1. Introduction
In recent years, a new lipoglycopeptide antibiotic named DBV has been approved for the treatment of ABSSSI [
1]. DBV is a semi-synthetic antibiotic with a structure very similar to that of teicoplanin: the most important improvement is the addition of an extended lipophilic side chain that allows DBV dimerization and anchoring to the bacterial cell membrane, thus enhancing its potency, prolonging its half-life and allowing for extended dosing intervals [
2,
3,
4,
5].
As with other glycopeptides, DBV acts by inhibiting the formation of peptidoglycan cross-linking in Gram-positive bacteria through interaction with terminal D-alanyl-D-alanine residues of peptidoglycan precursors, causing a natural resistance mechanism in Gram-negative ones. This antibiotic was shown to be particularly effective even against methicillin-resistant Staphylococcus aureus (MRSA), and its breakpoint MIC (Minimum Inhibitory Concentration) for both staphylococci of various species and streptococci is reported to be 0.125 mg/L [
6,
7]. Recent studies reported challenges related to the management of vancomycin-based treatments, including its inconvenient dosing (intravenous administration every 6 or 12 h), the occurrence of side effects (in particular nephrotoxity) and the increasing emergence of MRSA strains with reduced susceptibility to this drug [
2,
4,
5]; conversely, up-to-date DBV has never been related to nephrotoxity, it has proved more effective in particular circumstances (e.g., osteomyelitis [
8,
9]), it showed an extremely long half-life, thus allowing a weekly intravenous dosage, and demonstrated a very high AUC (Area Under the Curve)/MIC ratio, reducing the onset of resistant isolates and, sometimes, overcoming eventual resistance to vancomycin [
2,
5,
10,
11].
Several studies accurately described DBV pharmacokinetics both in healthy volunteers and in patients with renal/hepatic impairment [
12,
13,
14,
15]. Assuming a common regimen consisting of a first administration of 1000 mg on day 1 followed by a second dose of 500 mg on day 8 [
13], the expected mean C
max after the first infusion is nearly 250 mg/L for patients with normal renal function, and it was shown to increase in the case of renal dysfunctions [
14]; the same trend has been observed for AUC
0–7days [
14]. All these data reflect a CL reduction in the presence of renal impairment, even more pronounced after day 7; in particular, in the case of severe renal damage, the CL is reduced by half, causing the area under curve AUC
0–∞ to double [
14]. Conversely, patients who undergo regular intermittent hemodialysis (IHD) seem to not require dose adjustment [
5,
14]. Conversely, data about the impact of continuous renal replacement therapy, and in particular, veno-venous hemodiafiltration (CVVHDF) on DBV exposure are still few and controversial [
16,
17,
18].
Considering the peculiar features and the evidence of an AUC/MIC-dependent effect for DBV against both staphylococci and streptococci [
11,
19,
20], TDM could be beneficial in order to optimize its clinical effectiveness.
Nevertheless, several difficulties related to the quantification of DBV in plasma samples have been previously reported: in fact, it is well known that DBV shows extensive plasma protein binding (approximately 93%) that may cause drug co-precipitation during the extraction procedure; moreover, in the same work, a non-specific binding (NSB) to plastics has been reported [
21]. Moreover, the currently published methods for DBV quantification are based on the use of highly expensive platforms and reagents, such as Ultra-High-Performance Liquid Chromatography (UHPLC) and tandem mass spectrometry (MS/MS), using isotope-labeled internal standards or analogues that are not commercially available [
12,
15,
21,
22,
23]. On the other hand, some of these methods were only validated on rat plasma [
22,
23] and/or their described validation process lacked recovery or matrix effect data [
12,
15,
22], which can be extremely critical for the ruggedness of a method, particularly considering a molecule such as DBV.
