
Multi-Target Analysis and Suspect Screening of Xenobiotics in Milk by UHPLC-HRMS/MS

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Milk Samples
2.3. Extraction of Xenobiotics
2.4. Clean-Up
2.5. UHPLC-qOrbitrap Analysis
2.6. Target Analysis and Suspect Screening
2.7. Method Validation
3. Results and Discussion
3.1. Protein Precipitation Optimization
3.2. Fluoroquinolones, EDTA and Oasis HLB Optimization
3.3. Addition of Salts and EDTA to the Extraction Solvent
3.4. Validation
3.4.1. Instrumental and Procedural Limits of Quantification (LOQs)
3.4.2. Linearity-Ranges and Determination-Coefficients (r2)
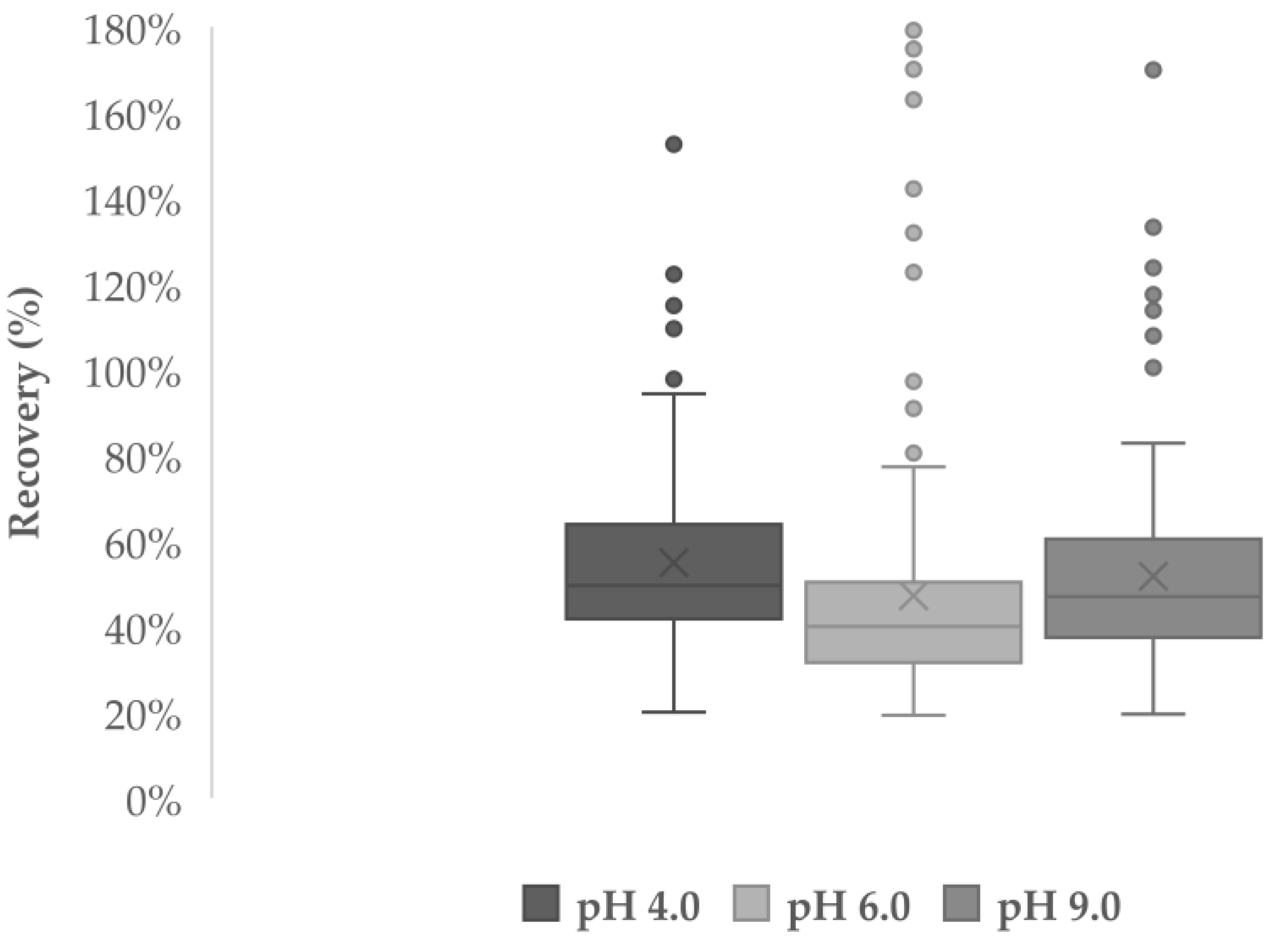
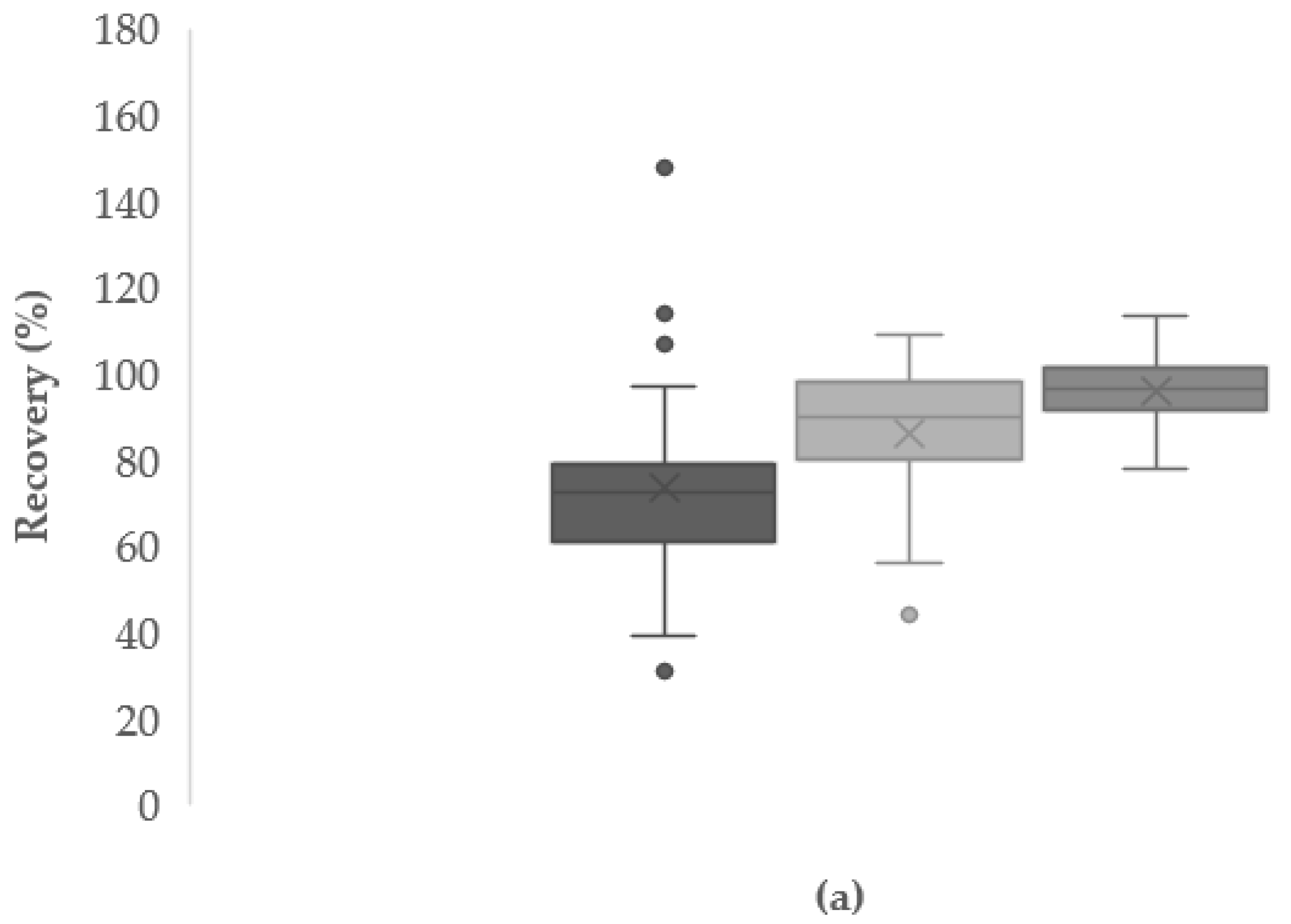
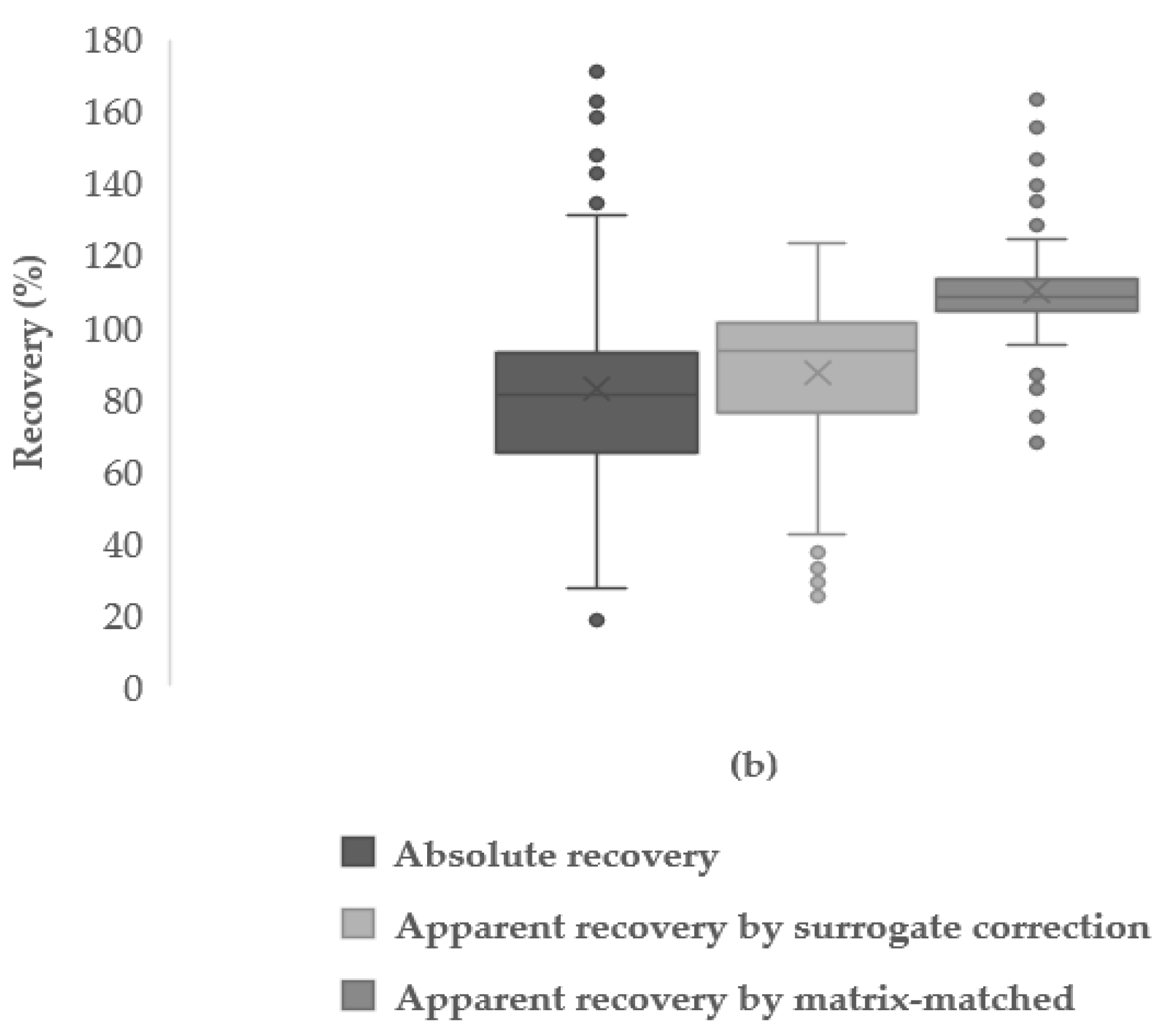
3.4.3. Absolute and Apparent Recoveries
3.4.4. Instrumental and Procedural Repeatability
3.4.5. Instrumental and Procedural Limits of Identification (LOIs)
3.5. Target Analysis of Commercial and Breast Milk Samples
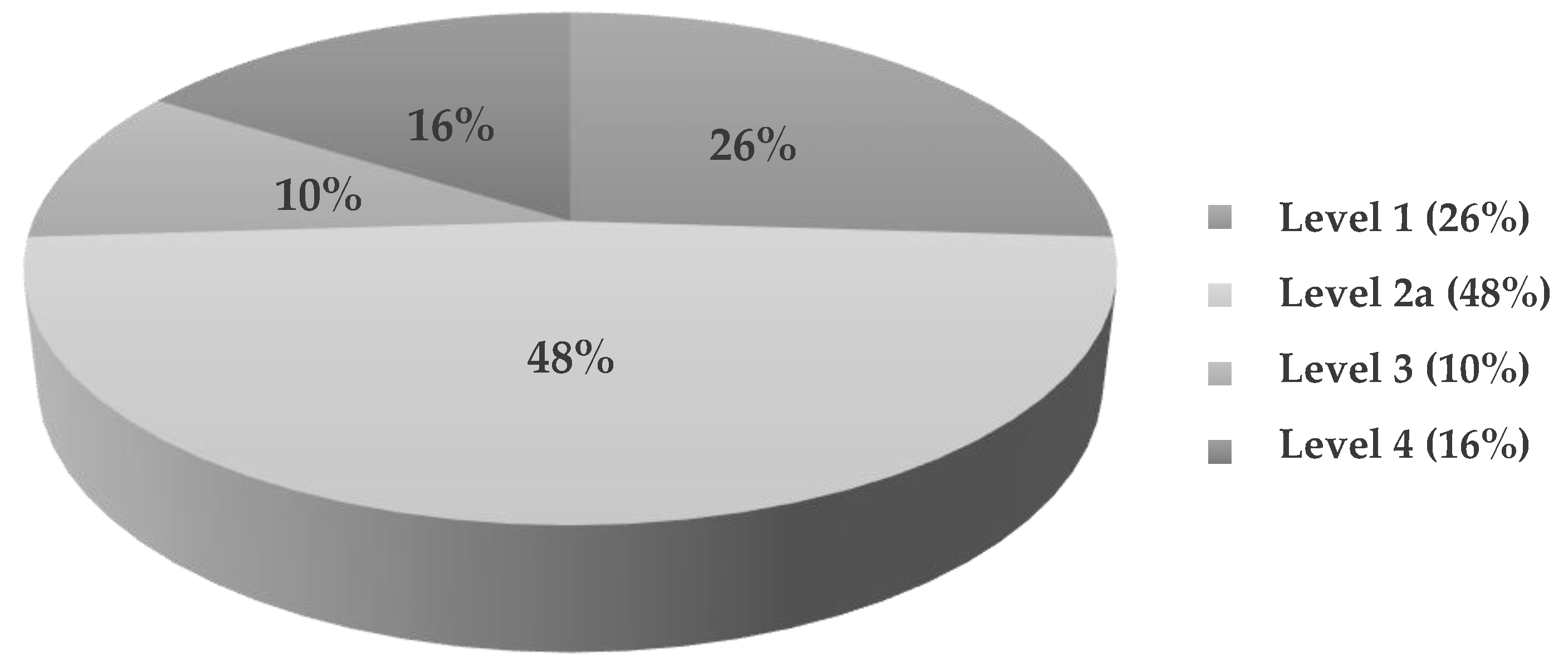
3.6. Suspect Analysis of Commercial and Breast Milk Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Aceña, J.; Heuett, N.; Gardinali, P.; Pérez, S. Chapter 12: Suspect screening of pharmaceuticals and related bioactive compounds, their metabolites and their transformation products in the aquatic