Separation and Determination of Some of the Main Cholesterol-Related Compounds in Blood by Gas Chromatography-Mass Spectrometry (Selected Ion Monitoring Mode)


 ,
,
Abstract
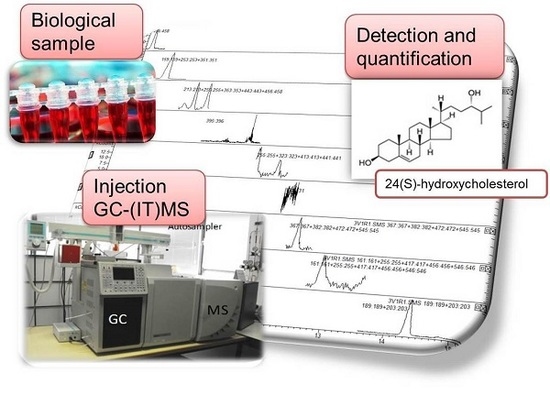
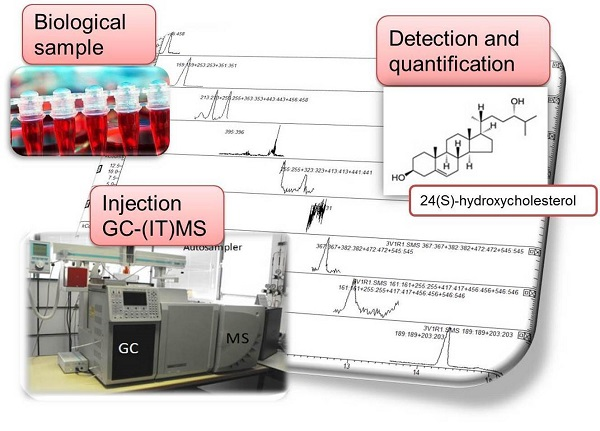
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Standard Solutions
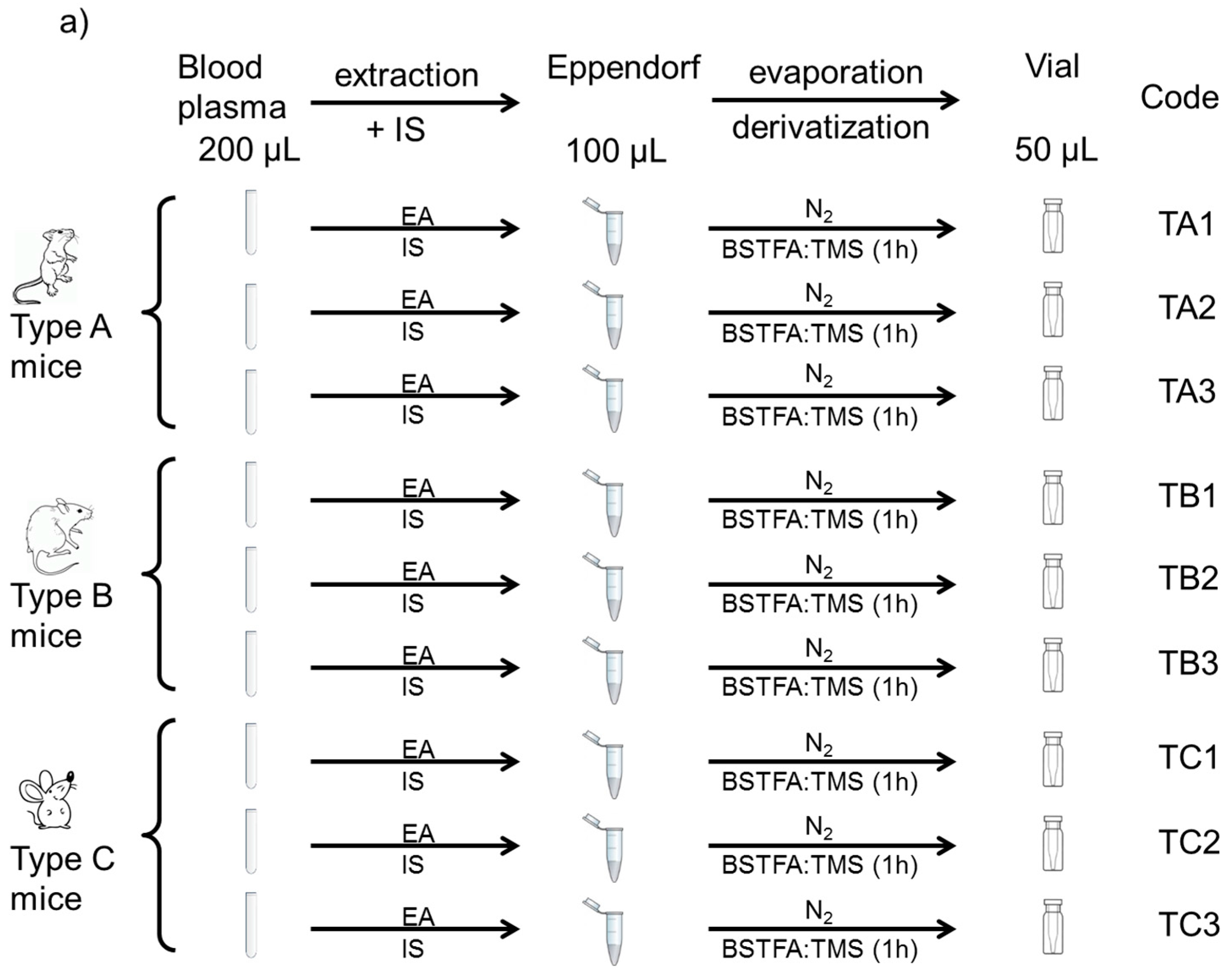
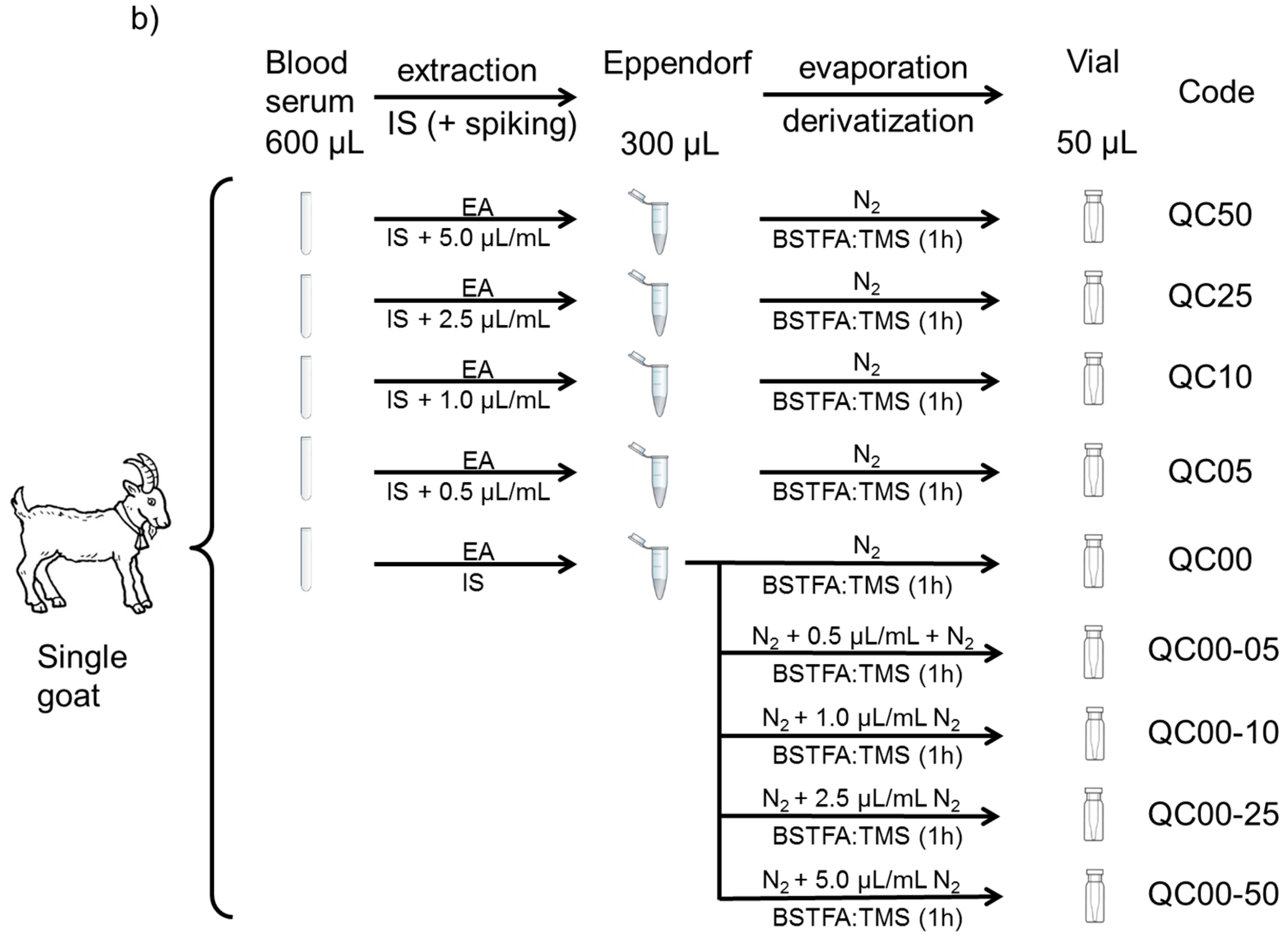
2.3. Samples and Sample Preparation
- (i)
- Mouse plasma as test samples
- (ii)
- Goat serum as quality control samples
2.4. GC-(EI/IT)MS Instrument and Method Conditions
3. Results and Discussion
3.1. Optimization of Detection Conditions
3.2. Validation of the Developed Method
3.3. Application of the Method to the Analysis of Biological Samples
4. Conclusions
Acknowledgments
Ethical Conduct of Research
Author Contributions
Conflicts of Interest
References
- Garenc, C.; Julien, P.; Levy, E. Oxysterols in biological systems: The gastrointestinal tract, liver, vascular wall and central nervous system. Free Radical Res. 2010, 44, 47–73. [Google Scholar] [CrossRef] [PubMed]
- Bjökhem, I.; Heverin, M.; Leoni, V.; Meaney, S.; Diczfalusy, U. Oxysterols and Alzheimer’s disease. Acta Neurol. Scand. 2006, 114, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Terao, J. Cholesterol hydroperoxides and their degradation mechanism. In Lipid Hydroperoxide-Derived Modification of Biomolecules; Kato, Y., Ed.; Springer: London, UK, 2014; pp. 83–91. [Google Scholar]
- Mulder, M. Sterols in the central nervous system. Curr. Opin. Clin. Nutr. 2009, 12, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Biasi, F.; Leonarduzzi, G. Oxysterol in the pathogenesis of major chronic diseases. Redox Biol. 2013, 1, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, W.J.; Wang, Y. Analysis of oxysterol metabolomes. Biochim. Biophys. Acta 2011, 1811, 784–799. [Google Scholar]