In this context, considering the challenges related to DBV determination in plasma and the wide need for a cheap yet reliable quantification method, a novel High-Performance Liquid Chromatography method coupled to single mass spectrometry (HPLC-MS) has been developed and validated in accordance with FDA and EMA guidelines [
24,
25] and finally applied for use in TDM on incurred samples from patients receiving an off-label posology (two intravenous infusions of 1500 mg, once weekly for two weeks, instead of the usual 1000 + 500 mg schedule [
26]) to treat osteoarticular infections, with normal or altered renal function.
2. Materials and Methods
2.1. Chemicals
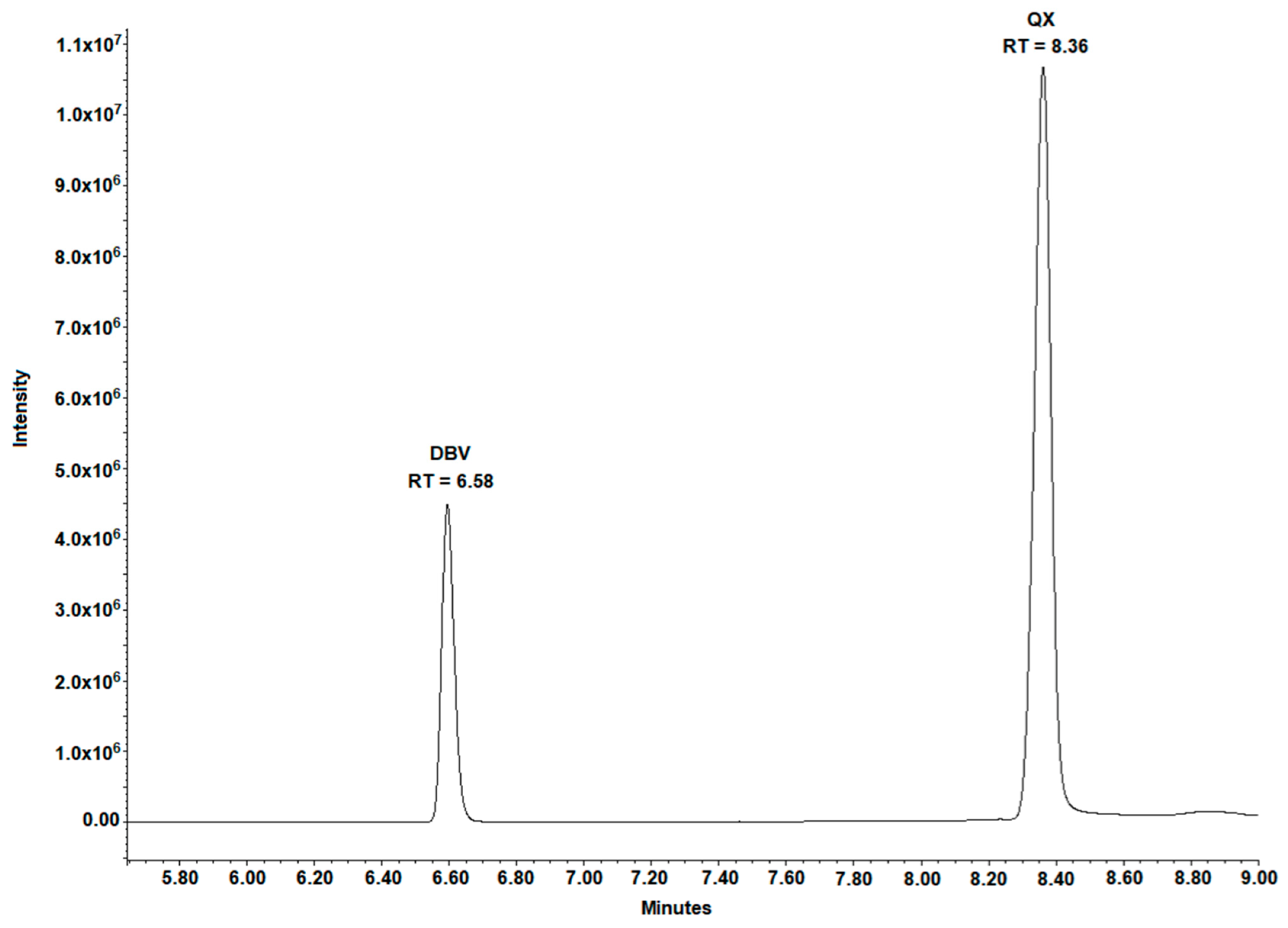
HPLC-grade acetonitrile (ACN) and methanol (MeOH) were purchased from VWR (Milan, Italy). HPLC-grade H2O was produced using a Milli-DI system coupled with a Synergy 185 system by Millipore (Milan, Italy). Blank plasma from healthy donors was acquired from the Blood Bank of ASL Città della Salute di Torino (Turin, Italy). Formic and phosphoric acids, 6,7-dimethyl-2,3-di(2-pyridyl) quinoxaline (QX) (purity 99.9%), the Internal Standard (IS), L-arginine (purity 99%), and Triton-X-100 were purchased from Sigma-Aldrich Corporation (Milan, Italy). DBV (purity 99%) was kindly donated by CoQua Lab (Turin, Italy). DBV pure powder was stored at −20 °C, QX was stored at +4 °C, and L-arginine at room temperature, in the dark in order to prevent any possible degradation.
2.2. Preliminary Experiments
In this work, we aimed to develop a sample preparation method with an optimized protein precipitation solution: this was performed by testing different combinations and proportions of ACN and MeOH. Moreover, in order to evaluate pH-dependent DBV solubility and protein binding, different percentages