environment, biota and humans using LC-HR-MS techniques. In Applications of Time-of-Flight and Orbitrap Mass Spectrometry in Environmental, Food, Doping, and Forensic Analysis; Pérez, S., Eichhorn, P., Barceló, D., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 357–378. [Google Scholar]
- Chiaia-Hernandez, A.C.; Krauss, M.; Hollender, J. Screening of lake sediments for emerging contaminants by liquid chromatography atmospheric pressure photoionization and electrospray ionization coupled to high resolution mass spectrometry. Environ. Sci. Technol. 2013, 47, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Rappaport, S.M. Implications of the exposome for exposure science. J. Expo. Sci. Environ. Epidemiol. 2011, 21, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Wild, C.P. Complementing the genome with an “exposome”: The outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1847–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennis, K.K.; Jones, D.P. The exposome: A new frontier for education. Am. Biol. Teach. 2016, 78, 542–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Balshaw, D.M.; Kwok, R.K.; Thompson, C.L.; Collman, G.W.; Birnbaum, L.S. The exposome: Embracing the complexity for discovery in environmental health. Environ. Health Perspect. 2016, 124, A137–A140. [Google Scholar] [CrossRef] [Green Version]
- Escher, B.I.; Hackermüller, J.; Polte, T.; Scholz, S.; Aigner, A.; Altenburger, R.; Böhme, A.; Bopp, S.K.; Brack, W.; Busch, W.; et al. From the exposome to mechanistic understanding of chemical-induced adverse effects. Environ. Int. 2017, 99, 97–106. [Google Scholar] [CrossRef]
- Patel, C.J.; Bhattacharya, J.; Butte, A.J. An Environment-Wide Association Study (EWAS) on type 2 diabetes mellitus. PLoS ONE 2010, 5, e10746. [Google Scholar] [CrossRef]
- Vineis, P.; Chadeau-Hyam, M.; Gmuender, H.; Gulliver, J.; Herceg, Z.; Kleinjans, J.; Kogevinas, M.; Kyrtopoulos, S.; Nieuwenhuijsen, M.; Phillips, D.H.; et al. The exposome in practice: Design of the EXPOsOMICS project. Int. J. Hyg. Environ. Health 2017, 220, 142–151. [Google Scholar] [CrossRef]
- Andra, S.S.; Austin, C.; Patel, D.; Dolios, G.; Awawda, M.; Arora, M. Trends in the application of high-resolution mass spectrometry for human biomonitoring: An analytical primer to studying the environmental chemical space of the human exposome. Environ. Int. 2017, 100, 32–61. [Google Scholar] [CrossRef] [Green Version]
- Exposure and Health. HBM4EU—Science and Policy for a Healthy Future. Available online: https://www.hbm4eu.eu/the-project/exposure-and-health/ (accessed on 2 July 2020).