- Griffiths, W.J.; Abdel-Khalik, J.; Yutuc, E.; Morgan, A.H.; Gilmore, I.; Hearn, T.; Wang, Y. Cholesterolomics: An update. Anal. Biochem. 2017, 524, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Trushina, E.; Mielke, M.M. Recent advances in the application of metabolomics to Alzheimer’s Disease. Biochim. Biophys. Acta 2014, 1842, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Leoni, V.; Caccia, C. Review. Oxysterols as biomarkers in neurodegenerative diseases. Chem. Phys. Lipids 2011, 164, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Baila-Rueda, L.; Cenarro, A.; Cofán, M.; Orera, I.; Barcelo-Batllori, S.; Pocoví, M.; Ros, E.; Civeira, F.; Domeño, C. Simultaneous determination of oxysterols, phytosterols and cholesterol precursors by high performance liquid chromatography tándem mass spectrometry in human serum. Anal. Methods 2013, 5, 2249–2257. [Google Scholar] [CrossRef]
- Heverin, M.; Bogdanovic, N.; Lütjohann, D.; Bayer, T.; Pikuleva, I.; Bretillon, L.; Dicfalusy, U.; Winblad, B.; Bjökhem, I. Changes in the levels of cerebral and extracerebral sterols in the brain of patients with Alzheimer’s disease. J. Lipid Res. 2004, 45, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.L.F.; Martins, I.J.; Martins, R.N. The involvement of lipids in Alzheimer’s disease. J. Genet. Genom. 2014, 41, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Kölsch, H.; Heun, R.; Kerksiek, A.; Bergmann, K.V.; Maier, W.; Lütjohann, D. Altered levels of plasma 24S- and 27-hydroxycholesterol in demented patients. Neurosci. Lett. 2004, 368, 303–308. [Google Scholar] [CrossRef] [PubMed]
- De la Luz-Hdez, K. Metabolomics and mammalian cell culture. In Metabolomics; Roessner, U., Ed.; InTech: Rijeka, Croatia, 2012; pp. 3–18. [Google Scholar]
- Nielsen, J. Metabolomics in functional genomics and systems biology. In Metabolome Analysis. An Introduction; Villas-Bôas, S.G., Roessner, U., Hansen, M.A.E., Smedsgaard, J., Nielsen, J., Eds.; John Wiley & Sons: Hoboken, NY, USA, 2007; pp. 3–14. [Google Scholar]
