Time-Resolved Fluorescence in Photodynamic Therapy


Abstract
:1. Introduction
1.1. Limitation Factors in PDT Dosimetry

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dose Factors | Interdependencies | Results |
| Photosensitizer | Individual variability | Variation in uptake, local location, and concentration; |
| Light fluence | Large extinction coefficient leads to self-shielding from light | |
| Oxygen | Chemically deplete oxygen molecules | |
| Light Fluence | Individual variability | Variation in distribution |
| Photosensitizers | High fluence rate can photobleach photosensitizers | |
| Oxygen | High fluence rate can deplete oxygen molecules [33] | |
| Oxygen | Individual variability | Variation in vasculature, perfusion, and oxygen saturation |
| Photosensitizer | Variation in photobleaching rate | |
| PDT treatment | Potential vasculature occlusion that reduces oxygen supply |

| Dosimetric | Measured Parameters | Limitations |
|---|---|---|
| Explicit | Main dose factors: Photosensitizer concentration; light fluence; oxygen concentration | Difficult to acquire complete data set and require a model to combine all these for the effective dose; ignored all microdosimetric changes induced from interdependencies of dose factors (as listed in Table 1) [32] |
| Implicit | Photobleaching | Need photosensitizers or a second reporter that can be photobleached; need to know the degree of photosensitizer coupling to cytotoxic photoproduct (e.g., 1O2); accurate modeling in tissue optics is required |
| Direct | Singlet oxygen phosphorescence at 1270 nm | Low SNR and technically difficult, e.g., requires photodetectors sensitive in the NIR region. Also, it does not account for effects from free radicals and other dose interdependencies [39,40] |
1.2. The Potential Role of Time-Resolved Fluorescence (TRF) for PDT Dosimetry
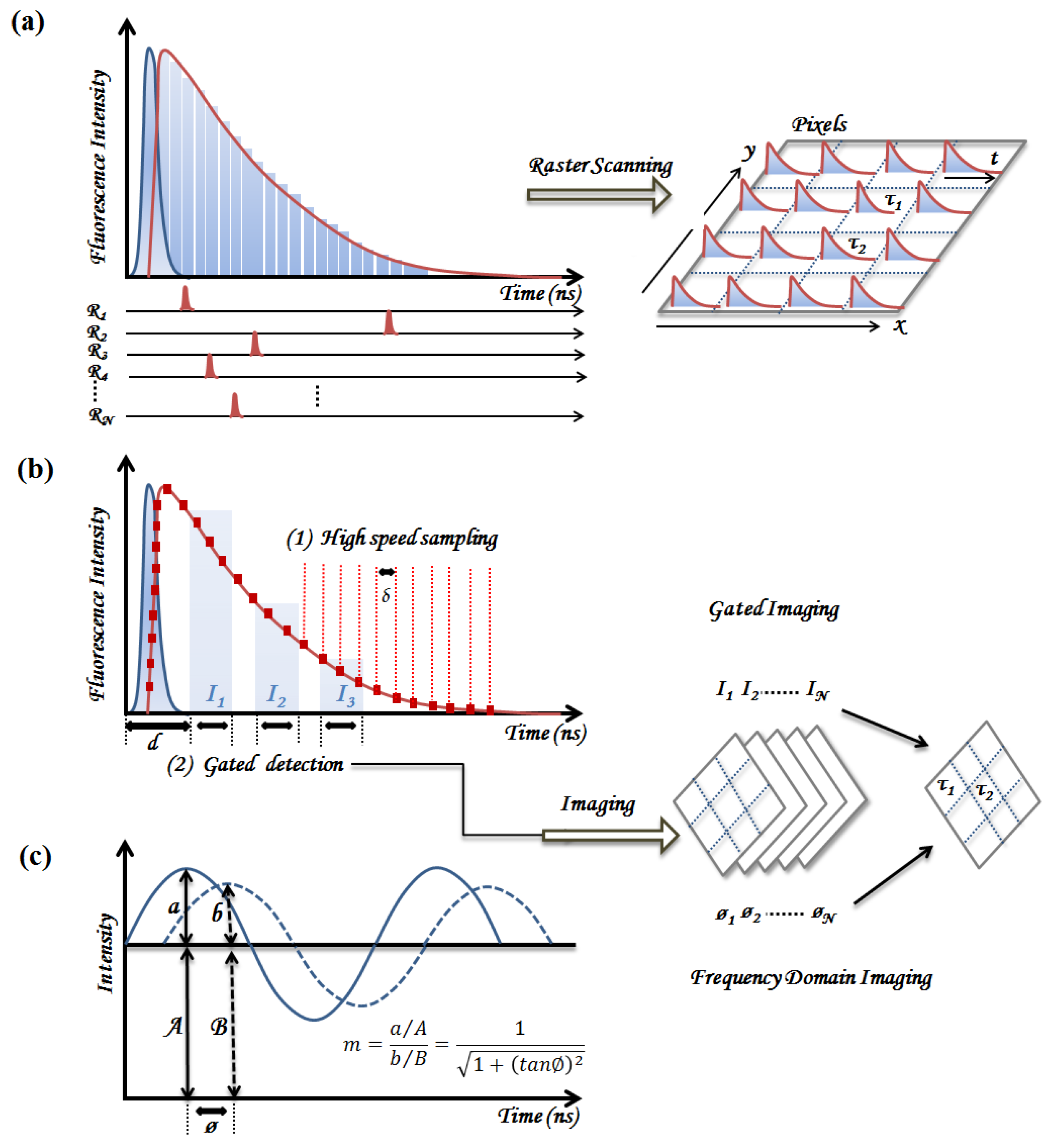
2. Principles of TRF Spectroscopy and Imaging
2.1. Time-Domain Fluorescence Spectroscopy and Imaging
2.1.1. Time-Correlated Single Photon Counting (TCSPC)

2.1.2. Pulse Sampling Techniques
2.2. Frequency Domain Fluorescence Spectroscopy
2.3. Summary of Instrumentation Requirements for Clinical Implementation
| Advantages | Disadvantages |
|---|---|
| TCSPC High sensitivity and temporal resolution Low systematic errors Suitable for resolving complex decays Easy implementation to existing scanning system Low cost | Very slow data acquisition to achieve desired signal Requires post-processing to correct distortions in long fluorescence lifetimes [67,68] Cannot tolerate ambient light |