of phosphoric acid were added to the IS working solution and tested. Finally, in order to prevent DBV NSB to vial walls, both Triton-X-100 at 1% and different concentrations of L-arginine were evaluated. Optimization of the target single-ion recording (SIR) traces for DBV and IS was performed through direct continuous infusion into the MS source.
2.3. Stock Solutions, Internal Standard, Standards, and Quality Controls
DBV stock solution was prepared at the concentration of 5 mg/mL by diluting the powder in a mixture of dimethyl sulfoxide (DMSO) and MeOH 50:50 (v:v), while QX was prepared at the final concentration of 1 mg/mL in a mixture of MeOH:H2O 90:10 (v:v).
DBV and QX stock solutions were stored at −80 °C and +4 °C, respectively, for a maximum of six months. Standard 9 (STD 9) and quality control (QC) aliquots were prepared by independently spiking blank plasma from healthy donors with DBV stock solution and were stored at −80 °C for a maximum of six months. Calibration ranges and QC concentrations, chosen considering literature-derived data [
7], are reported in
Table 1. IS working solution was prepared before each analytical session by diluting 5 µL of QX stock solution (final concentration: 1250 ng/mL) and 40 µL of phosphoric acid (final concentration: 1%) in 4 mL of H
2O: MeOH 50:50 (
v:v).
2.4. Sample Preparation
The extraction protocol consisted of protein precipitation followed by dilution. In detail, 50 µL of IS was added to 50 µL of plasma sample and 300 µL of ACN:MeOH 50:50 (v:v). All the samples were then vortex mixed and centrifuged: 200 µL of the supernatant was then diluted with 800 µL of pure water. Finally, 5 µL of L-arginine stock solution (50 mg/mL in H2O) was added to each sample before injection into the HPLC-MS system. The injection volume was 5 µL.