- Gouveia, M.J.; Brindley, P.J.; Santos, L.L.; Correia da Costa, J.M.; Gomes, P.; Vale, N. Mass spectrometry techniques in the survey of steroid metabolites as potential disease biomarkers: A review. Metabolis 2013, 62, 1206–1217. [Google Scholar] [CrossRef] [PubMed]
- Want, E.J.; Cravatt, B.F.; Siuzdak, G. The expanding role of mass spectrometry in metabolite profiling and characterization. ChemBioChem 2005, 6, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
- Dzeletovic, S.; Breuer, O.; Lund, E.; Diczfalusy, U. Determination of cholesterol oxidation products in human plasma by isotope dilution-mass spectrometry. Anal. Biochem. 1995, 225, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Ahmida, H.S.M.; Bertucci, P.; Franzò, L.; Massoud, R.; Cortese, C.; Lala, A.; Federici, G. Simultaneous determination of plasmatic phytosterols and cholesterol precursors using gas chromatography-mass spectrometry (GC-MS) with selective ion monitoring (SIM). J. Chromatogr. B 2006, 842, 43–47. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.G.; Smith, D.D.; Stiles, A.R.; Rusell, D.W. A comprehensive method for extraction and quantitative analysis of sterols and steroids from human plasma. J. Lipid Res. 2012, 53, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, D.; Semedo, R.; Castilho, M.C.; Silva, J.M.; Ramos, F. Selection of the derivatization reagent—The case of human blood colesterol, its precursors and phytosterols GC-MS analyses. J. Chromatogr. B 2011, 879, 3806–3811. [Google Scholar] [CrossRef] [PubMed]
- Andrade, I.; Santos, L.; Ramos, F. Advances in analytical methods to study cholesterol metabolism: The determination of serum noncholesterol sterol. Biomed. Chromatogr. 2013, 27, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, W.J.; Crick, P.J.; Wang, Y. Methods for oxysterol analysis: Past, present and future. Biochem. Pharmacol. 