- Yusa, V.; Millet, M.; Coscolla, C.; Roca, M. Analytical methods for human biomonitoring of pesticides. A review. Anal. Chim. Acta 2015, 891, 15–31. [Google Scholar] [CrossRef]
- Siddique, S.; Kubwabo, C.; Harris, S.A. A review of the role of emerging environmental contaminants in the development of breast cancer in women. Emerg. Contam. 2016, 2, 204–219. [Google Scholar] [CrossRef]
- García Lara, N.R.; Peña Caballero, M. Riesgos asociados al uso no controlado de la leche materna donada. An. Pediatría 2017, 86, 237–239. [Google Scholar] [CrossRef] [PubMed]
- Irazusta, A. High-pressure homogenization and high hydrostatic pressure processing of human milk: Preservation of immunological components for human milk banks. J. Dairy Sci. 2020, 103, 14. [Google Scholar] [CrossRef] [PubMed]
- Stefanidou, M.; Maravelias, C.; Spiliopoulou, C. Human exposure to endocrine disruptors and breast milk. Endocr. Metab. Immune Disord. Drug Targets 2009, 9, 269–276. [Google Scholar] [CrossRef]
- Jadhav, M.R.; Pudale, A.; Raut, P.; Utture, S.; Shabeer, T.A.; Banerjee, K. A unified approach for high-throughput quantitative analysis of the residues of multi-class veterinary drugs and pesticides in bovine milk using LC-MS/MS and GC–MS/MS. Food Chem. 2019, 272, 292–305. [Google Scholar] [CrossRef]
- Beyene, T. Veterinary drug residues in food-animal products: Its risk factors and potential effects on public health. J. Vet. Sci. Technol. 2015, 7. [Google Scholar] [CrossRef]
- Díaz, R.; Ibáñez, M.; Sancho, J.V.; Hernández, F. Target and non-target screening strategies for organic contaminants, residues and illicit substances in food, environmental and human biological samples by UHPLC-QTOF-MS. Anal. Methods 2012, 4, 196–209. [Google Scholar] [CrossRef] [Green Version]
- Plassmann, M.M.; Brack, W.; Krauss, M. Extending analysis of environmental pollutants in human urine towards screening for suspected compounds. J. Chromatogr. A 2015, 1394, 18–25. [Google Scholar] [CrossRef]
- Nickerson, K. Environmental contaminants in breast milk. J. Midwifery Women Health 2006, 51, 26–34. [Google Scholar] [CrossRef]
- Cano-Sancho, G.; Alexandre-Gouabau, M.-C.; Moyon, T.; Royer, A.-L.; Guitton, Y.; Billard, H.; Darmaun, D.; Rozé, J.-C.; Boquien, C.-Y.; Le Bizec, B.; et al. Simultaneous exploration of nutrients and pollutants in human milk and their impact on preterm infant growth: An integrative cross-platform approach. Environ. Res. 2020, 182, 109018. [Google Scholar] [CrossRef]
- Tran, C.D.; Dodder, N.G.; Quintana, P.J.E.; Watanabe, K.; Kim, J.H.; Hovell, M.F.; Chambers, C.D.; Hoh, E. Organic contaminants in human breast milk identified by non-targeted analysis. Chemosphere 2020, 238, 124677. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Gómez, R.; Jiménez-Díaz, I.; Zafra-Gómez, A.; Ballesteros, O.; Navalón, A. A multiresidue method for the determination of selected endocrine disrupting chemicals in human breast milk based on a simple extraction procedure. Talanta 2014, 130, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Baduel, C.; Mueller, J.F.; Tsai, H.; Gomez Ramos, M.J. Development of sample extraction and clean-up strategies for target and non-target analysis of environmental contaminants in biological matrices. J. Chromatogr. A 2015, 1426, 33–47. [Google Scholar] [CrossRef]
- Adenuga, A.A.; Ayinuola, O.; Adejuyigbe, E.A.; Ogunfowokan, A.O. Biomonitoring of phthalate esters in breast-milk and urine samples as biomarkers for neonates’ exposure, using modified quechers method with agricultural biochar as dispersive solid-phase extraction absorbent. Microchem. J. 2020, 152, 104277. [Google Scholar] [CrossRef]
- Pourchet, M.; Debrauwer, L.; Klanova, J.; Price, E.J.; Covaci, A.; Caballero-Casero, N.; Oberacher, H.; Lamoree, M.; Damont, A.; Fenaille, F.; et al. Suspect and non-targeted screening of chemicals of emerging concern for human biomonitoring, environmental health studies and support to risk assessment: From promises to challenges and harmonisation issues. Environ. Int. 2020, 139, 105545. [Google Scholar] [CrossRef]
- Shishov, A.; Nechaeva, D.; Bulatov, A. HPLC-MS/MS determination of non-steroidal anti-inflammatory drugs in bovine milk based on simultaneous deep eutectic solvents formation and its solidification. Microchem. J. 2019, 150, 104080. [Google Scholar] [CrossRef]
- Herrera-Herrera, A.V.; Hernández-Borges, J.; Rodríguez-Delgado, M.Á. Fluoroquinolone antibiotic determination in bovine, ovine and caprine milk using solid-phase extraction and high-performance liquid chromatography-fluorescence detection with ionic liquids as mobile phase additives. J. Chromatogr. A 2009, 1216, 7281–7287. [Google Scholar] [CrossRef]
- Meng, Z.; Shi, Z.; Liang, S.; Dong, X.; Li, H.; Sun, H. Residues investigation of fluoroquinolones and sulphonamides and their metabolites in bovine milk by quantification and confirmation using ultra-performance liquid chromatography–tandem mass spectrometry. Food Chem. 2015, 174, 597–605. [Google Scholar] [CrossRef]
- Kantiani, L.; Farré, M.; Barceló, D. Rapid residue analysis of fluoroquinolones in raw bovine milk by online solid phase extraction followed by liquid chromatography coupled to tandem mass spectrometry. J. Chromatogr. A 2011, 1218, 9019–9027. [Google Scholar] [CrossRef]