2.5. HPLC-MS Instrument and Chromatographic Conditions
The HPLC-MS instrument consisted of a Waters (Milan, Italy) 1525 binary pump, a degasser, a CTC PAL autosampler and an Altus
® SQ detector (a single quadrupole mass spectrometer, Perkin Elmer, Milan, Italy). Empower 3 Pro
® software (year 2007 version, Waters, Milan, Italy) was used for data processing. Chromatographic separation was performed on an Atlantis
® T3 5 µm 4.6 × 150 mm column (Waters, Milan, Italy), protected by a SecurityGuard
® with a C18 (4.0 mm × 3.0 mm) pre-column (Phenomenex, Torrance, CA, USA) and maintained at 45 °C using a column oven. Separation was obtained with a gradient (
Table 2) of mobile phases A (MP-A, 0.05%
v/
v formic acid in HPLC-grade H
2O) and B (MP-B 0.05%
v/
v formic acid in HPLC-grade ACN). A volumetric “T-switch” was used to discard 70% of the flow from the analytical column, only allowing 300 µL/min to enter the MS source. The instrument was settled in positive electrospray ionization mode (ESI+) for DBV and IS (QX). The general mass settings are presented in
Table 3.
2.6. Accuracy, Precision, Limits of Quantification, Detection and Dilution Integrity
Accuracy and inter-day precision were evaluated, as requested by the FDA and EMA guidelines [
24,
25]. Six validation sessions were performed and three different QCs samples were quantified in duplicate during each analysis; intra-day precision was evaluated in five replicates during a single analytical session. Inter-day and intra-day imprecision were expressed as the relative standard deviation (RSD%) from the nominal value at each QC concentration. Integration was performed considering peak areas. The Upper Limit of Quantification (ULOQ) corresponds to the highest point (STD 9) of the calibration curve; the Limit of Quantification (LOQ) is the lowest amount (STD 1) of each calibration curve; the Lower Limit of Quantification (LLOQ) and the Limit of Detection (LOD) were considered as the lowest dilution of STD 1, which yielded a signal-to-noise ratio higher than 10 and 3, respectively. Moreover, the LLOQ was tested in five intraday and interday replicates in order to test percentage bias and RSD% lower than 20%. Dilution integrity was evaluated by analyzing a QC sample prepared at a concentration of 500 mg/mL (twice higher than the STD 9) after a 3-fold dilution in 5 intra-day replicates.
2.7. Recovery and Extraction Efficiency
Recovery was estimated during six validation sessions at high, medium, and low concentrations by comparing peak areas obtained from the injection of samples spiked with analytes before the extraction (pre-spike) with those originated from the chemical mix spiked with analytes at the same concentration and not extracted. Extraction efficiency was measured through the comparison of pre-spike and other samples spiked at the same concentration after the extraction (post-spike).
2.8. Matrix Effect
The matrix effect was evaluated by comparing the “post-spike” signal at high, medium, and low QC levels in 6 different blank plasma lots with those from the direct injection of the same concentration of analytes in a solution of water–ACN–MeOH (70:15:15
v:v:v), as described by Taylor [
27]. Moreover, the IS-normalized matrix effect (IS-nME), defined as the difference in the “area of the analyte/area of IS” ratio between the same analyte concentrations in matrix extracts and in pure solvents, was evaluated as previously described [
28,
29].
2.9. Carry Over
Carry over was determined by injecting blank samples after the ULOQ calibration standard. Carry over signal in these samples had to be lower than 20% of the LOQ for DBV and 5% for the IS.
2.10. Stability
Stock solutions’ stability was tested after 3 months by comparing the peak areas of fresh stock solutions with old stock solution stored at −80 °C, even considering the NSB phenomenon through the direct injection of diluted (1:1000 v:v) stock solutions in a UHPLC-PDA system (Acquity H-Class® system, Waters, Milan, Italy).
Conversely, the stability in human plasma was evaluated at three QC concentrations using the presented HPLC-MS method. Multiple aliquots of QCs were prepared, analyzed once, and then stored at −80 °C. The effect of “freeze-and-thaw” cycles on the stability of plasma samples was determined by analyzing 2 aliquots at each QC concentration, previously frozen at −80 °C, thawed up to three times and then compared with freshly prepared/extracted QC samples; post-extraction autosampler stability was evaluated through the comparison between QCs analyzed immediately after the extraction and their reanalysis after 7 days in the autosampler (10 °C). The long-term stability of DBV in human plasma was tested up to 2 months at −80 °C.