| Time-gated and pulse sampling Capable of single-shot detection Rapid data acquisition of fluorescence decays Good for background subtraction Immune to ambient lighting | Difficult to predict instrument noise Low sensitivity so requires sufficient quantum yield Time resolution is subject to the gate window High instrumentation cost |
| General Broad excitation spectra of short pulsed lasers Can be operated at room light ( f *< 10 Hz ) | Complex opto-electronic systems for detector and light sources compared to FD–FLIM |
3. Applications of TRF on PDT Photosensitizers
3.1. Time-Resolved Studies of PDT Photosensitizers in Solution and In Vitro
| PS. | Lifetimes (τ) / Localization | PS. Conc. | Ex. (nm) | Em. (nm) |
|---|---|---|---|---|
| HpD/ Photofrin | 14 ns (Organic solution) [43] | 5 µg/mL | 364 | 615 |
| 10 ns (Organic solution) [70] | 0.06–6 µg/mL | 405 | > 580 | |
| 5.5 ns (Mitochondria, MLL) [70] | 10 µg/mL | 810 | 600–750 | |
| 13.3 ns (Monomer, mitochondria) [70] | 10 µg/mL | 810 | 600–750 | |
| 13.6 ns (Monomer, mitochondria) [45] | 2 µg/mL | 398 | 627–651 | |
| 8.5 ns (Aggregates, mitochondria) [45] | 2 µg/mL | 398 | 651–687 | |
| 8.0 ns (Aggregates, mitochondria) [78] | 5 µg/mL | 514 | 600–700 | |
| 4.8 ns (Cell membrane) [78] | 5 µg/mL | 514 | 600–700 | |
| 1.0 ns (Aggregates, mouse model) [31] | 20 mg/kg | 514 | 630 | |
| 13 ns (Monomers, mouse model) [31] | 20 mg/kg | 514 | 630 | |
| PpIX | 16.4 ns (Organic solution) [70] ; | 10 mM | 810 | 600–750 |
| 6.3 ns (Averaged. mitochondria) [70] | 10 mM | 810 | 600–750 | |
| 7.5 ns (Averaged, mitochondria) [79] | 1 mM | 398 | 610–640 | |
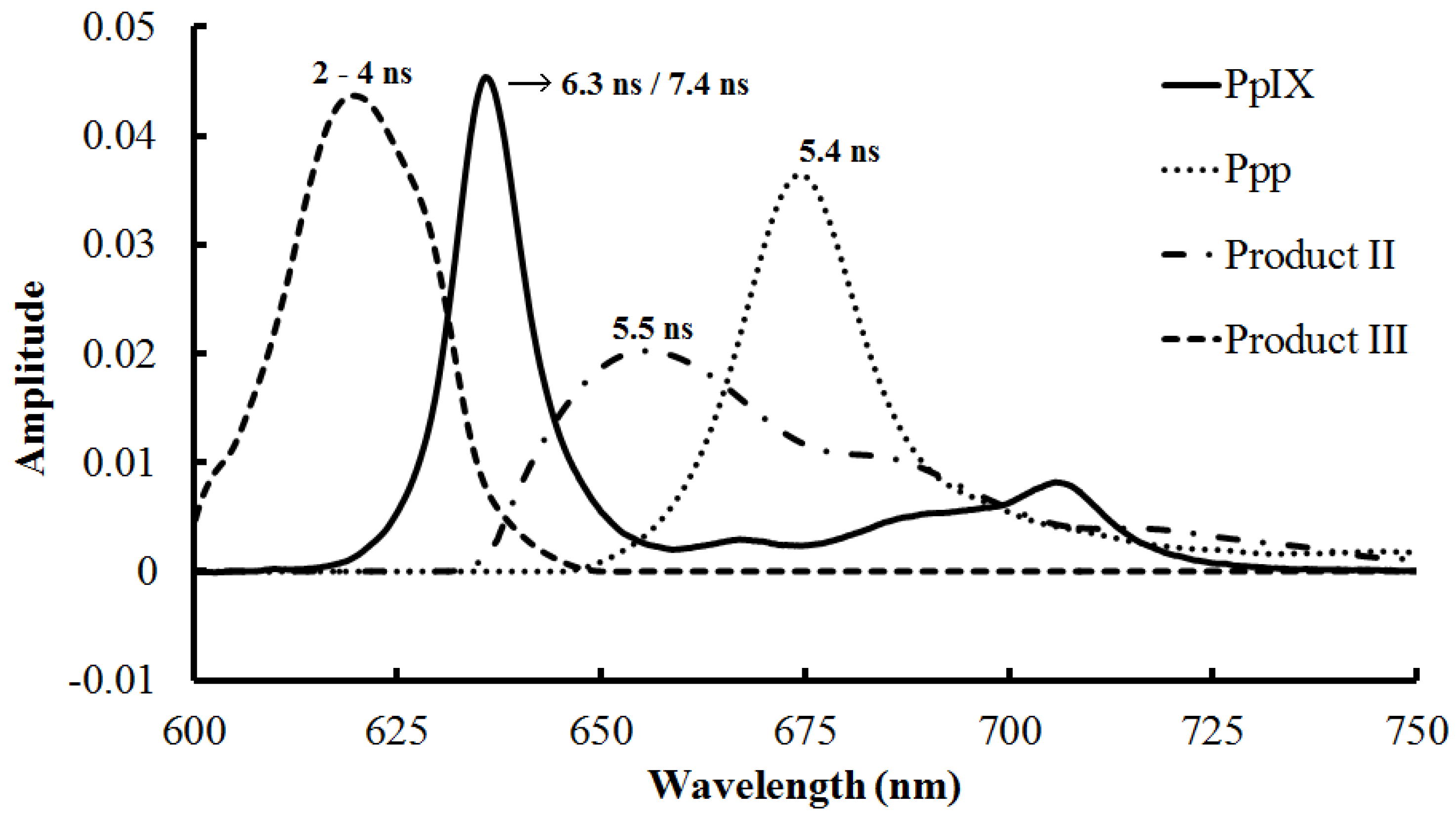
| 2–4 ns (Photoproducts, cytoplasm) [69] | 1 mM | 398 | >590 | |
| 5.4 ns (Ppp, Mitochondria) [71] | 20 µM | 670 | 674 | |
| mTHPC | 10 ns (Ethanol solution) [72] | 40 µM | 355 | 456–794 |
| 8.4 ns (Methanol) [73] | 15 µM | 590 | > 630 | |
| 4.8 ns (Macrophages, V79) [73] | 15 µM | 380–450 | >630 | |
| AlPcS2 ZnPPC | 4.0 ns (Macrophages, V79) [73] | 100 µM | 380–450 | >630 |
| 2.5–3 ns (Macrophages, V79) [73] | 10–50 µM | 380–450 | >630 | |
| HPPH | 5.7 ns (PBS) [74] | 100 µM | 400 | 670–710 |
| 7.6 ns (liposome confined) [74] | 100 µM | 400 | 670–710 | |
| 6.4 ns (tissue phantom) [75] | 0.5 µM | 660 | 720 | |
| 4.3 ns (mouse tumor, before PDT) [75] | 3 µM/kg | 660 | 720 | |
| 5.0 ns (mouse tumor, after PDT) [75] | 3 µM/kg | 660 | 720 | |
| Chlorin-e6 | 4.5 ns (Monomer, methanol) [76] | 5 µM | 800 | 635–740 |
| ~ 0.5 ns (Aggregates, methanol) [76] | 5 µM | 800 | 740 | |
| 0.5–3 ns (Part. aggregated, Lysosome) [76] | 5 µM | 430 | >710 | |