- Lopes, B.R.; Barreiro, J.C.; Cass, Q.B. Bioanalytical challenge: A review of environmental and pharmaceuticals contaminants in human milk. J. Pharm. Biomed. Anal. 2016, 130, 318–325. [Google Scholar] [CrossRef]
- Jiménez-Díaz, I.; Vela-Soria, F.; Rodríguez-Gómez, R.; Zafra-Gómez, A.; Ballesteros, O.; Navalón, A. Analytical methods for the assessment of endocrine disrupting chemical exposure during human fetal and lactation stages: A review. Anal. Chim. Acta 2015, 892, 27–48. [Google Scholar] [CrossRef] [PubMed]
- Devanathan, G.; Subramanian, A.; Sudaryanto, A.; Takahashi, S.; Isobe, T.; Tanabe, S. Brominated flame retardants and polychlorinated biphenyls in human breast milk from several locations in India: Potential contaminant sources in a municipal dumping site. Environ. Int. 2012, 39, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Asante, K.A.; Adu-Kumi, S.; Nakahiro, K.; Takahashi, S.; Isobe, T.; Sudaryanto, A.; Devanathan, G.; Clarke, E.; Ansa-Asare, O.D.; Dapaah-Siakwan, S.; et al. Human exposure to PCBs, PBDEs and HBCDs in Ghana: Temporal variation, sources of exposure and estimation of daily intakes by infants. Environ. Int. 2011, 37, 921–928. [Google Scholar] [CrossRef] [PubMed]
- López-García, E.; Mastroianni, N.; Postigo, C.; Valcárcel, Y.; González-Alonso, S.; Barceló, D.; López de Alda, M. Simultaneous LC–MS/MS determination of 40 legal and illegal psychoactive drugs in breast and bovine milk. Food Chem. 2018, 245, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Calafat, A.M.; Slakman, A.R.; Silva, M.J.; Herbert, A.R.; Needham, L.L. Automated solid phase extraction and quantitative analysis of human milk for 13 phthalate metabolites. J. Chromatogr. B 2004, 805, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Malarvannan, G.; Kunisue, T.; Isobe, T.; Sudaryanto, A.; Takahashi, S.; Prudente, M.; Subramanian, A.; Tanabe, S. Organohalogen compounds in human breast milk from mothers living in Payatas and Malate, the Philippines: Levels, accumulation kinetics and infant health risk. Environ. Pollut. 2009, 157, 1924–1932. [Google Scholar] [CrossRef]
- Jamin, E.L.; Bonvallot, N.; Tremblay-Franco, M.; Cravedi, J.-P.; Chevrier, C.; Cordier, S.; Debrauwer, L. Untargeted profiling of pesticide metabolites by LC-HRMS: An exposomics tool for human exposure evaluation. Anal. Bioanal. Chem. 2014, 406, 1149–1161. [Google Scholar] [CrossRef]
- Gago-Ferrero, P.; Schymanski, E.L.; Hollender, J.; Thomaidis, N.S. Chapter 13—Nontarget analysis of environmental samples based on Liquid Chromatography Coupled to High Resolution Mass Spectrometry (LC-HRMS). In Comprehensive Analytical Chemistry; Pérez, S., Eichhorn, P., Barceló, D., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 381–403. [Google Scholar] [CrossRef]
- Hermo, M.P.; Nemutlu, E.; Kır, S.; Barrón, D.; Barbosa, J. Improved determination of quinolones in milk at their MRL levels using LC–UV, LC–FD, LC–MS and LC–MS/MS and validation in line with regulation 2002/657/EC. Anal. Chim. Acta 2008, 613, 98–107. [Google Scholar] [CrossRef]
- Gross, J.H. Mass spectrometry. A textbook. Anal. Bioanal. Chem. 2005, 381, 1319–1320. [Google Scholar]
- Cortéjade, A.; Kiss, A.; Cren, C.; Vulliet, E.; Buleté, A. Development of an analytical method for the targeted screening and multi-residue quantification of environmental contaminants in urine by liquid chromatography coupled to high resolution mass spectrometry for evaluation of human exposures. Talanta 2016, 146, 694–706. [Google Scholar] [CrossRef]
- Menger, F.; Ahrens, L.; Wiberg, K.; Gago-Ferrero, P. Suspect screening based on market data of polar halogenated micropollutants in river water affected by wastewater. J. Hazard. Mater. 2021, 401, 123377. [Google Scholar] [CrossRef]
- Musatadi, M.; González-Gaya, B.; Irazola, M.; Prieto, A.; Etxebarria, N.; Olivares, M.; Zuloaga, O. Focused ultrasound-based extraction for target analysis and suspect screening of organic xenobiotics in fish muscle. Sci. Total Environ. 2020, 740, 139894. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Zubiaguirre, L.; Zabaleta, I.; Usobiaga, A.; Prieto, A.; Olivares, M.; Zuloaga, O.; Elizalde, M.P. Target and suspect screening of substances liable to migrate from food contact paper and cardboard materials using liquid chromatography-high resolution tandem mass spectrometry. Talanta 2020, 208, 120394. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.M.; Simon, E.; Stroomberg, G.J.; de Boer, J.; de Boer, R.; van der Linden, S.C.; Leonards, P.E.G.; Lamoree, M.H. Identification strategy for unknown pollutants using high-resolution mass spectrometry: Androgen-disrupting compounds identified through effect-directed analysis. Anal. Bioanal. Chem. 2011, 400, 3141–3149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agilent Captiva ND and Captiva ND Lipids Method Guide. Available online: https://www.crawfordscientific.com/media/wysiwyg//Literature/Sample_Prep/Agilent/Captiva-ND-and-ND-Lipids-Method-Guide.pdf (accessed on 25 April 2019).
- Dualde, P.; Pardo, O.; Fernández, S.F.; Pastor, A.; Yusà, V. Determination of four parabens and bisphenols A, F and S in human breast milk using QuEChERS and liquid chromatography coupled to mass spectrometry. J. Chromatogr. B 2019, 1114–1115, 154–166. [Google Scholar] [CrossRef]
- Available online: Http://europa.eu.int/eur-lex/pri/en/oj/dat/2002/l_221/l_22120020817en00080036.pdf (accessed on 19 January 2021).