2.11. Selectivity
The method was tested for its ability to differentiate DBV and IS signal from endogenous components in the matrix or other sample components. Moreover, 12 antiretrovirals (rilpivirine, dolutegravir, cobicistat, darunavir, elvitegravir, atazanavir, tenofovir alafenamide, tenofovir, abacavir, lamivudine, zidovudine, and emtricitabine), 13 antihypertensives (atenolol, doxazosin, ramipril, telmisartan, amlodipine, sacubitril, indapamide, hydrochlorothiazide, valsartan, nebivolol, chlorthalidone, olmesartan, and clonidine), 4 antifungal drugs (fluconazole, voriconazole, posaconazole, and itraconazole), and 3 glycopeptide antibiotics (vancomycin, teicoplanin, and daptomycin) were added to blank samples and to a QC H in order to investigate the presence of interfering peaks at DBV and QX retention times (RTs) or the evidence of significant changes in their signal, respectively.
2.12. Clinical Application
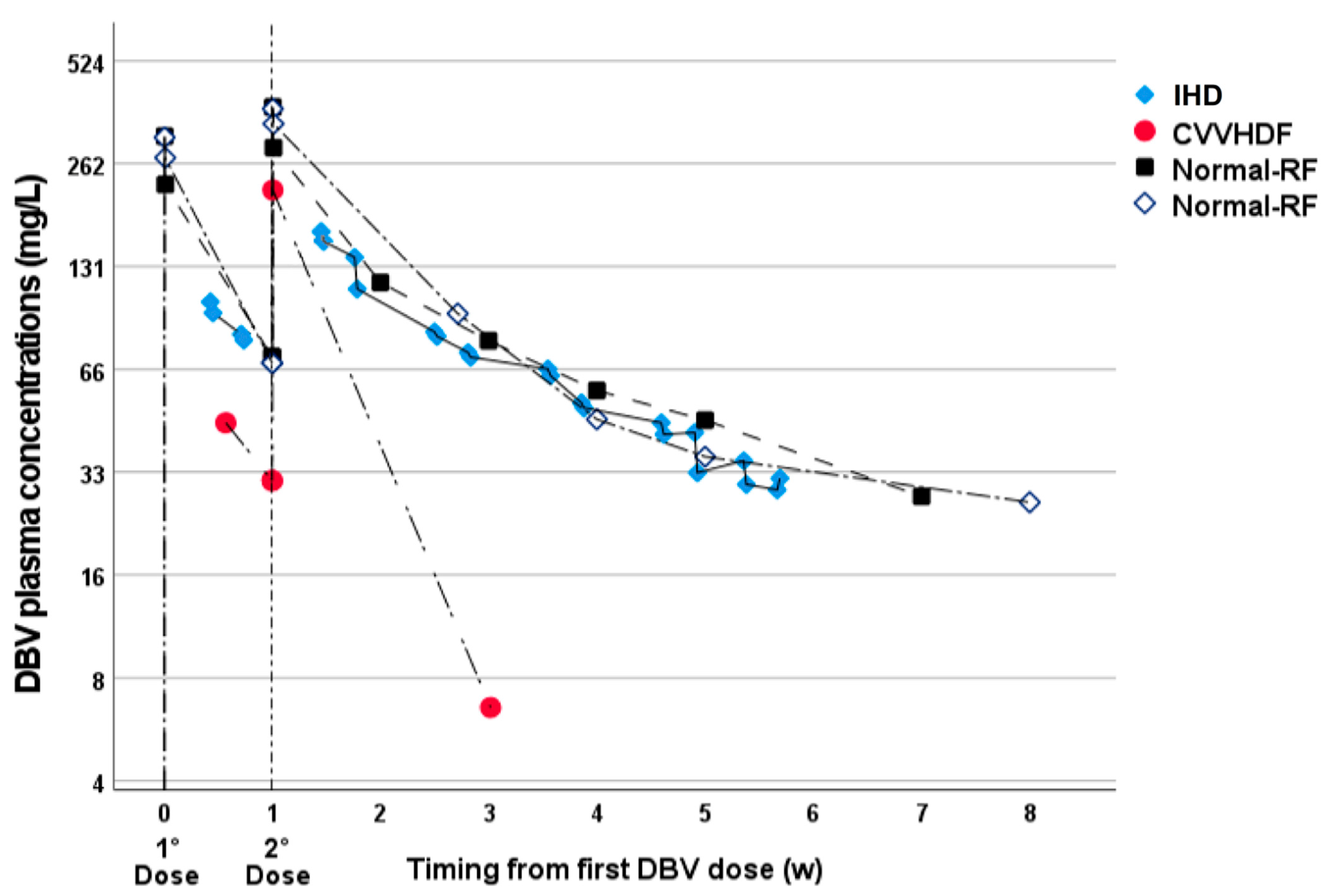
The validated method was applied for routine use in TDM in patients who underwent treatment with two high 1500 mg weekly doses of DBV, in the context of the observational study “Appropriatezza terapeutica della terapia antinfettiva” (ethical approval n. 0040388 23 April 2020). Fifty-two samples were received at the Laboratory of Clinical Pharmacology and Pharmacogenetics of the University of Turin for TDM, from 4 male patients who gave informed consent for the use of TDM data. The mean patients characteristics were: age of 65 years old (range 40–74), weight of 73.5 kg (range 70–85), BMI of 23.2 (range 19.4–25.5); all patients received two high 1500 mg DBV doses in two weeks. For 2 patients, the reason for TDM was altered renal function and renal replacement therapy (eGFR before dialysis 6.2 and 17.4 mL/min), with osteoarticular meticillin-resistant