2013, 86, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Karuna, R.; Christen, I.; Sailer, A.W.; Bitsch, F.; Zhang, J. Detection of dihydroxycholesterol in human plasma using HPLC-ESI-MS/MS. Steroids 2015, 99, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Kakehi, M.; Satomi, Y.; Kamiguchi, H.; Jinno, F. Method development for the determination of 24S-hydroxycholesterol in human plasma without derivatization by high-performance liquid chromatography with tandem mass spectrometry in atmospheric pressure chemical ionization mode. J. Sep. Sci. 2015, 38, 3516–3524. [Google Scholar] [CrossRef] [PubMed]
- Pataj, Z.; Liebisch, G.; Schmitz, G.; Matysik, S. Quantification of oxysterols in human plasma and red blood cells by liquid chromatography high-resolution tandem mass spectrometry. J. Chromatogr. A 2016, 1439, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, W.J.; Abdel-Khalik, J.; Crick, P.J.; Yutuc, E.; Wang, Y. New methods for analysis of oxysterols and related compounds by LC-MS. J. Steroid Biochem. 2016, 162, 4–26. [Google Scholar] [CrossRef] [PubMed]
- Menéndez-Carreño, M.; García-Herreros, C.; Astiasarán, I.; Ansorena, D. Validation of a gas chromatography-mass spectrometry method for the analysis of sterol oxidation products in serum. J. Chromatogr. B 2008, 864, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Schött, H.F.; Lütjohann, D. Validation of an isotope dilution gas chromatography-mass spectrometry method for combined analysis of oxysterols and oxyphytosterols in serum samples. Steroids 2015, 99, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Matysik, S.; Klünemann, H.H.; Schmitz, G. Gas chromatography-tandem mass spectrometry method for the simultaneous determination of oxysterols, plant sterols, and cholesterol precursors. Clin. Chem. 2012, 58, 1557–1564. [Google Scholar] [CrossRef] [PubMed]
- Matysik, S.; Schmitz, G. Application of gas chromatography-triple quadrupole mass spectrometry to the determination of sterol components in biological samples in consideration of the ionization mode. Biochimie 2013, 95, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, W.J.; Wang, Y. Analysis of neurosterols by GC-MS and LC-MS/MS. J. Chromatogr. B 2009, 877, 2778–2805. [Google Scholar] [CrossRef] [PubMed]