- Solliec, M.; Roy-Lachapelle, A.; Sauvé, S. Development of a suspect and non-target screening approach to detect veterinary antibiotic residues in a complex biological matrix using liquid chromatography/high-resolution mass spectrometry. Rapid Commun. Mass Spectrom. 2015, 29, 2361–2373. [Google Scholar] [CrossRef]
- Peng, T.; Zhu, A.-L.; Zhou, Y.-N.; Hu, T.; Yue, Z.-F.; Chen, D.-D.; Wang, G.-M.; Kang, J.; Fan, C.; Chen, Y.; et al. Development of a simple method for simultaneous determination of nine subclasses of non-steroidal anti-inflammatory drugs in milk and dairy products by ultra-performance liquid chromatography with tandem mass spectrometry. J. Chromatogr. B 2013, 933, 15–23. [Google Scholar] [CrossRef]
- Lin, J.; Chen, W.; Zhu, H.; Wang, C. Determination of free and total phthalates in commercial whole milk products in different packaging materials by gas chromatography-mass spectrometry. J. Dairy Sci. 2015, 98, 8278–8284. [Google Scholar] [CrossRef] [Green Version]
- Schymanski, E.L.; Jeon, J.; Gulde, R.; Fenner, K.; Ruff, M.; Singer, H.P.; Hollender, J. Identifying small molecules via high resolution mass spectrometry: Communicating confidence. Environ. Sci. Technol. 2014, 48, 2097–2098. [Google Scholar] [CrossRef]
- PubChem. Hazardous Substances Data Bank (HSDB): 599. Available online: https://pubchem.ncbi.nlm.nih.gov/source/hsdb/599#section=Interactions-(Complete) (accessed on 6 December 2020).
- Yalçin, S.S.; Güneş, B.; Yalçin, S. Incredible pharmaceutical residues in human milk in a cohort study from Şanlıurfa in Turkey. Environ. Toxicol. Pharmacol. 2020, 80, 103502. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | Without Oasis HLB | With Oasis HLB | |||
|---|---|---|---|---|---|
| Detected (%) | Recoveries (%) * | Detected (%) | Recoveries (%) * | ||
| (i) | AcN + NaCl | 88 | 63–92 (80) | 88 | 9–93 (46) |
| (ii) | AcN + Na2SO4 + NaCl | 92 | 68–110 (90) | 82 | 9–122 (44) |
| (iii) | AcN + MgSO4 + NaCl | 84 | 50–110 (78) | 78 | 30–111 (69) |
| (iv) | 1. AcN, 2. AcN + MgSO4 + NaCl | 89 | 58–101 (81) | 79 | 26–118 (50) |
| (v) | AcN:Milli-Q water 0.1% EDTA (95:5, v/v) | 81 | 7–85 (47) | Not performed | Not performed |
| Experiment | Without Oasis HLB | With Oasis HLB | |
|---|---|---|---|
| Recoveries (%) * | Recoveries (%) * | ||
| (i) | AcN + NaCl | 85–115 (109) | 72–108 (97) |
| (ii) | AcN + Na2SO4 + NaCl | 92–119 (109) | 81–112 (101) |
| (iv) | 1. AcN, 2. AcN + MgSO4 + NaCl | 95–128 (114) | 83–113 (102) |
| Repeatability | Concentration | Number of Analytes | ||||
|---|---|---|---|---|---|---|
| <10% | 10–20% | 20–50% | >50% | |||
| Instrumental | 50 ng/g (final extract) | 234 | 8 | 2 | 1 | |
| Procedural | 10 ng/g (milk) | Abs. rec. | 183 | 37 | 16 | 9 |
| App. rec. | 191 | 34 | 13 | 7 | ||
| 40 ng/g (milk) | Abs. rec. | 197 | 26 | 15 | 7 | |
| App. rec. | 195 | 28 | 15 | 7 | ||
| Compound | Whole Bovine Milk (ng/g) | Raw Bovine Milk (ng/g) | Milk Powder (ng/g) |
|---|---|---|---|
| Acetaminophen m | n.d | 3.2 ± 0.4 | n.d |
| Benzothiazole a | n.d | n.d | 11 ± 6 |
| Caffeine c | n.d | n.d | 2.0 ± 0.1 |
| EHDAB m | n.d | n.d | 0.70 ± 0.04 |
| Genistein | n.d | n.d | 20.1 ± 0.3 |
| Genistin | n.d | n.d | 29 ± 1 |
| Glycitin | n.d | n.d | 4.7 ± 0.6 |
| 2-Hydroxybenzothiazole | <LOQ | n.d | <LOQ |
| Azoxystrobin | n.d | <LOQ | n.d |
| Benzethonium | n.d | <LOQ | n.d |
| Bicalutamide | <LOQ | <LOQ | <LOQ |
| BEHA | n.d | <LOQ | n.d |
| BEHP | n.d | <LOQ | n.d |
| Butylparaben | <LOQ | <LOQ | <LOQ |
| Carbendazim | <LOQ | n.d | n.d |
| Cortisone | <LOQ | <LOQ | <LOQ |
| Cotinine | <LOQ | n.d | <LOQ |
| Crotamiton | n.d | <LOQ | n.d |
| Diethyl Toluamide | <LOQ | <LOQ | <LOQ |
| DOP m | n.d | <LOQ | n.d |
| Enrofloxacin | <LOQ | n.d | n.d |
| Fenpropimorph | <LOQ | <LOQ | <LOQ |
| Finasteride | n.d | n.d | <LOQ |
| Ifosfamide | n.d | <LOQ | <LOQ |
| Methylparaben | <LOQ | n.d | n.d |
| Pirimicarb | <LOQ | <LOQ | <LOQ |
| Pirimiphos-methyl | n.d | n.d | <LOQ |
| Primidone | <LOQ | <LOQ | <LOQ |