Staphilococcus aureus (MRSA) infection: one underwent intermittent hemodialysis (IHD, 4 h every 2–3 days, 24 samples, 6 weeks of TDM follow-up) and the other one continuous veno-venous hemodiafiltration (CVVHDF, 4 samples, 3 weeks of follow-up). The other two patients were in treatment for osteoarticular MRSA infection with normal renal function (eGFR 107.5 and 113.8 mL/min). They underwent TDM in order to evaluate the overall exposure over a period of 8 weeks, considering the already-known AUC/MIC-dependent PK/PD effect for DBV [
19,
20]. Blood sampling in lithium heparin tubes for TDM purposes was scheduled before and after DBV infusion and then, for IHD, it was repeated before and after each dialysis session; all other samples were taken at variable timings, based on clinical needs.
2.13. Incurred Sample Reanalysis
In order to assess method reproducibility in real-life specimens, 32 incurred samples were reanalyzed after a period of storage at −80 °C of 2–4 weeks. According to EMA guidelines [
24], percent deviation between the first and second analysis, compared with their mean value, had to fall within at least ±20% for at least 67% of reanalyzed samples.
4. Discussion
Nowadays, TDM has a crucial role in the management of those therapies characterized both by a narrow therapeutic index or by the need to constantly maintain adequate drug concentrations in blood, especially higher than the MIC, such as in the case of antibiotics.
Currently, very few data about DBV pharmacokinetics, and its off-label applications, have been reported in the literature. Furthermore, analytical procedures reported in the literature are sometimes discordant due to the chemical–physical properties of the molecule.