- Narváez-Rivas, M.; Pham, A.J.; Schilling, M.W.; León-Camacho, M. A new SPE/GC-fid method for the determination of cholesterol oxidation products. Application to subcutaneous fat from Iberian dry-curred ham. Talanta 2014, 122, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Toivo, J.; Piironen, V.; Kalo, P.; Varo, P. Gas chromatographic determination of major sterols in edible oils and fast using solid-phase extraction in simple preparation. Chromatographia 1998, 48, 745–750. [Google Scholar] [CrossRef]
- Campoy, S.; Sierra, S.; Suarez, B.; Ramos, M.C.; Velasco, J.; Burgos, J.S.; Adrio, J.L. Semisynthesis of novel monacolin J derivatives: Hypocholesterolemic and neuroprotective activities. J. Antibiot. 2010, 63, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, R.; Nakagawa, S.; Tanabe, A.; Ikeuchi, T.; Miida, T.; Yamato, S. Determination of 24S-hydroxycholesterol in human cerebrospinal fluid by gas chromatography/mass spectrometry. Bunseki Kagaku 2008, 57, 707–713. [Google Scholar] [CrossRef]
- EURACHEM/CITAC Guide 2014. The Fitness for Purpose of Analytical Methods, 2nd ed.; 2014; Available online: www.eurachem.org (accessed on 10 September 2017).
- Thompson, M.; Ellison, S.R.; Wood, R. Harmonized Guidelines for Single Laboratory Validation of Methods of Analysis; IUPAC Technical Report 2002; International Union of Pure and Applied Chemistry: Research Triangle Park, NC, USA, 2002; Volume 74, pp. 835–855. [Google Scholar]
- Cuadros-Rodríguez, L.; García-Campaña, A.M.; Bosque-Sendra, J.M. Statistical Estimation of Linear Calibration Range. Anal. Lett. 1996, 29, 1231–1239. [Google Scholar] [CrossRef]
- International Organization for Standardization. Capability of Detection—Part 2: Methodology in the Linear Calibration Case; ISO 11843-2:2000; International Organization for Standardization: Geneva, Switzerland, 2000. [Google Scholar]