| Progesterone | <LOQ | n.d | n.d |
| Propiconazole | n.d | <LOQ | n.d |
| Pyrazophos | <LOQ | n.d | n.d |
| Sulfamethazine | <LOQ | n.d | n.d |
| Sulfamethoxazole | <LOQ | n.d | n.d |
| Terbutryn | <LOQ | <LOQ | <LOQ |
| Compound | A (ng/g) | B (ng/g) | C (ng/g) | D (ng/g) |
|---|---|---|---|---|
| Benzophenone | n.d | n.d | 4 ± 1 | n.d |
| Benzothiazole | 45 ± 4 | n.d | n.d | n.d |
| BEHP m | 45 ± 30 | n.d | 48 ± 29 | n.d |
| Caffeine c | 988 ± 127 | 35 ± 2 | 33 ± 3 | 1011 ± 210 |
| Caprolactam | 27 ± 2 | n.d | n.d | n.d |
| DOP m | 39 ± 27 | n.d | 41 ± 26 | n.d |
| EHDAB m | n.d | n.d | n.d | 0.58 ± 0.01 |
| MBHB c | <LOQ | n.d | 4 ± 2 | n.d |
| Orlistat | n.d | n.d | 2.3 ± 0.4 | n.d |
| Triethyl phosphate | n.d | n.d | 20 ± 8 | n.d |
| 2,4-Dinitrophenol | <LOQ | n.d | n.d | n.d |
| Azoxystrobin | n.d | <LOQ | n.d | n.d |
| Benzethonium | n.d | <LOQ | n.d | n.d |
| Bicalutamide | n.d | <LOQ | <LOQ | <LOQ |
| BEHA | n.d | n.d | <LOQ | n.d |
| BPA | <LOQ | n.d | n.d | n.d |
| Butylparaben | <LOQ | <LOQ | <LOQ | <LOQ |
| Cotinine | <LOQ | <LOQ | <LOQ | n.d |
| Crotamiton | <LOQ | n.d | <LOQ | <LOQ |
| DBP | n.d | n.d | <LOQ | n.d |
| Diethyl Toluamide | <LOQ | n.d | <LOQ | <LOQ |
| Exemestane | n.d | n.d | <LOQ | n.d |
| Fluvoxamine | n.d | n.d | <LOQ | n.d |
| MBP | <LOQ | n.d | n.d | n.d |
| Medroxyprogesterone | <LOQ | <LOQ | <LOQ | n.d |
| Methylparaben | <LOQ | n.d | n.d | n.d |
| Norfloxacin | n.d | n.d | <LOQ | n.d |
| Pindolol | n.d | <LOQ | n.d | <LOQ |
| Pirimicarb | <LOQ | <LOQ | <LOQ | <LOQ |
| Propiconazole | <LOQ | <LOQ | <LOQ | <LOQ |
| Sotalol | <LOQ | n.d | n.d | <LOQ |
| Sulfamethoxazole | <LOQ | n.d | n.d | n.d |
| Tamoxifen | <LOQ | n.d | <LOQ | n.d |
| Trimethoprim | n.d | n.d | <LOQ | n.d |
| Triphenylphosphate | <LOQ | n.d | n.d | n.d |
| Commercial Milk | Breast Milk | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Annotated Compound | Molecular Formula | Exact Mass | tR (min) | 1 | 2 | 3 | A | B | C | D |
| [M+H]+ | ||||||||||
| Level 1 | ||||||||||
| Acetaminophen | C8H9NO2 | 151.06333 | 1.77 | X | ✓ | X | X | ✓ | X | X |
| BEHP | C24H38O4 | 390.27701 | 15.90 | X | X | X | ✓ | X | ✓ | X |
| Benzophenone | C13H10O | 182.07316 | 11.75 | X | X | X | X | X | ✓ | X |
| Benzothiazole | C7H5NS | 135.01427 | 6.50 | X | X | ✓ | ✓ | X | X | X |
| Caffeine | C8H10N4O2 | 194.08038 | 2.50 | X | X | ✓ | ✓ | ✓ | ✓ | ✓ |
| Caprolactam | C6H11NO | 113.08406 | 2.35 | X | X | X | ✓ | X | X | X |
| DOP | C24H38O4 | 390.27701 | 15.90 | X | <LOI | X | <LOI | X | <LOI | X |
| EHDAB | C17H27NO2 | 277.20418 | 14.50 | X | X | <LOI | X | X | X | <LOI |
| Genistein * | C15H10O5 | 270.05282 | 7.55 | X | X | ✓ | X | X | X | X |
| Genistin * | C21H20O10 | 432.10565 | 4.30 | X | X | ✓ | X | X | X | X |
| Glycitin * | C22H22O10 | 446.12130 | 3.43 | X | X | <LOI | X | X | X | X |
| MBHB | C16H24O3 | 264.17256 | 13.60 | X | X | X | <LOI | X | <LOI | X |
| Triethyl phosphate | C6H15O4P | 182.0708 | 5.70 | X | X | X | X | X | ✓ | X |
| Level 2a | ||||||||||
| 1,2,3,9-tetrahydro-4H-carbazol-4-one oxime | C12H12N2O | 200.09496 | 4.99 | ✓ | X | X | X | X | X | X |
| 3-Acetyl-2,5-dimethylfuran | C8H10O2 | 138.06808 | 3.12 | X | X | X | X | X | X | ✓ |
| 3-amino-2-phenyl-2H-pyrazolo [4,3-c]pyridine-4,6-diol | C12H10N4O2 | 242.08038 | 5.07 | ✓ | ✓ | ✓ | ✓ | X | X | X |
| 3-Methyl-5-(5,5,8a-trimethyl-2-methylene-7-oxodecahydro-1-naphthalenyl)pentyl acetate | C22H36O3 | 348.26645 | 13.70 | X | ✓ | X | ✓ | ✓ | ✓ | X |
| 4-Indolecarbaldehyde * | C9H7NO | 145.05276 | 3.05 | ✓ | ✓ | ✓ | X | ✓ | ✓ | X |
| 4-Methoxycinnamic acid * | C10H10O3 | 178.06299 | 7.04 | X | X | X | ✓ | X | X | X |
| 5-Hydroxymethyl-2-furaldehyde | C6H6O3 | 126.03169 | 1.63 | X | X | ✓ | X | X | X | X |
| 7-Methyl-2-phenylquinoline-4-carboxylic acid | C17H13NO2 | 263.09463 | 12.09 | ✓ | ✓ | X | X | X | X | X |
| Amfepramone | C13H19NO | 205.14666 | 5.66 | X | X | X | ✓ | X | X | X |
| Avobenzone | C20H22O3 | 310.15689 | 14.40 | X | X | X | X | ✓ | ✓ | X |