| ~0.1 ns (Aggregates, Lysosome) [76] | 5 µM | 430 | >710 |
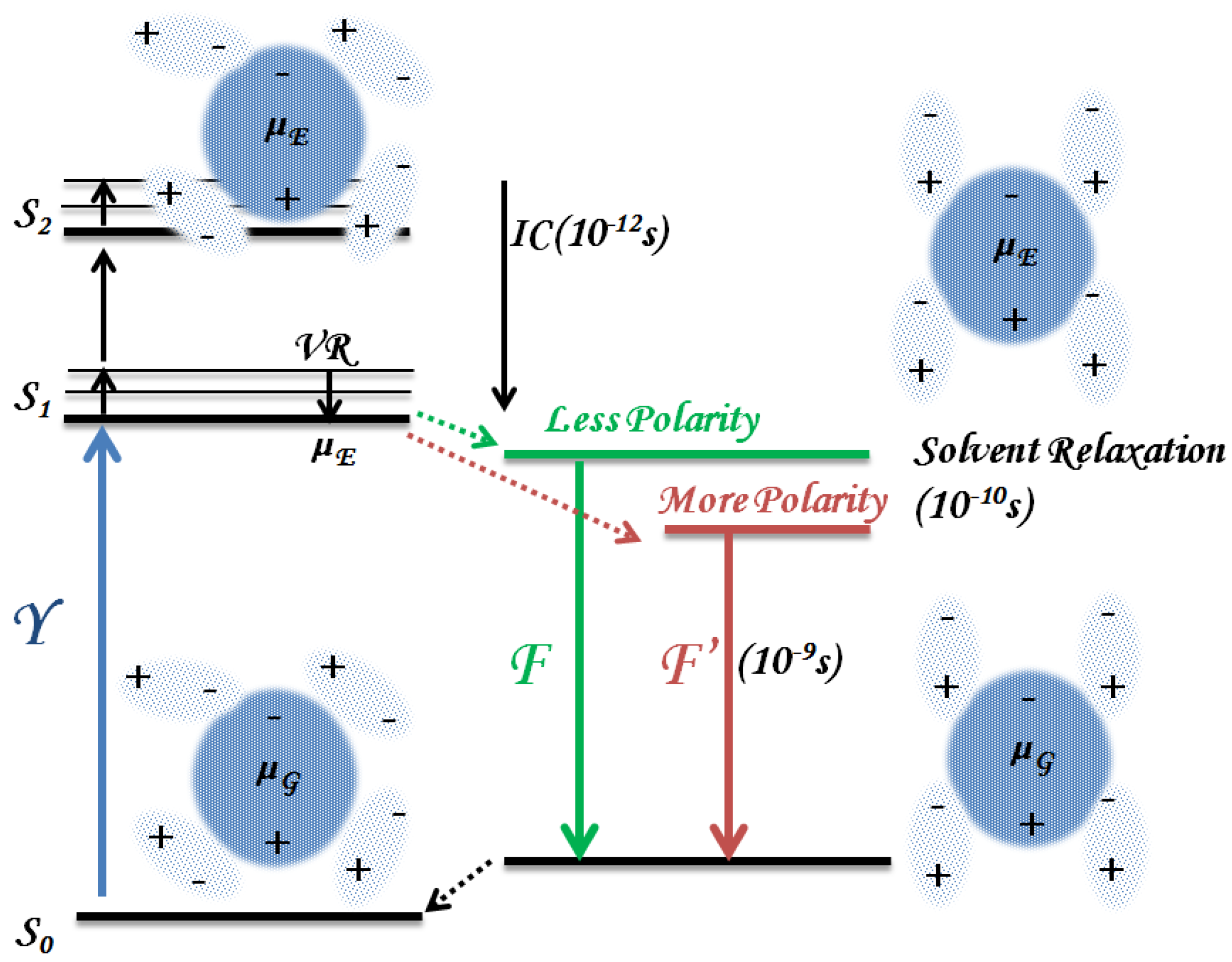
3.1.1. Solvent Effect

3.1.2. Binding to Biomolecules
Mitochondrial Localization
Lysosome Localization
Cell Membrane Localization
3.1.3. Photoproducts and Self-Aggregation

3.1.4. Prolonged Irradiation
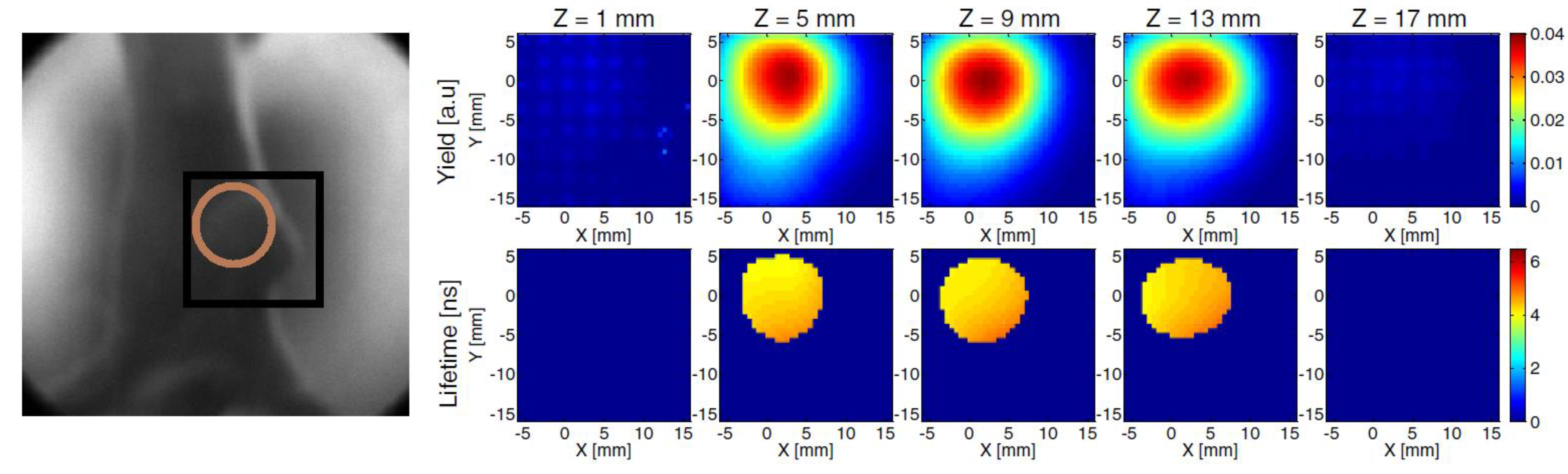
3.2. Time-Resolved Studies of PDT Photosensitizer in Vivo

3.2.1. Endogenous Fluorophores
3.2.2. Microenvironment—Oxygen Level, Vascularization, and pH
3.3. Discussion
| Key Information for Dosimetry | Information Yielded by PDT-FLIM |
|---|---|
| Drug interactions with subcellular organelles (Section 3.1.2) | Quenching of photosensitizer fluorescence lifetime while binding to biomolecules [73,115]. Changes of fluorescence lifetimes through the drug uptake process [78]. |
| Photoproducts (Section 3.1.3) | FLIM is able to determine photoproduct species although significant spectral overlap exists. Understanding of photoproduct contribution is essential to avoid over- or underdose estimation [31,45,69]. |
| Apoptosis (Section 3.2.1) | NADH fluorescence lifetime would increase immediately after the initiation of apoptosis. This can be applicable to apoptosis-mediated PDT [105,116,117]. |
| Necrosis (Section 3.2.1) | NADH fluorescence lifetime does not change through the necrosis procedures. This can be related to necrosis-mediated PDT (plasma membrane as a target) [116,117]. |
| Cell function (Section 3.2.1) | Cell metabolism and mitochondrial malfunction can be revealed by the ratio of free NADH (short lifetime) and bound NADH [44,104,117]. |
| Oxygen Sensing (Section 3.2.2) | Decreased oxygen level lead to increased lifetime [75]. An instrument with low laser repetition rate and CCD detection is under development for sensing oxygen concentration and fluorescence lifetime [118]. |
4. Challenges and Advances in Using TRF for Clinical Applications
- (i)
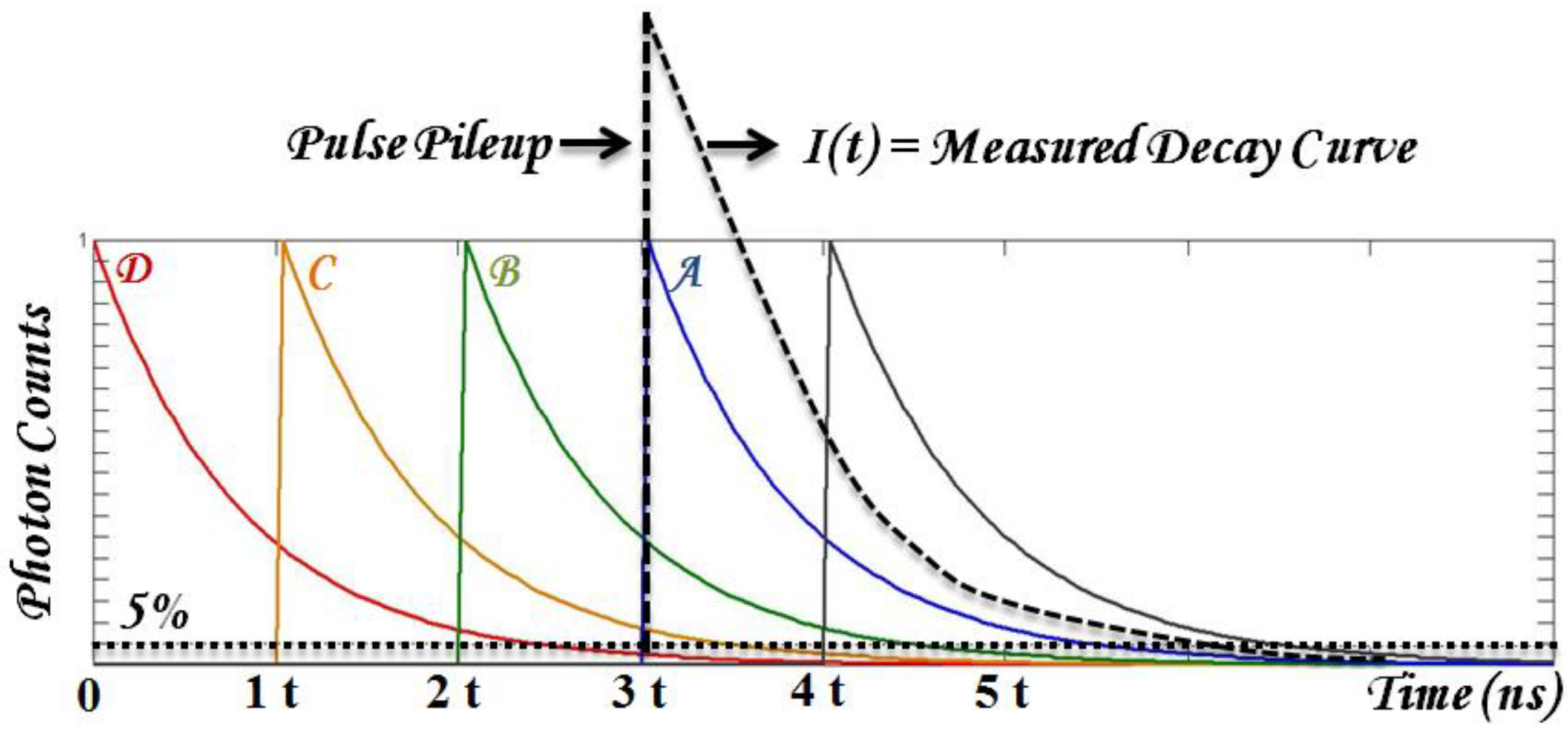
- Robust data analysis that deals with substantial biological variables and low SNR. Time-domain parameters are typically retrieved from fitting the results with known decay dynamics. Fitting accuracy may be reduced in multiple exponential decay and low SNR from photosensitizer fluorescence. In addition, the typical fluorescence lifetime range of photosensitizers can be long enough to introduce pulse pile-up and incomplete decay problems using time-domain TRF techniques. To be practical, the first challenge to overcome is to have fast and robust algorithms to retrieve time-resolved parameters, τi (lifetime) and Ai (coefficients).