- Ostrowski, S.M.; Johnson, K.; Siefert; Shank, M.S.; Sironi, L.; Wolozin, B.; Landreth, G.E.; Ziady, A.G. Simvastain inhibits protein isoprenylation in the brain. Neuroscience 2016, 329, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Sallustio, F.; Studer, V. Targeting new pharmacological approaches for Alzheimer’s disease: Potential for statins and phosphodiesterase inhibitors. CNS Neurol. Disord. 2016, 15, 647–659. [Google Scholar] [CrossRef]
- McGuinness, B.; Craig, D.; Bullock, R.; Malouf, R.; Passmore, P. Statins for the treatment of dementia. Cochrane DB. Syst. Rev. 2004, 7, 1–74. [Google Scholar]
- Refolo, L.M.; Pappolla, M.A.; LaFrancois, J.; Malester, B.; Schmidt, S.D.; Thomas-Bryant, T.; Tint, G.S.; Wang, R.; Mercken, M.; Pentanceska, S.S.; et al. A cholesterol-lowering drug reduces β-amyloid pathology in a transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis. 2001, 8, 890–899. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
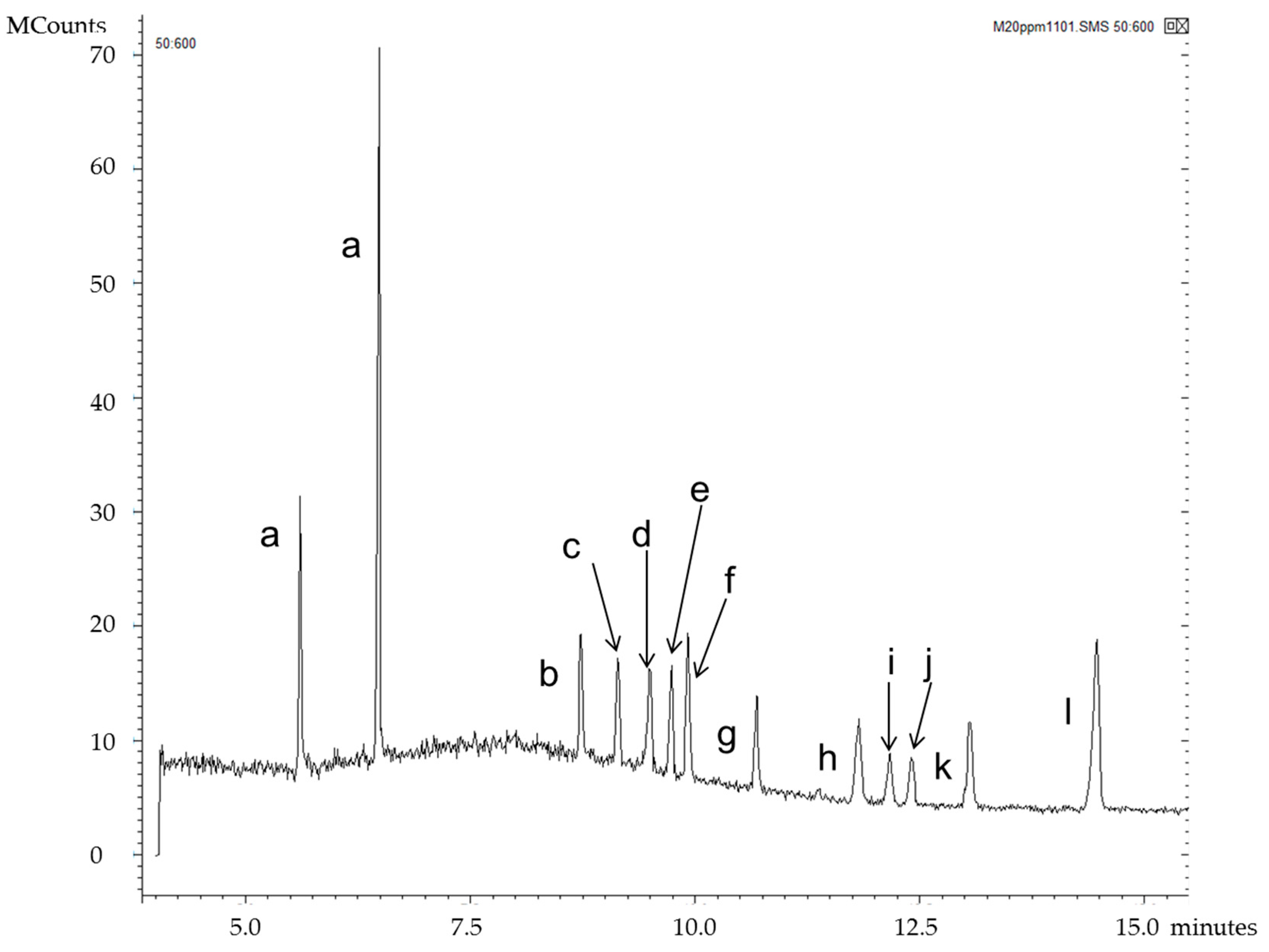
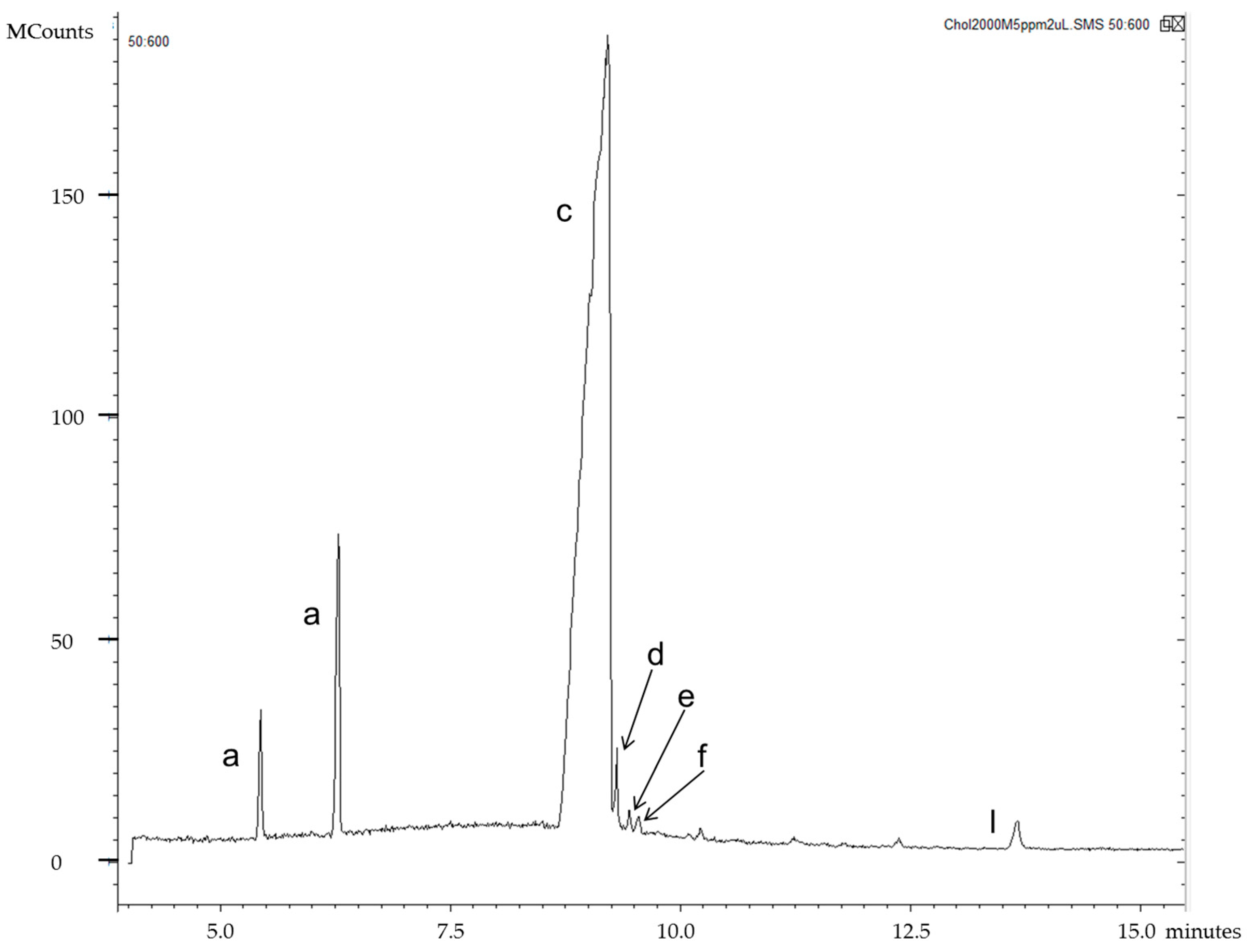
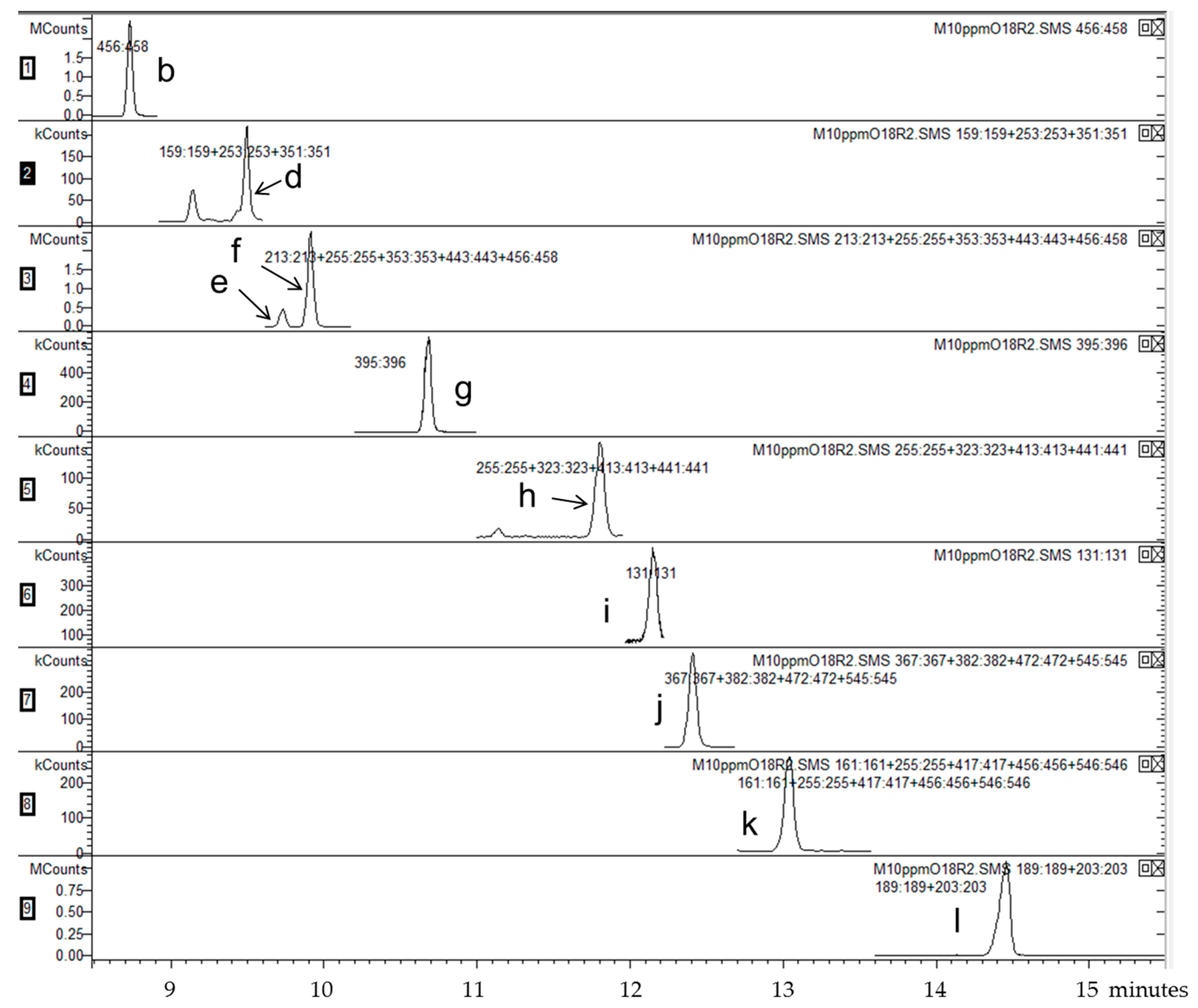
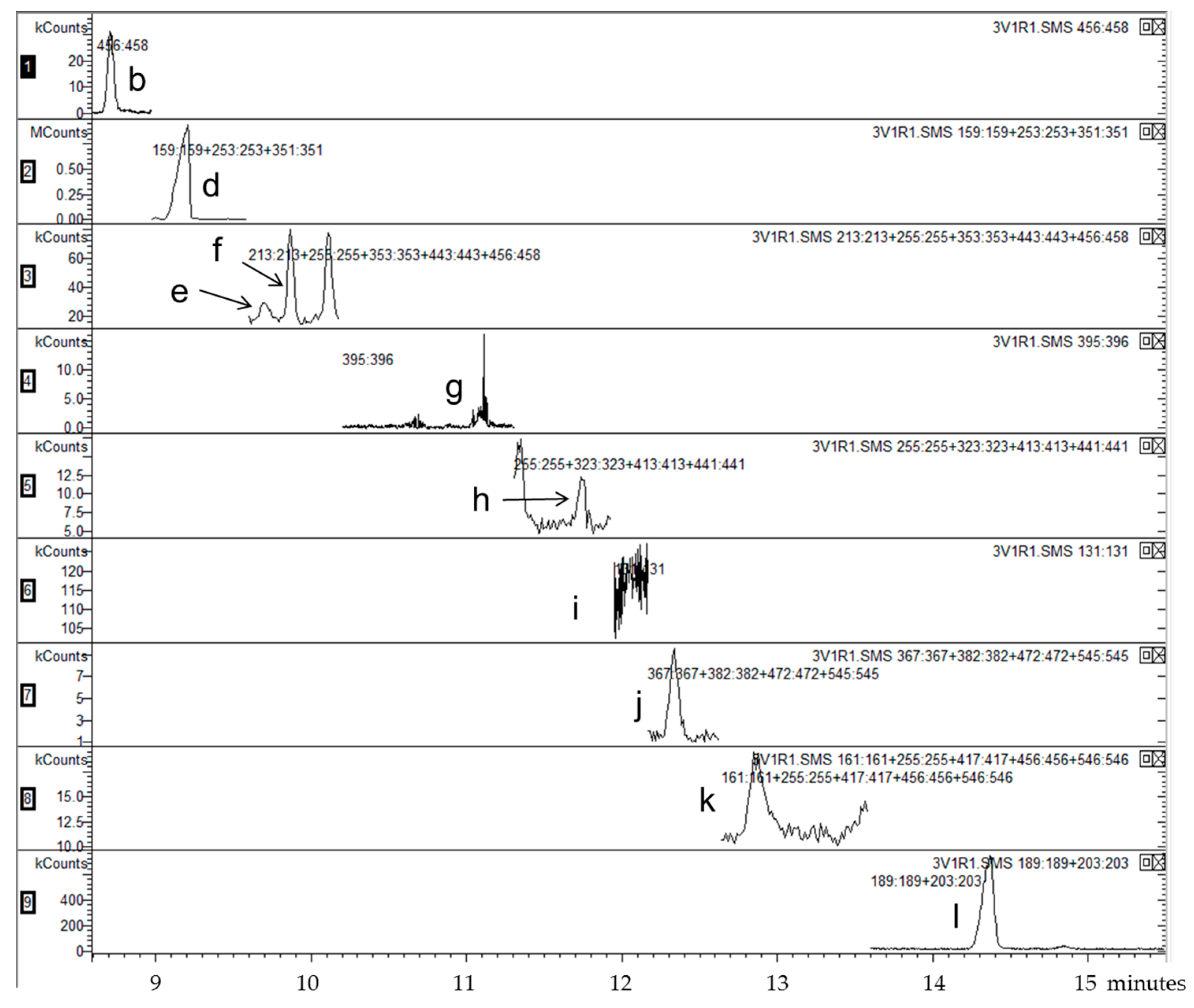
| Segment | Time Range (min) | m/z Selected | Analyte |
|---|---|---|---|
| 1 | 4.00–8.98 | 456–458 | 7αOHChol |
| 2 | 8.98–9.64 | 159, 253, 351 | Desm |
| 3 | 9.64–10.20 | 213, 255, 353, 443, 456–458 | Latho, 7βOHChol |
| 4 | 10.20–11.31 | 395–396 | Lano |
| 5 | 11.31–11.99 | 255, 323, 413, 441 | 24OHChol |
| 6 | 11.99–12.25 | 131 | 25OHChol |
| 7 | 12.25–12.70 | 367, 382, 472, 545 | 7KetoChol |
| 8 | 12.70–13.60 | 161, 255, 417, 456, 546 | 27OHChol |
| 9 | 13.60–15.50 | 189, 203 | betulin (IS) |
| Standard | m/z | tR (min) |
|---|---|---|
| 7α-hydroxycholesterol | 456, 457 | 8.73 |
| cholesterol | 368, 329, 133, 353, 145 | 9.14 |
| desmosterol | 253, 351, 159, 456 | 9.49 |
| lathosterol | 255, 458, 213, 443, 353 | 9.74 |
| 7β-hydroxycholesterol | 456, 457 | 9.92 |
| lanosterol | 395, 396 | 10.68 |
| 24(S)-hydroxycholesterol | 145, 323, 413, 159, 441, 255, 546 | 11.82 |
| 25-hydroxycholesterol | 131, 145 | 12.17 |
| 7-ketocholesterol | 472, 367, 382, 545, 161 | 12.40 |
| 27-hydroxycholesterol | 456, 417, 161, 546, 255, 441, 129 | 13.06 |