In this study, a new HPLC-MS method was validated in accordance with FDA and EMA guidelines, for DBV quantification in human plasma. This analytical protocol was designed in order to warrant the maximum adaptability to a clinical routine context, even in laboratories which lack expensive UHPLC-MS/MS instrumentation or cannot afford the use of isotope-labeled IS. In fact, to our knowledge, to date, only
2H
6-DBV is available on the market and it is extremely expensive (more than 1000 euros for 1 mg); moreover, for molecules characterized by a very high molecular weight, biprotonation with ESI+ and two chlorine atoms (polyalogens), the use of isotope-linked IS has to be managed with high caution due to the high risk of significant cross-talk when MS analysis with unit mass resolution is adopted (natural isotopes of the unlabeled analyte can partially overlap with the MS spectrum of the labeled IS). The choice of the concentration range used for the calibration curve and QCs was challenging; we focused on published data [
7] reporting that C
max of the two-dose regimen (1000 mg on day 1 followed by 500 mg on day 8) can reach 278,000 ng/mL, and chose 250,000 ng/mL for CAL 9.
Since DBV has a strong affinity with plasma proteins, which could cause its co-precipitation during sample preparation, a previous work proposed the dilution of samples instead of protein precipitation [
21]. Nevertheless, this approach could result in the suboptimal release of DBV from plasma proteins before LC-MS analysis which, other than drastically reducing analyte concentration in the final extract, could potentially increase the effect of NSB to the vials.
Our preliminary experiments evaluating the best precipitating solution indicated that ACN:MeOH 50:50 (v:v) and 100%MeOH yield superior performance in terms of analyte recovery compared to ACN%; in turn, ACN:MeOH 50:50 (v:v) resulted in a clearer supernatant, with a reduced impact on analyte recovery and was therefore selected as the precipitating solution. The experiments regarding the effect of phosphoric acid revealed that the addition of 1% yielded a 20% increase in the analyte’s recovery compared with samples without phosphoric acid, while higher percentages (2% and 5%) did not result in a further increase in the chromatographic signal. Finally, comparative experiments to contrast DBV NSB showed that the addition of 1% Triton-X-100 had a similar effect to that of 5 µL of L-arginine (50 mg/mL). Nevertheless, significantly higher background noise was observed in the chromatograms of samples treated with Triton-X-100, suggesting a possible increase in soluble contaminants within the extracts. For this reason, L-arginine was chosen as an additive to competitively decrease DBV NSB. Regarding the optimization of SIR traces, adducts with two and one proton were selected for DBV and IS, respectively. Concerning DBV, the effect of the presence of two chlorine atoms within its chemical structure caused the exact mass +2 daltons to be the most represented and stable ion to be recorded; therefore, the final selected SIR corresponded to the formula (1814.6 + 2 + 2)/2 = 909.3.
All validation parameters were satisfactory according to the guidelines. Accuracy and precision evaluations totally fulfilled the validation purposes. The relatively low extraction efficiency could be partially explained by the extensive binding of DBV to plasma proteins. Other previous works which described DBV PK, using the LC-MS/MS technique for quantification, used acetonitrile for protein precipitation and did not report all the extraction efficiency and recovery data [
12,
15,
22], while another recent work evidenced poor recovery by using acetonitrile as a precipitating solution, in perfect accordance with our results [
23]. In detail, only Zhu et al. [
23] reported better recoveries (nearly 75%) with MeOH as a precipitating solution, while Alebich-Kolbah et al. [
21] chose to avoid protein precipitation, using an extreme dilution for sample preparation; nevertheless, this approach is viable only with instruments with a very high sensitivity and, moreover, could lead to problems in the long term due to the effect of remaining proteins in the final extract. Concerning the present study, the reproducibility of the extraction efficiency with ACN-: MeOH indicated that the impact on the analytical result remained acceptable, according to EMA and FDA guidelines, since the calibration curve and QC samples were prepared in the same matrix as the real-life specimens. Similarly, ion suppression was present but well reproducible among samples, even at different concentrations. The evaluation of IS-nME showed an even higher reproducibility, indicating that, despite the low chemical similarity between DBV and QX, this can be used as an acceptable IS, particularly according to EMA guidelines and previous works [
24,
28]. In any case, the described protocol can be easily modified in the future with the adoption of a stable isotope-linked IS or with a suitable analogue IS in order to further improve its analytical performance.