- (ii)
- Tissue optics that affects light focusing and drug targeting efficiency. This can be approached by modeling light transport using diffuse optical tomography.
- (iii)
- Compatible instrumentation for clinical implementation. It is desired to have compact instrumentation with accessibility to desired tissue target (e.g., coupled to endoscopy). In addition, spectral-resolved analysis (e.g., hyperspectral TRF) may provide additional advantages in terms of interpreting multiple sources of fluorophores.
4.1. Data Analysis

4.2. Tissue Optics in PDT and Lifetime Measurement
4.3. Instrumentation
4.3.1. FLIM Endoscopy
4.3.2. Hyperspectral TRF Imaging
5. Conclusion and Outlook
Acknowledgments
Conflict of Interests
References
- Kalka, K.; Merk, H.; Mukhtar, H. Photodynamic therapy in dermatology. J. Am. Acad. Dermatol. 2000, 42, 389–413. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, R.; Cotlier, E.; Schmidt-erfurth, U.; Hasan, T. Mechanisms of action of photodynamic therapy with verteporfin for the treatment of age-related macular degeneration. Surv. Ophthalmol. 2000, 45, 195–214. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.C.; Patterson, M.S. The physics, biophysics and technology of photodynamic therapy. Phys. Med. Biol. 2008, 53, R61–R109. [Google Scholar] [CrossRef] [PubMed]
- Nseyo, U.O.; Shumaker, B.; Klein, E.; Sutherland, K. Photodynamic therapy using porfimer sodium as an alternative to cystectomy in patients with refractory transitional cell carcinoma in situ of the bladder. Bladder Photofrin Study Group. J. Urol. 1998, 160, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.B.; Brown, E.A.; Walker, I. The present and future role of photodynamic therapy in cancer treatment Photodynamic therapy. Lancet Oncol. 2004, 5, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Usuda, J.; Kato, H.; Okunaka, T.; Furukawa, K.; Yamada, K.; Suga, Y.; Honda, H.; Nagatsuka, Y.; Ohira, T.; Tsuboi, M.; et al. Photodynamic Therapy (PDT) for lung cancers. J. Thorac. Oncol. 2006, 1, 489–493. [Google Scholar] [CrossRef]
- Overholt, B.F.; Wang, K.K.; Burdick, J.S.; Lightdale, C.J.; Kimmey, M.; Nava, H.R.; Sivak, M.V; Nishioka, N.; Barr, H.; et al. Five-year efficacy and safety of photodynamic therapy with Photofrin in Barrett’s high-grade dysplasia. Gastrointest. Endosc. 2007, 66, 460–468. [Google Scholar] [CrossRef]
- Eggener, S.E.; Scardino, P.T.; Carroll, P.R.; Zelefsky, M.J.; Sartor, O.; Hricak, H.; Wheeler, T.M.; Fine, S.W.; Trachtenberg, J.; et al. Focal therapy for localized prostate cancer: A critical appraisal of rationale and modalities. J. Urol. 2007, 178, 2260–2267. [Google Scholar] [CrossRef]
- Molpus, K.L.; Kato, D.; Hamblin, M.R.; Lilge, L.; Bamberg, M.; Hasan, T. Intraperitoneal photodynamic therapy of human epithelial ovarian carcinomatosis in a xenograft murine model. Cancer Res. 1996, 56, 1075–1082. [Google Scholar] [PubMed]
- Hopper, C.; Kübler, A.; Lewis, H.; Tan, I.B.; Putnam, G. mTHPC-mediated photodynamic therapy for early oral squamous cell carcinoma. Int. J. Cancer 2004, 111, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Nyst, H.J.; van Veen, R.L.P.; Tan, I.B.; Peters, R.; Spaniol, S.; Robinson, D.J.; Stewart, F.; Levendag, P.C.; Sterenborg, H.J.C.M. Performance of a dedicated light delivery and dosimetry device for photodynamic therapy of nasopharyngeal carcinoma: Phantom and volunteer experiments. Lasers Surg. Med. 2007, 39, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, T.J.; Gomer, C.J.; Henderson, B.W.; Jori, G.; Kessel, D.; Korbelik, M.; Moan, J.; Peng, Q. Photodynamic therapy. J. Natl. Cancer Inst. 1998, 90, 889–905. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, I.J.; Dougherty, T.J. Basic principles of photodynamic therapy. J. Porphyr. Phthalocyanines 2001, 5, 105–129. [Google Scholar] [CrossRef]
- Portale, G.; Hagen, J.A.; Peters, J.H.; Chan, L.S.; DeMeester, S.R.; Gandamihardja, T.A.K.; DeMeester, T.R. Modern 5-year survival of resectable esophageal adenocarcinoma: single institution experience with 263 patients. J. Am. Coll. Surg. 2006, 202, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.J.; Wilson, B.C. Photodynamic therapy of brain tumors–A work in progress. Lasers Surg. Med. 2006, 38, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.; Palte, G.; Schnell, O.; Tonn, J.-C.; Herms, J.; Stepp, H. 5-Aminolevulinic acid-induced protoporphyrin IX levels in tissue of human malignant brain tumors. Photochem. Photobiol. 2010, 86, 1373–1378. [Google Scholar] [CrossRef] [PubMed]
- Oleinick, N.L.; Morris, R.L.; Belichenko, I. The role of apoptosis in response to photodynamic therapy: what, where, why, and how. Photochem. Photobiol. Sci. 2002, 1, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Kessel, D.; Luo, Y.; Deng, Y.; Chang, C.K. The role of subcellular localization in initiation of apoptosis by photodynamic therapy. Photochem. Photobiol. 1997, 65, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Price, M.; Heilbrun, L.; Kessel, D. Effects of the oxygenation level on formation of different reactive oxygen species during photodynamic therapy. Photochem. Photobiol. 2013, 89, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Vanlangenakker, N.; Parthoens, E.; Deckers, W.; Devos, M.; Festjens, N.; Guerin, C.J.; Brunk, U.T.; Declercq, W.; Vandenabeele, P. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010, 17, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Calzavara-Pinton, P.G.; Venturini, M.; Sala, R. Photodynamic therapy: Update 2006. Part 1: Photochemistry and photobiology. J. Eur. Acad. Dermatol. Venereol. 2007, 21, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Dwarakanath, B.S.; Muralidhar, K.; Jain, V. Cellular uptake, localization and photodynamic effects of haematoporphyrin derivative in human glioma and squamous carcinoma cell lines. J. Photochem. Photobiol. B. 2003, 69, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Mojzisova, H.; Bonneau, S.; Vever-Bizet, C.; Brault, D. Cellular uptake and subcellular distribution of chlorin e6 as functions of pH and interactions with membranes and lipoproteins. Biochim. Biophys. Acta 2007, 1768, 2748–2756. [Google Scholar]
- Kiesslich, T.; Berlanda, J.; Plaetzer, K.; Krammer, B.; Berr, F. Comparative characterization of the efficiency and cellular pharmacokinetics of Foscan- and Foslip-based photodynamic treatment in human biliary tract cancer cell lines. Photochem. Photobiol. Sci. 2007, 6, 619–627. [Google Scholar]
- Sandell, J.L.; Zhu, T.C. A review of in vivo optical properties of human tissues and its impact on PDT. 2012; 4, 773–787. [Google Scholar]
