, R.; et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat. Commun. 2014, 5, 3116. [Google Scholar] [CrossRef] [Green Version]
- Yeh, I.; Botton, T.; Talevich, E.; Shain, A.H.; Sparatta, A.J.; de la Fouchardière, A.; Mully, T.W.; North, J.P.; Garrido, M.C.; Gagnon, A.; et al. Activating MET kinase rearrangements in melanoma and Spitz tumours. Nat. Commun. 2015, 6, 7174. [Google Scholar] [CrossRef] [Green Version]
- Yeh, I.; Tee, M.G.; Botton, T.; Shain, A.H.; Sparatta, A.J.; Gagnon, A.; Vemula, S.S.; Garrido, M.C.; Nakamaru, T.; Isoyama, T.; et al. NTRK3 kinase fusions in Spitz tumours. J. Pathol. 2016, 240, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Newman, S.; Fan, L.; Pribnow, A.; Silkov, A.; Rice, S.V.; Lee, S.; Shao, Y.; Shaner, B.; Mulder, H.; Nakitandwe, J.; et al. Clinical genome sequencing uncovers potentially targetable truncations and fusions of MAP3K8 in spitzoid and other melanomas. Nat. Med. 2019, 25, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Cappellesso, R.; Nozzoli, F.; Marino, F.Z.; Simi, S.; Castiglione, F.; De Giorgi, V.; Cota, C.; Senetta, R.; Scognamiglio, G.; Anniciello, A.M.; et al. NTRK Gene Fusion Detection in Atypical Spitz Tumors. Int. J. Mol. Sci. 2021, 22, 12332. [Google Scholar] [CrossRef] [PubMed]
- Quan, V.L.; Zhang, B.; Zhang, Y.; Mohan, L.S.; Shi, K.; Wagner, A.; Kruse, L.; Taxter, T.; Beaubier, N.; White, K.; et al. Integrating Next-Generation Sequencing with Morphology Improves Prognostic and Biologic Classification of Spitz Neoplasms. J. Investig. Dermatol. 2020, 140, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Kerckhoffs, K.G.P.; Aallali, T.; Ambarus, C.A.; Sigurdsson, V.; Jansen, A.M.L.; Blokx, W.A.M. Expanding spectrum of “spitzoid” lesions: A small series of 4 cases with MAP2K1 mutations. Virchows Arch. 2021, 479, 195–202. [Google Scholar] [CrossRef]
- Bastian, B.C.; de la Fouchardière, A.; Elder, D.E.; Gerami, P.; Lazar, A.J.; Massi, D.; Nagore, E.; Scolyer, R.A.; Yun, S.J. Genomic landscape of melanoma. In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; IARC: Lyon, France, 2018; pp. 72–75. [Google Scholar]
- Urso, C. Melanocytic Skin Neoplasms: What Lesson From Genomic Aberrations? Am. J. Dermatopathol. 2019, 41, 623–629. [Google Scholar] [CrossRef]
- Elder, D.E.; Bastian, B.C.; Cree, I.A.; Massi, D.; Scolyer, R.A. The 2018 World Health Organization Classification of Cutaneous, Mucosal, and Uveal Melanoma: Detailed Analysis of 9 Distinct Subtypes Defined by Their Evolutionary Pathway. Arch. Pathol. Lab. Med. 2020, 144, 500–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnhill, R.L.; Bahrami, A.; Bastian, B.C.; Busam, K.J.; Cerroni, L.; de la Fouchardière, A.; Elder, D.E.; Gerami, P.; Lazova, R.; Schmidt, B.; et al. Malignant Spitz tumour (Spitz melanoma). In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; IARC: Lyon, France, 2018; pp. 108–110. [Google Scholar]
- Barnhill, R.L.; Bahrami, A.; Bastian, B.C.; Busam, K.J.; Cerroni, L.; de la Fouchardière, A.; Elder, D.E.; Gerami, P.; Lazova, R.; Schmidt, B.; et al. Spitz naevus. In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; IARC: Lyon, France, 2018; pp. 111–113. [Google Scholar]