| betulin (IS) | 189, 203, 496 | 14.47 |
| Compound | RF | LIN (%) | LOD (µg/mL) | LOQ (µg/mL) | Recovery Factor (%) | Matrix Effect (%) | Repeatability | |
|---|---|---|---|---|---|---|---|---|
| Consecutive Measurements | Overall Process | |||||||
| 7αOH Chol | 0.115 | 98.2 | 0.01 | 0.03 | 83.8 | 97.3 | 0.106 | 0.200 |
| Desm | 0.013 | 98.7 | 0.01 | 0.03 | 74.2 | 96.9 | 0.073 | 0.166 |
| Latho | 0.025 | 98.0 | 0.12 | 0.37 | 67.7 | 104.8 | 0.088 | 0.277 |
| 7βOH Chol | 0.100 | 98.8 | 0.01 | 0.03 | 67.0 | 100.1 | 0.159 | 0.271 |
| Lano | 0.040 | 98.6 | 0.02 | 0.07 | 72.7 | 98.0 | 0.148 | 0.170 |
| 24OH Chol | 0.0053 | 98.0 | 0.14 | 0.44 | 71.0 | 96.5 | 0.181 | 0.228 |
| 25OH Chol | 0.022 | 96.5 | 0.35 | 1.06 | 97.8 | 99.4 | 0.146 | 0.294 |
| 7Keto Chol | 0.020 | 97.9 | 0.07 | 0.20 | 73.2 | 95.5 | 0.114 | 0.197 |
| 27OH Chol | 0.023 | 98.5 | 0.02 | 0.07 | 65.8 | 97.7 | 0.059 | 0.203 |
| Data | 7αOH Chol | Desm | Latho | 7βOH Chol | Lano | 24OH Chol | 25OH Chol | 7Keto Chol | 27OH Chol | |
|---|---|---|---|---|---|---|---|---|---|---|
| TA1 | Average (μg/mL) | 0.76 | 0.25 | d (0.13) | 0.80 | nd | 1.28 | nd | 2.92 | 0.39 |
| RSD (%) | 5.51 | 30.08 | 33.67 | 0.89 | - | 14.75 | - | 3.90 | 14.74 | |
| TA2 | Average (μg/mL) | 0.22 | 0.28 | 0.46 | 0.41 | nd | 1.06 | nd | 1.62 | 0.87 |
| RSD (%) | 3.80 | 31.19 | 22.59 | 18.89 | - | 8.50 | - | 8.10 | 33.02 | |
| TA3 | Average (μg/mL) | 0.70 | 0.39 | 0.39 | 0.76 | nd | 0.99 | nd | 2.99 | 0.84 |
| RSD (%) | 9.48 | 7.61 | 27.30 | 9.16 | - | 11.98 | - | 9.14 | 22.16 | |
| TB1 | Average (μg/mL) | 0.14 | 0.19 | 0.49 | 0.19 | nd | 0.76 | nd | d (0.17) | 0.51 |
| RSD (%) | 19.53 | 30.19 | 21.59 | 21.76 | - | 17.01 | - | 27.23 | 26.05 | |
| TB2 | Average (μg/mL) | 0.42 | nd | d (0.19) | 0.35 | nd | 2.01 | nd | 1.70 | 0.42 |
| RSD (%) | 0.24 | - | 30.19 | 10.11 | - | 13.94 | - | 11.17 | 10.57 | |
| TB3 | Average (μg/mL) | 0.11 | nd | nd | 0.11 | nd | 1.04 | nd | 0.53 | 0.35 |
| RSD (%) | 5.24 | - | - | 11.97 | - | 1.36 | - | 9.00 | 28.14 | |