- Abels, C. Targeting of the vascular system of solid tumours by photodynamic therapy (PDT). Photochem. Photobiol. Sci. 2004, 3, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Sun, C.H.; Liaw, L.H.; Berns, M.W.; Nelson, J.S. In vitro and in vivo photosensitizing capabilities of 5-ALA versus photofrin in vascular endothelial cells. Lasers Surg. Med. 1999, 24, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Georgakoudi, I.; Foster, T.H. Singlet oxygen- versus nonsinglet oxygen-mediated mechanisms of sensitizer photobleaching and their effects on photodynamic dosimetry. Photochem. Photobiol. 1998, 67, 612–625. [Google Scholar] [CrossRef] [PubMed]
- Dysart, J.S.; Patterson, M.S. Photobleaching kinetics, photoproduct formation, and dose estimation during ALA induced PpIX PDT of MLL cells under well oxygenated and hypoxic conditions. Photochem. Photobiol. Sci. 2006, 5, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Schneckenburger, H.; Steiner, R.; Rueck, A. In vivo photoproduct formation during PDT with ALA-induced endogenous porphyrins. J Photochem. Photobiol. B. 1993, 18, 287–290. [Google Scholar] [CrossRef] [PubMed]
- König, K.; Wabnitz, H.; Dietel, W. Variation in the fluorescence decay properties of haematoporphyrin derivative during its conversion to photoproducts. J. Photochem. Photobiol. B. 1990, 8, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.C.; Patterson, M.S.; Lilge, L. Implicit and explicit dosimetry in photodynamic therapy : a new paradigm. Lasers Med. Sci. 1997, 12, 182–199. [Google Scholar] [PubMed]
- Weston, M.A.; Patterson, M.S. Monitoring oxygen concentration during photodynamic therapy using prompt photosensitizer fluorescence. Phys. Med. Biol. 2013, 58, 7039–7059. [Google Scholar] [CrossRef] [PubMed]
- Diamond, K.R.; Malysz, P.P.; Hayward, J.E.; Patterson, M.S. Quantification of fluorophore concentration in vivo using two simple fluorescence-based measurement techniques. J. Biomed. Opt. 2005, 10, 024007. [Google Scholar] [CrossRef] [PubMed]
- Diamond, K.R.; Patterson, M.S.; Farrell, T.J. Quantification of fluorophore concentration in tissue-simulating media by fluorescence measurements with a single optical fiber. Appl. Opt. 2003, 42, 2436–2342. [Google Scholar] [CrossRef]
- Patterson, M.S.; Wilson, C.; Graff, R. In vivo tests of the concept of photodynamic threshold dose in normal rat liver photosensitized by aluminum pthalocyanine. Photochem. Photobiol. 1990, 51, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Sheng, C.; Hoopes, P.J.; Hasan, T.; Pogue, B.W. Photobleaching-based dosimetry predicts deposited dose in ALA-PpIX PDT of rodent esophagus. Photochem. Photobiol. 2007, 83, 738–748. [Google Scholar] [PubMed]
- Dysart, J.S.; Patterson, M.S. Characterization of Photofrin photobleaching for singlet oxygen dose estimation during photodynamic therapy of MLL cells in vitro. Phys. Med. Biol. 2005, 50, 2597–2616. [Google Scholar] [CrossRef] [PubMed]
- Niedre, M.; Patterson, M.S.; Wilson, B.C. Direct near-infrared luminescence detection of singlet oxygen generated by photodynamic therapy in cells in vitro and tissues in vivo. Photochem. Photobiol. 2002, 75, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Jarvi, M.T.; Niedre, M.J.; Patterson, M.S.; Wilson, B.C. Singlet oxygen luminescence dosimetry (SOLD) for photodynamic therapy: Current status, challenges and future prospects. Photochem. Photobiol. 2006, 82, 1198–1210. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescnece Spectroscopy, 3rd ed.; Springer: New York, NY, USA, 2006. [Google Scholar]
- Suhling, K.; French, P.M.W.; Phillips, D. Time-resolved fluorescence microscopy. Photochem. Photobiol. Sci. 2005, 4, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Cubeddu, R.; Ramponi, R.; Bottiroli, G. Time-resolved fluorescence spectroscopy of hematoporphyrin derivative in micelles. Chem. Phys. Lett. 1986, 128, 439–442. [Google Scholar] [CrossRef]
- Wu, Y.; Zheng, W.; Qu, J.Y. Sensing cell metabolism by time-resolved autofluorescence. Opt. Lett. 2006, 31, 3122–3124. [Google Scholar] [CrossRef] [PubMed]
- Ruck, A.; Hulshoff, C.; Kinzler, I.; Becker, W.; Steiner, R. SLIM : A new method for molecular imaging. Microsc. Res. Tech. 2007, 492, 485–492. [Google Scholar] [CrossRef]
- Verveer, P.J.; Squire, A.; Bastiaens, P.I. Global analysis of fluorescence lifetime imaging microscopy data. Biophys. J. 2000, 78, 2127–2137. [Google Scholar] [CrossRef] [PubMed]
- Pelet, S.; Previte, M.J.R.; Laiho, L.H.; So, P.T.C. A fast global fitting algorithm for fluorescence lifetime imaging microscopy based on image segmentation. Biophys. J. 2004, 87, 2807–2817. [Google Scholar] [CrossRef]
- Bright, F.V; Munson, C.A. Time-resolved fluorescence spectroscopy for illuminating complex systems. Anal. Chim. Acta 2003, 500, 71–104. [Google Scholar] [CrossRef]
- Marcu, L. Fluorescence lifetime techniques in medical applications. Ann. Biomed. Eng. 2012, 40, 304–331. [Google Scholar] [PubMed]
- Becker, W.; Bergmann, A.; Biskup, C. Multispectral fluorescence lifetime imaging by TCSPC. Microsc. Microanal. 2007, 70, 403–409. [Google Scholar]
- Fang, Q.; Papaioannou, T.; Jo, J.A.; Vaitha, R.; Shastry, K.; Marcu, L. Time-domain laser-induced fluorescence spectroscopy apparatus for clinical diagnostics. Rev. Sci. Instrum. 2004, 75, 151. [Google Scholar] [CrossRef]
- Papaioannou, T.; Preyer, N.W.; Fang, Q.; Brightwell, A.; Carnohan, M.; Cottone, G.; Ross, R.; Jones, L.R.; Marcu, L. Effects of fiber-optic probe design and probe-to-target distance on diffuse reflectance measurements of turbid media: an experimental and computational study at 337 nm. Appl. Opt. 2004, 43, 2846–2860. [Google Scholar] [CrossRef] [PubMed]
- Pfefer, T.J.; Paithankar, D.Y.; Poneros, J.M.; Schomacker, K.T.; Nishioka, N.S. Temporally and spectrally resolved fluorescence spectroscopy for the detection of high grade dysplasia in Barrett’s esophagus. Lasers Surg. Med. 2003, 32, 10–16. [Google Scholar] [PubMed]
- Mycek, M.-A.; Schomacker, K.T.; Nishioka, N.S. Colonic polyp differentiation using time-resolved autofluorescence spectroscopy. Gastroenterol. Endosc. 1998, 48, 390–394. [Google Scholar] [CrossRef]
- Butte, P.V.; Fang, Q.; Jo, J.A.; Yong, W.H.; Pikul, B.K.; Black, K.L.; Marcu, L. Intraoperative delineation of primary brain tumors using time-resolved fluorescence spectroscopy. J. Biomed. Opt. 2010, 15, 027008. [Google Scholar] [CrossRef]