- Yeh, I.; Busam, K.J. Spitz melanocytic tumours: A review. Histopathology 2022, 80, 122–134. [Google Scholar] [CrossRef]
- Massi, D.; Tomasini, C.; Senetta, R.; Paglierani, M.; Salvianti, F.; Errico, M.E.; Donofrio, V.; Collini, P.; Tragni, G.; Sementa, A.R.; et al. Atypical spitz tumors in patients younger than 18 years. J. Am. Acad. Dermatol. 2015, 72, 37–46. [Google Scholar] [CrossRef]
- De Giorgi, V.; Venturi, F.; Silvestri, F.; Trane, L.; Savarese, I.; Scarfì, F.; Cencetti, F.; Pecenco, S.; Tramontana, M.; Maio, V.; et al. Atypical Spitz tumors: An epidemiological, clinical and dermoscopic multicenter study with 16-year follow-up. Clin. Exp. Dermatol. 2022. [Google Scholar] [CrossRef]
- Benton, S.; Zhao, J.; Zhang, B.; Bahrami, A.; Barnhill, R.L.; Busam, K.; Cerroni, L.; Cook, M.G.; de la Fouchardière, A.; Elder, D.E.; et al. Impact of Next-generation Sequencing on Interobserver Agreement and Diagnosis of Spitzoid Neoplasms. Am. J. Surg. Pathol. 2021, 45, 1597–1605. [Google Scholar] [CrossRef]
- de la Fouchardière, A.; Blokx, W.; van Kempen, L.C.; Luzar, B.; Piperno-Neumann, S.; Puig, S.; Alos, L.; Calonje, E.; Massi, D.; ESP Dermatopathology Working Group; et al. ESP, EORTC, and EUROCAN expert opinion: Practical recommendations for the pathological diagnosis and clinical management of intermediate melanocytic tumors and rare related melanoma variants. Virchows Arch. 2021, 479, 3–11. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
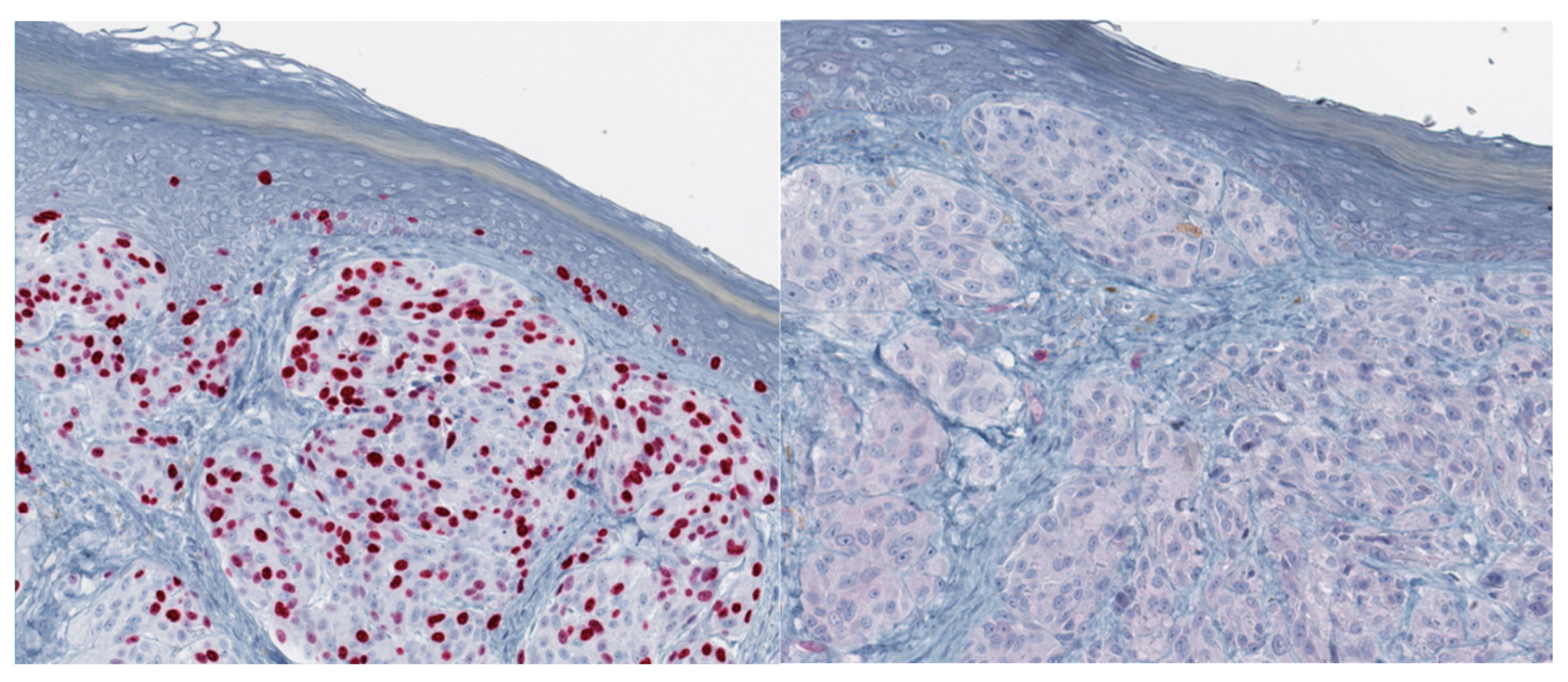{kind=link}
| Spitz Tumors | HRAS Mutation; MAP2K1 Mutation; ALK, ROS1, NTRK1, NTRK3, MAP3K8, BRAF, RET, MET, NTRK2, ERB4, FGFR1, MAP3K3, PRKDC Kinase Fusions |
| Conventional nevi and melanomas | BRAF/NRAS mutations |
| Blue tumors | GNAQ/GNA11 mutations |
| Deep penetrating tumors | BRAF mutation + CTNNB1 mutation or APC loss |
| Pigmented epithelioid melanocytomas | BRAF mutation + PRKAR1A mutation or PRKCA fusion |
| BAP1-inactivated tumors | BRAF mutation + BAP1 mutations |
| Spitz Nevi | CGH: Isolated Gains 7p and 11p; Tetraploidy; HRAS Mutation |
| Atypical Spitz tumors | CGH: ≥1 chromosomal abnormality; PTEN mutation; heterozygous loss of 9p21 |
| Spitz melanoma | CGH: ≥1 chromosomal abnormality; PTEN mutation; heterozygous loss of 9p21; TERT promoter mutations |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urso, C.; De Giorgi, V.; Massi, D. Conceptual Evolution and Current Approach to Spitz Tumors. Dermatopathology 2022, 9, 136-142. https://doi.org/10.3390/dermatopathology9020017
Urso C, De Giorgi V, Massi D. Conceptual Evolution and Current Approach to Spitz Tumors. Dermatopathology. 2022; 9(2):136-142. https://doi.org/10.3390/dermatopathology9020017
Chicago/Turabian StyleUrso, Carmelo, Vincenzo De Giorgi, and Daniela Massi. 2022. "Conceptual Evolution and Current Approach to Spitz Tumors" Dermatopathology 9, no. 2: 136-142. https://doi.org/10.3390/dermatopathology9020017
APA StyleUrso, C., De Giorgi, V., & Massi, D. (2022). Conceptual Evolution and Current Approach to Spitz Tumors. Dermatopathology, 9(2), 136-142. https://doi.org/10.3390/dermatopathology9020017