| TC1 | Average (μg/mL) | 0.061 | nd | nd | 0.04 | nd | 0.72 | nd | d (0.16) | 0.26 |
| RSD (%) | 9.74 | - | - | 10.26 | - | 20.86 | - | 11.56 | 23.14 | |
| TC2 | Average (μg/mL) | 0.15 | nd | d (0.20) | 0.13 | nd | 0.85 | nd | 0.87 | 0.24 |
| RSD (%) | 5.33 | - | 29.67 | 22.95 | - | 19.91 | - | 2.01 | 30.00 | |
| TC3 | Average (μg/mL) | 0.057 | nd | d (0.34) | d (0.016) | nd | 0.98 | nd | 0.56 | 0.31 |
| RSD (%) | 1.99 | - | 24.09 | 21.72 | - | 19.98 | - | 21.57 | 22.01 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valverde-Som, L.; Carrasco-Pancorbo, A.; Sierra, S.; Santana, S.; Ruiz-Samblás, C.; Navas, N.; Burgos, J.S.; Cuadros-Rodríguez, L. Separation and Determination of Some of the Main Cholesterol-Related Compounds in Blood by Gas Chromatography-Mass Spectrometry (Selected Ion Monitoring Mode). Separations 2018, 5, 17. https://doi.org/10.3390/separations5010017
Valverde-Som L, Carrasco-Pancorbo A, Sierra S, Santana S, Ruiz-Samblás C, Navas N, Burgos JS, Cuadros-Rodríguez L. Separation and Determination of Some of the Main Cholesterol-Related Compounds in Blood by Gas Chromatography-Mass Spectrometry (Selected Ion Monitoring Mode). Separations. 2018; 5(1):17. https://doi.org/10.3390/separations5010017
Chicago/Turabian StyleValverde-Som, Lucia, Alegría Carrasco-Pancorbo, Saleta Sierra, Soraya Santana, Cristina Ruiz-Samblás, Natalia Navas, Javier S. Burgos, and Luis Cuadros-Rodríguez. 2018. "Separation and Determination of Some of the Main Cholesterol-Related Compounds in Blood by Gas Chromatography-Mass Spectrometry (Selected Ion Monitoring Mode)" Separations 5, no. 1: 17. https://doi.org/10.3390/separations5010017
APA StyleValverde-Som, L., Carrasco-Pancorbo, A., Sierra, S., Santana, S., Ruiz-Samblás, C., Navas, N., Burgos, J. S., & Cuadros-Rodríguez, L. (2018). Separation and Determination of Some of the Main Cholesterol-Related Compounds in Blood by Gas Chromatography-Mass Spectrometry (Selected Ion Monitoring Mode). Separations, 5(1), 17. https://doi.org/10.3390/separations5010017