- Butte, P.V; Mamelak, A.N.; Nuno, M.; Bannykh, S.I.; Black, K.L.; Marcu, L. Fluorescence lifetime spectroscopy for guided therapy of brain tumors. Neuroimage 2011, 54, S125–S135. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Hwang, J.; Krishnamoorthy, M.; Ye, K.; Zhang, Y.; Ning, J.; Wang, R.C.; Deen, M.J.; Fang, Q. High-throughput acousto-optic-tunable-filter-based time-resolved fluorescence spectrometer for optical biopsy. Opt. Lett. 2009, 34, 1132–1134. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Papaioannou, T.; Fang, Q. Single-shot acquisition of time-resolved fluorescence spectra using a multiple delay optical fiber bundle. Opt. Lett. 2008, 33, 791–793. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; An, R.; Hayward, J.E.; Farrell, T.J.; Fang, Q. Hyperspectral fluorescence lifetime imaging for optical biopsy. J. Biomed. Opt. 2013, 18, 1–7. [Google Scholar] [CrossRef]
- Sun, Y.; Chaudhari, A.J.; Lam, M.; Xie, H.; Yankelevich, D.R.; Phipps, J.; Liu, J.; Fishbein, M.C.; Cannata, J.M.; Shung, K.K.; et al. Multimodal characterization of compositional, structural and functional features of human atherosclerotic plaques. Biomed. Opt. Express 2011, 2, 2288–2298. [Google Scholar] [CrossRef] [PubMed]
- Munro, I.; McGinty, J.; Galletly, N.; Requejo-Isidro, J.; Lanigan, P.M.P.; Elson, D.S.; Dunsby, C.; Neil, M.A.A.; Lever, M.J.; Stamp, G.W.H.; et al. Toward the clinical application of time-domain fluorescence lifetime imaging. J. Biomed. Opt. 2005, 10, 051403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agronskaia, A.V; Tertoolen, L.; Gerritsen, H.C. Fast fluorescence lifetime imaging of calcium in living cells. J. Biomed. Opt. 2004, 9, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Sharman, K.K.; Periasamy, A.; Ashworth, H.; Demas, J.N. Error analysis of the rapid lifetime determination method for double-exponential decays and new windowing schemes. Anal. Chem. 1999, 71, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Booth, M.J.; Wilson, T. Low-cost, frequency-domain, fluorescence lifetime confocal microscopy. J. Microsc. 2004, 214, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Phipps, J.E.; Meier, J.; Hatami, N.; Poirier, B.; Elson, D.S.; Farwell, D.G.; Marcu, L. Endoscopic fluorescence lifetime imaging for in vivo intraoperative diagnosis of oral carcinoma. Microsc. Microanal. 2013, 4, 1–8. [Google Scholar]
- Celli, A.; Sanchez, S.; Behne, M.; Hazlett, T.; Gratton, E.; Mauro, T. The epidermal Ca(2+) gradient: Measurement using the phasor representation of fluorescent lifetime imaging. Biophys. J. 2010, 98, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Barber, P.R.; Ameer-Beg, S.M.; Gilbey, J.; Carlin, L.; Keppler, M.; Ng, T.; Vojnovic, B. Multiphoton time-domain fluorescence lifetime imaging microscopy: Practical application to protein-protein interactions using global analysis. J. R. Soc. Interface 2009, 6, S93–S105. [Google Scholar]
- Leung, R.W.K.; Yeh, S.-C.A.; Fang, Q. Effects of incomplete decay in fluorescence lifetime estimation. Biomed. Opt. Express 2011, 2, 2517–2531. [Google Scholar] [PubMed]
- Ruck, A. Fluorescence lifetime imaging in PDT. An overview. Med. Laser Appl. 2005, 20, 125–129. [Google Scholar] [CrossRef]
- Russell, J.A.; Diamond, K.R.; Collins, T.J.; Tiedje, H.F.; Hayward, J.E.; Farrell, T.J.; Patterson, M.S.; Fang, Q. Characterization of fluorescence lifetime of Photofrin and delta-aminolevulinic acid induced protoporphyrin IX in living cells using single and two-photon excitation. IEEE J. Quantum Electron. 2008, 14, 158–166. [Google Scholar] [CrossRef]
- Theodossiou, T.; MacRobert, A.J. Comparison of the photodynamic effect of exogenous photoprotoporphyrin and protoporphyrin IX on PAM 212 murine keratinocytes. Photochem. Photobiol. 2002, 76, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Howe, L.; Sucheta, A.; Einarsdottir, O.; Zhang, J.Z. Time-resolved studies of the excited-state dynamics of meso-Tetra (hydroxylphenyl) cholorin in Solution. Photochem. Photobiol. 1999, 69, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Connelly, J.P.; Botchway, S.W.; Kunz, L.; Pattison, D.; Parker, A.W.; MacRobert, A.J. Time-resolved fluorescence imaging of photosensitiser distributions in mammalian cells using a picosecond laser line-scanning microscope. J. Photochem. Photobiol. A Chem. 2001, 142, 169–175. [Google Scholar] [CrossRef]
- Mermut, O.; Noiseux, I.; Bouchard, J.-P.; Cormier, J.-F.; Desroches, P.; Fortin, M.; Gallant, P.; Leclair, S.; Vernon, M.L.; et al. Effect of liposomal confinement on photothermal and photo-oximetric fluorescence lifetimes of photosensitizers with varying hydrophilicity. J. Biomed. Opt. 2008, 13, 041314. [Google Scholar] [CrossRef] [PubMed]
- Mo, W.; Rohrbach, D.; Sunar, U. Imaging a photodynamic therapy photosensitizer in vivo with a time-gated fluorescence tomography system. J. Biomed. Opt. 2012, 17, 071306. [Google Scholar] [CrossRef] [PubMed]
- Kelbauskas, L.; Dietel, W. Internalization of aggregated photosensitizers by tumor cells : subcellular yime-resolved fluorescence spectroscopy on derivatives of pyropheophorbide-a ethers and chlorin e6 under femtosecond one- and two-photon excitation. Photochem. Photobiol. 2002, 76, 686–694. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, A.E.; Gallagher, W.M.; Byrne, A.T. Porphyrin and nonporphyrin photosensitizers in oncology: preclinical and clinical advances in photodynamic therapy. Photochem. Photobiol. 2009, 85, 1053–1074. [Google Scholar] [CrossRef] [PubMed]
- Yeh, S.-C.A.; Diamond, K.R.; Patterson, M.S.; Nie, Z.; Hayward, J.E.; Fang, Q. Monitoring photosensitizer uptake using two photon fluorescence lifetime imaging microscopy. Theranostics 2012, 2, 817–826. [Google Scholar] [PubMed]
- Kress, M.; Meier, T.; Steiner, R.; Dolp, F.; Erdmann, R.; Ortmann, U.; Rück, A. Time-resolved microspectrofluorometry and fluorescence lifetime imaging of photosensitizers using picosecond pulsed diode lasers in laser scanning microscopes. J. Biomed. Opt. 2003, 8, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Hilf, R. Mitochondria are targets of photodynamic therapy. J. Bioenerg. Biomembr. 2007, 39, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.; Oseroff, A. Mitochondria-based photodynamic anti-cancer therapy. Adv. Drug Deliv. Rev. 2001, 49, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Saczko, J.; Mazurkiewicz, M.; Chwiłkowska, A.; Kulbacka, J.; Kramer, G.; Ługowski, M.; Snietura, M.; Banaś, T. Intracellular distribution of Photofrin in malignant and normal endothelial cell lines. Folia Biol. (Praha). 2007, 53, 7–12. [Google Scholar] [PubMed]
- Singh, G.; Jeeves, W.P.; Wilson, B.C.; Jang, D. Mitochondrial photosensitization by Photofrin II. Photochem. Photobiol. 1987, 46, 645–649. [Google Scholar] [CrossRef]