| Bis(2-ethylhexyl) amine | C16H35N | 241.27695 | 11.34 | ✓ | X | ✓ | X | X | X | X |
| Carvone | C10H14O | 150.10447 | 11.72 | X | X | X | X | X | ✓ | X |
| Citroflex A-4 | C20H34O8 | 402.22537 | 14.00 | X | ✓ | X | X | X | X | X |
| Didecyldimethylammonium | C22H47N | 325.37085 | 12.60 | X | X | X | ✓ | X | X | X |
| Isoquinoline * | C9H7N | 129.05785 | 3.80 | ✓ | ✓ | ✓ | X | X | X | X |
| Nootkatone | C15H22O | 218.16707 | 12.95 | X | X | X | X | X | ✓ | X |
| OPEO | C16H26O2 | 250.19328 | 13.07 | X | X | X | X | ✓ | ✓ | ✓ |
| Piperine * | C17H19NO3 | 285.13649 | 11.81 | X | X | ✓ | X | X | ✓ | ✓ |
| Shogaol * | C17H24O3 | 276.17254 | 10.70 | X | ✓ | X | ✓ | ✓ | ✓ | X |
| Tetramethylene sulfoxide | C4H8OS | 104.02959 | 1.46 | X | X | X | ✓ | X | X | X |
| Level 3 | ||||||||||
| 2-Oxindole/4-Hydroxyindole/ 5-Hydroxyindole | C8H7NO | 133.05276 | 1.54 | ✓ | ✓ | ✓ | X | X | X | X |
| 2-Hydroxyquinoline/ 8-Hydroxyquinoline * | C9H7NO | 145.05276 | 3.95 | ✓ | X | ✓ | X | X | X | X |
| 1,5-Isoquinolinediol/ 2,4-Quinolinediol | C9H7NO2 | 161.04768 | 4.17 | ✓ | ✓ | ✓ | X | X | X | X |
| 1,5-Isoquinolinediol/ 2,4-Quinolinediol | C9H7NO2 | 161.04768 | 4.48 | ✓ | X | X | X | X | X | X |
| Paraxanthine/Theophylline/Theobromine * | C7H8N4O2 | 180.06473 | 1.69 | X | X | X | ✓ | ✓ | ✓ | ✓ |
| Level 4 | ||||||||||
| 2-Oxindole/4-Hydroxyindole/ 5-Hydroxyindole/ 6-Methylbenzoxazole | C8H7NO | 133.05276 | 4.68 | ✓ | X | ✓ | ✓ | ✓ | ✓ | ✓ |
| Pulegone/D,L-Camphor/Citral * | C10H16O | 152.12012 | 7.81 | X | X | X | ✓ | ✓ | ✓ | X |
| DL-2-(acetylamino)-3-phenylpropanoic acid/ 4-morpholinobenzoic acid | C11H13NO3 | 207.08954 | 3.86 | X | ✓ | X | X | X | X | X |
| 2-(8-Hydroxy-4a,8-dimethyldecahydro-2-naphthalenyl) acrylic acid/ 2-[(2R,4aR,8R,8aR)-8-hydroxy-4a,8-dimethyl-decahydronaphthalen-2-yl] prop-2-enoic acid/Polygodial | C15H24O3 | 252.17254 | 11.83 | X | X | X | X | X | ✓ | X |
| 5-O-Methylgenistein/5,7-dihydroxy-3-(4-methoxyphenyl)-4H-chromen-4-one/Biochanin A/Glycitein/Wogonin | C16H12O5 | 284.06847 | 6.46 | X | X | ✓ | X | X | X | X |
| 1,4a-dimethyl-9-oxo-7-(propan-2-yl)-1,2,3,4,4a,9,10,10a-octahydrophenanthrene-1-carboxylic acid/Kahweol | C20H26O3 | 314.18819 | 12.45 | X | X | ✓ | X | X | ✓ | X |
| N-Benzylformamide/Phenacylamine | C8H9NO | 135.06841 | 3.13 | ✓ | ✓ | ✓ | X | X | ✓ | ✓ |
| [M-H]− | ||||||||||
| Level 2a | ||||||||||
| 2-(8-Hydroxy-4a,8-dimethyldecahydro-2-naphthalenyl)acrylic acid | C15H24O3 | 252.17254 | 10.82 | X | X | X | ✓ | X | ✓ | X |
| 2,5-di-tert-Butylhydroquinone | C14H22O2 | 222.16198 | 12.49 | X | X | X | ✓ | ✓ | ✓ | ✓ |
| Myristyl sulfate | C14H30O4S | 294.18648 | 12.22 | X | X | X | ✓ | X | X | ✓ |
| Saccharin | C7H5NO3S | 182.99901 | 1.38 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | X |
| Level 4 | ||||||||||
| Chrysin/Daidzein/Abietic acid | C15H10O4 | 254.05791 | 6.09 | X | X | ✓ | X | X | X | X |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musatadi, M.; González-Gaya, B.; Irazola, M.; Prieto, A.; Etxebarria, N.; Olivares, M.; Zuloaga, O. Multi-Target Analysis and Suspect Screening of Xenobiotics in Milk by UHPLC-HRMS/MS. Separations 2021, 8, 14. https://doi.org/10.3390/separations8020014
Musatadi M, González-Gaya B, Irazola M, Prieto A, Etxebarria N, Olivares M, Zuloaga O. Multi-Target Analysis and Suspect Screening of Xenobiotics in Milk by UHPLC-HRMS/MS. Separations. 2021; 8(2):14. https://doi.org/10.3390/separations8020014
Chicago/Turabian StyleMusatadi, Mikel, Belén González-Gaya, Mireia Irazola, Ailette Prieto, Nestor Etxebarria, Maitane Olivares, and Olatz Zuloaga. 2021. "Multi-Target Analysis and Suspect Screening of Xenobiotics in Milk by UHPLC-HRMS/MS" Separations 8, no. 2: 14. https://doi.org/10.3390/separations8020014
APA StyleMusatadi, M., González-Gaya, B., Irazola, M., Prieto, A., Etxebarria, N., Olivares, M., & Zuloaga, O. (2021). Multi-Target Analysis and Suspect Screening of Xenobiotics in Milk by UHPLC-HRMS/MS. Separations, 8(2), 14. https://doi.org/10.3390/separations8020014