Interestingly, the quantification by single-MS spectrometry was possible even at high concentration ranges due to the high molecular mass of DBV (and therefore the lower molar concentration), the adoption of a strong dilution factor during sample preparation (40×), and the use of a low injection volume with high mobile phases flow (1 mL/min), providing further analyte dilution before the MS source. The use of a high flow was successfully adapted for use with ESI+ through the diversion of nearly 70% of the mobile phase flow, allowing a final flow 300 µL/min to enter the source, within the optimal range for ESI.
Unfortunately, to date, no acknowledged therapeutic ranges are known for DBV, since it shows AUC/MIC-dependent kinetics and because no concentration-dependent toxicity was described in the literature, although higher incidence of rash was described at very high doses. Currently, the protein-binding adjusted breakpoint MIC (0.125 mg/L divided by 7% of free fraction = 1786 mg/L) can serve as a reference value under which the growth of DBV-sensitive bacteria could be not successfully inhibited, as reported in some works and in accordance with experimental data by Leighton et al. [
20,
30]. Nevertheless, it is important to note that the vast majority of the staphylococci and streptococci isolates show MIC values abundantly lower than 0.125 ng/mL (most often 0.060 and sometimes 0.030 mg/L); therefore, total DBV higher than 450 or 900 ng/mL could still be active.
The observed LLOQ and LOD allow one to even measure concentrations considerably lower than DBV MIC adjusted by plasma protein binding for the vast majority of bacterial strains, confirming the suitability of this method for the long-term evaluation of DBV concentrations. Moreover, the absence of significant carry over allows one to analyze both peak and trough concentrations within the same analytical session; it is worth noting that, in the majority of cases, the 1500 mg dose administration resulted in DBV Cmax higher than the ULOQ (250 mg/L). In these cases, accurate DBV quantification was possible after 3-fold dilution, as confirmed by the dilution integrity experiments. Concerning stability, the observed data were crucial to prove DBV robustness in several analytical settings.
Compared with previous methods [
12,
15,
21,
22,
23], the one proposed in this work provides an extremely cheap and reliable alternative, being based on HPLC and single-MS detection, without the use of labeled IS or unavailable analogues, but also provides a very thorough description of the validation process, with particular attention to the determination of the variability in extraction efficiency and matrix effect. Obviously, the drawback of the use of HPLC is a generally longer runtime for each injection, but this drawback is well balanced with the ruggedness of the validation process in human plasma and the potential for wider use in laboratories which lack UHPLC-MS/MS instrumentation.
The application of this method on incurred clinical samples for use in TDM showed a clinically relevant impact of CVVH on DBV exposure. On the other hand, IHD of 4 h every 2–4 days resulted in an overall exposure comparable with the one observed in patients with normal renal function. Based on these results, the application of a dual high dose of DBV appeared to be suitable for use with IHD, while particular attention should be paid to CVVHDF. Deepening this issue, a recent study showed evidence that the clearance of DBV can vary significantly among patients who undergo continuous renal replacement therapy, particularly based on the albuminemia [
16]. The observed data in CVVHDF in this work are in good accordance with the ones from Corona et al., but in this case, we highlighted that the increase in DBV clearance in CVVHDF are high enough to reduce the overall exposure in a clinically significant manner, even in the absence of hypoalbuminemia, compared with patients with normal renal function.
In this setting, the presented method was shown to be clinically useful for the rapid identification of the CVVHDF-related reduction in DBV exposure, promptly guiding a treatment switch before reaching a DBV concentration below the theoretical protein-binding adjusted breakpoint MIC.


 ,
, 



{kind=link}
{kind=link}