- Hsieh, Y.-J.; Wu, C.-C.; Chang, C.-J.; Yu, J.-S. Subcellular localization of Photofrin determines the death phenotype of human epidermoid carcinoma A431 cells triggered by photodynamic therapy: when plasma membranes are the main targets. J. Cell. Physiol. 2003, 194, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Lassalle, H.-P.; Wagner, M.; Bezdetnaya, L.; Guillemin, F.; Schneckenburger, H. Fluorescence imaging of Foscan and Foslip in the plasma membrane and in whole cells. J. Photochem. Photobiol. B. 2008, 92, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Khursid, A.; Atif, M.; Firdous, S.; Zaidi, S.S.Z.; Salman, R.; Ikram, M. Study of the efficacy of 5 ALA-mediated photodynamic therapy on human larynx squamous cell carcinoma (Hep2c) cell line. Laser Phys. 2010, 20, 1673–1678. [Google Scholar] [CrossRef]
- AlSalhi, M.S.; Atif, M.; AlObiadi, A.; Aldwayyan, A.S. Photodynamic damage study of HeLa cell line using ALA. Laser Phys. 2011, 21, 733–739. [Google Scholar] [CrossRef]
- Fakhar-e-Alam, M.; Atif, M.; Rehman, T.; Sadia, H.; Firdous, S. The role of sensitivity of ALA (PpIX)-based PDT on Human embryonic kidney cell line (HEK293T). Laser Phys. 2011, 21, 1428–1437. [Google Scholar] [CrossRef]
- Atif, M.; Fakhar-e-Alam, M.; Firdous, S.; Zaidi, S.S.Z.; Suleman, R.; Ikram, M. Study of the efficacy of 5-ALA mediated photodynamic therapy on human rhabdomyosarcoma cell line (RD). Laser Phys. Lett. 2010, 7, 757–764. [Google Scholar]
- Chwiłkowska, A.; Saczko, J.; Modrzycka, T.; Marcinkowska, A.; Malarska, A.; Bielewicz, J.; Patalas, D.; Banaś, T. Uptake of photofrin II, a photosensitizer used in photodynamic therapy, by tumour cells in vitro. Acta Biochim. Pol. 2003, 50, 509–513. [Google Scholar] [PubMed]
- Woodburn, K.W.; Vardaxis, N.J.; Hill, J.S.; Kaye, A.H.; Phillips, D.R. Subcellular localization of porphyrins using confocal laser scanning microscopy. Photochem. Photobiol. 1991, 54, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Geze, M.; Morli, P.; Mazi, J.C.; Smith, K.M.; Santw, R. Lysosomes, a key target of hydrophobic for photochemotherapeutic applications photosensitizers proposed. 1993, 20, 23–35. [Google Scholar]
- Høgset, A. Photochemical internalisation in drug and gene delivery. Adv. Drug Deliv. Rev. 2004, 56, 95–115. [Google Scholar] [CrossRef] [PubMed]
- Scully, A.; Ostler, R.B.; Phillips, D.; O’Neill, P.; Townsend, K.M.S.; Parker, A.W.; MacRobert, A.J. Application of fluorescence lifetime imaging microscopy to the investigation of intracellular PDT mechanisms. Bioimaging 1997, 5, 9–18. [Google Scholar] [CrossRef]
- Moan, J.; Berg, K.; Bommer, J.C. Action spectra of phthalocyanines with respect to photosensitization of cells. Photochem. Photobiol. 1992, 56, 171–175. [Google Scholar] [PubMed]
- Ma, L.; Bagdonas, S.; Moan, J. The photosensitizing effect of the photoproduct of protoporphyrin IX. J. Photochem. Photobiol. B. 2001, 60, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Nothdurft, R.E.; Patwardhan, S.V; Akers, W.; Ye, Y.; Achilefu, S.; Culver, J.P. In vivo fluorescence lifetime tomography. J. Biomed. Opt. 2009, 14, 024004. [Google Scholar]
- Kumar, A.T.N.; Raymond, S.B.; Dunn, A.K.; Bacskai, B.J.; Boas, D.A. A time domain fluorescence tomography system for small animal imaging. IEEE Trans. Med. Imaging 2008, 27, 1152–1163. [Google Scholar] [CrossRef] [PubMed]
- Milstein, A.B.; Oh, S.; Webb, K.J.; Bouman, C.A.; Zhang, Q.; Boas, D.A.; Millane, R.P. Fluorescence optical diffusion tomography. Appl. Opt. 2003, 42, 3081–3094. [Google Scholar] [CrossRef] [PubMed]
- Ntziachristos, V.; Weissleder, R. Experimental three-dimensional fluorescence reconstruction of diffuse media by use of a normalized Born approximation. Opt. Lett. 2001, 26, 893–895. [Google Scholar] [CrossRef] [PubMed]
- Nava, H.R.; Allamaneni, S.S.; Dougherty, T.J.; Michele, T.; Tan, W.; Wilding, G.; Henderson, B.W. Photodynamic therapy (PDT) using HPPH for the treatment of precancerous lesions associated with Barrett’s esophagus. Lasers Surg Med 2011, 43, 705–712. [Google Scholar]
- Sunar, U.; Rohrbach, D.; Rigual, N.; Tracy, E.; Keymel, K.; Cooper, M.T.; Baumann, H.; Henderson, B.H. Monitoring photobleaching and hemodynamic responses to HPPH-mediated photodynamic therapy of head and neck cancer: a case report. Opt. Express 2010, 18, 14969–14978. [Google Scholar] [CrossRef] [PubMed]
- Sud, D.; Zhong, W.; Beer, D.G.; Mycek, M.-A. Time-resolved optical imaging provides a molecular snapshot of altered metabolic function in living human cancer cell models. Opt. Express 2006, 14, 4412–4426. [Google Scholar] [CrossRef] [PubMed]
- Pogue, B.W.; Pitts, J.D.; Mycek, M.-A.; Sloboda, R.D.; Wilmot, C.M.; Brandsema, J.F.; O’Hara, J. In vivo NADH fluorescence monitoring as an assay for cellular damage in photodynamic therapy. Photochem. Photobiol. 2001, 74, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Ghukasyan, V.V; Kao, F. Monitoring cellular metabolism with fluorescence lifetime of reduced nicotinamide adenine dinucleotide. J. Phys. Chem. 2009, 113, 11532–11540. [Google Scholar]
- Wang, H.-W.; Gukassyan, V.; Chen, C.-T.; Wei, Y.-H.; Guo, H.-W.; Yu, J.-S.; Kao, F.-J. Differentiation of apoptosis from necrosis by dynamic changes of reduced nicotinamide adenine dinucleotide fluorescence lifetime in live cells. J. Biomed. Opt. 2008, 13, 054011. [Google Scholar] [CrossRef] [PubMed]
- Maeno, E.; Shimizu, T.; Okada, Y. Normotonic cell shrinkage induces apoptosis under extracellular low Cl conditions in human lymphoid and epithelial cells. Acta physiol 2006, 187, 217–222. [Google Scholar]
- Yu, J.-S.; Guo, H.-W.; Wang, C.-H.; Wei, Y.-H.; Wang, H.-W. Increase of reduced nicotinamide adenine dinucleotide fluorescence lifetime precedes mitochondrial dysfunction in staurosporine-induced apoptosis of HeLa cells. J. Biomed. Opt. 2011, 16, 036008. [Google Scholar] [CrossRef] [PubMed]
- Becker, T.L.; Paquette, A.D.; Keymel, K.R.; Henderson, B.W.; Sunar, U. Monitoring blood flow responses during topical ALA-PDT. Biomed. Opt. Express 2010, 2, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Niesner, R.; Peker, B.; Schlüsche, P.; Gericke, K.-H.; Hoffmann, C.; Hahne, D.; Müller-Goymann, C. 3D-resolved investigation of the pH gradient in artificial skin constructs by means of fluorescence lifetime imaging. Pharm. Res. 2005, 22, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Georgakoudi, I.; Nichols, M.G.; Foster, T.H. The mechanism of Photofrin photobleaching and its consequences for photodynamic dosimetry. Photochem. Photobiol. 1997, 65, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Foster, T.H. Photochemical oxygen consumption sensitized by a porphyrin phosphorescent probe in two model systems. Biophys. J. 2000, 78, 2597–2605. [Google Scholar] [CrossRef] [PubMed]
- Finlay, J.C.; Mitra, S.; Foster, T.H. In vivo mTHPC photobleaching in normal rat skin exhibits unique irradiance-dependent features. Photochem. Photobiol. 2002, 75, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Nichols, M.G.; Foster, T.H. Oxygen diffusion and reaction kinetics in the photodynamic therapy of multicell tumour spheroids. Phys. Med. Biol. 1994, 39, 2161–2181. [Google Scholar] [CrossRef] [PubMed]
- Errington, R.J.; Ameer-Beg, S.M.; Vojnovic, B.; Patterson, L.H.; Zloh, M.; Smith, P.J. Advanced microscopy solutions for monitoring the kinetics and dynamics of drug-DNA targeting in living cells. Adv. Drug Deliv. Rev. 2005, 57, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.-W.; Chen, C.-T.; Wei, Y.-H.; Lee, O.K.; Gukassyan, V.; Kao, F.-J.; Wang, H.-W. Reduced nicotinamide adenine dinucleotide fluorescence lifetime separates human mesenchymal stem cells from differentiated progenies. J. Biomed. Opt. 2008, 13, 050505. [Google Scholar] [PubMed]
- Su, G.-C.; Wei, Y.-H.; Wang, H.-W. NADH fluorescence as a photobiological metric in 5-aminolevlinic acid (ALA)-photodynamic therapy. Opt. Express 2011, 19, 21145–21154. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Urayama, P.; Mycek, M.-A. Imaging fluorescence lifetime modulation of a ruthenium-based dye in living cells: the potential for oxygen sensing. J. Phys. D. Appl. Phys. 2003, 36, 1689–1695. [Google Scholar] [CrossRef]
- Celli, J.P.; Spring, B.Q.; Rizvi, I.; Evans, C.L.; Samkoe, K.S.; Verma, S.; Pogue, B.W.; Hasan, T. Imaging and photodynamic therapy: Mechanisms, monitoring, and optimization. Chem. Rev. 2010, 110, 2795–2838. [Google Scholar] [PubMed]
- Lee, K.C.; Siegel, J.; Webb, S.E.; Lévêque-Fort, S.; Cole, M.J.; Jones, R.; Dowling, K.; Lever, M.J.; French, P.M. Application of the stretched exponential function to fluorescence lifetime imaging. Biophys. J. 2001, 81, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Budiman, M.; Stroshine, R.L.; Cornillon, P. Moisture measurement in cheese analogue using stretched and multi-exponential models of the magnetic resonance T2 relaxation curve. J. Dairy Res. 2002, 69, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.; Cotlet, M.; Hofkens, J.; Gensch, T.; De Schryver, F.C.; Schaffer, J.; Seidel, C.A. An experimental comparison of the maximum likelihood estimation and nonlinear least-squares fluorescence lifetime analysis of single molecules. Anal. Chem. 2001, 73, 2078–2086. [Google Scholar] [CrossRef] [PubMed]
- Maarek, J.M.; Marcu, L.; Snyder, W.J.; Grundfest, W.S. Time-resolved fluorescence spectra of arterial fluorescent compounds: Reconstruction with the Laguerre expansion technique. Photochem. Photobiol. 2000, 71, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.A.; Fang, Q.; Marcu, L. Ultrafast Method for the Analysis of Fluorescence Lifetime Imaging Microscopy Data Based on the Laguerre Expansion Technique. IEEE J. Quantum Electron. 2005, 11, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.A.; Fang, Q.; Papaioannou, T.; Baker, J.D.; Dorafshar, A.H.; Reil, T.; Qiao, J.-H.; Fishbein, M.C.; Freischlag, J.A.; Marcu, L. Laguerre-based method for analysis of time-resolved fluorescence data: application to in vivo characterization and diagnosis of atherosclerotic lesions. J. Biomed. Opt. 2006, 11, 021004. [Google Scholar] [CrossRef]
- Chen, Y.; Periasamy, A. Characterization of two-photon excitation fluorescence lifetime imaging microscopy for protein localization. Microsc. Res. Tech. 2004, 63, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Barber, P.R.; Ameer-Beg, S.M.; Gilbey, J.D.; Edens, R.J.; Ezike, I.; Vojnovic, B. Global and pixel kinetic data analysis for FRET detection by multi-photon time-domain FLIM. Proc. SPIE 2005, 5700, 171–181. [Google Scholar]
- Jacques, S.L. How tissue optics affect dosimetry of photodynamic therapy. J. Biomed. Opt. 2010, 15, 051608. [Google Scholar] [CrossRef] [PubMed]
- Patterson, M.S.; Pogue, B.W. Mathematical model for time-resolved and frequency-domain fluorescence spectroscopy in biological tissues. Appl. Opt. 1994, 33, 1963–1974. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Partovi, F.; Field, M.S.; Rava, R.P. Diffuse reflectance from turbid media: An analytical model of photon migration. Appl. Opt. 1993, 32, 1115–1121. [Google Scholar] [CrossRef]
- Vishwanath, K.; Pogue, B.; Mycek, M. Quantitative fluorescence lifetime spectroscopy in turbid media : Comparison of theoretical , experimental and computational methods. Phys. Med. Biol. 2002, 47, 3387–3405. [Google Scholar] [CrossRef] [PubMed]
- Vishwanath, K.; Mycek, M.-A. Time-resolved photon migration in bi-layered tissue models. Opt. Express 2005, 13, 7466–7482. [Google Scholar] [CrossRef] [PubMed]
- Pyka, T.; Schulz, R.; Ale, A.; Ntziachristos, V. Revisiting the normalized Born approximation: effects of scattering. Opt. Lett. 2011, 36, 4329–4331. [Google Scholar]
- Patwardhan, S.V; Culver, J.P. Quantitative diffuse optical tomography for small animals using an ultrafast gated image intensifier. J. Biomed. Opt. 2008, 13, 011009. [Google Scholar] [CrossRef] [PubMed]
- Andersen, A.H.; Kak, A.C. Simultaneous algebraic reconstruction technique (SART): A superior implementation of the art algorithm. Ultrason. Imaging 1984, 6, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.T.N.; Raymond, S.B.; Bacskai, B.J.; Boas, D.A. Comparison of frequency-domain and time-domain fluorescence lifetime tomography. Opt. Lett. 2008, 33, 470–472. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.T.; Raymond, S.B.; Boverman, G.; Boas, D.A.; Bacskai, B.J. Time resolved fluorescence tomography of turbid media based on lifetime contrast. Opt. Express 2006, 14, 12255–12270. [Google Scholar] [CrossRef] [PubMed]
- Fruhwirth, G.O.; Ameer-Beg, S.; Cook, R.; Watson, T.; Ng, T.; Festy, F. Fluorescence lifetime endoscopy using TCSPC for the measurement of FRET in live cells. Opt. Express 2010, 18, 11148–11158. [Google Scholar] [CrossRef] [PubMed]
- König, K. Clinical multiphoton tomography. J. Biophotonics 2008, 1, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Konig, K.; Riemann, I. High-resolution multiphoton tomography of human skin with subcellular spatial resolution and picosecond time resolution. J. Biomed. Opt. 2003, 8, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Bec, J.; Liu, J.; Sun, Y.; Lam, M.; Yankelevich, D.R.; Marcu, L. Multispectral scanning time-resolved fluorescence spectroscopy (TRFS) technique for intravascular diagnosis. Biomed. Opt. Express 2012, 3, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- De Beule, P.A.A.; Dunsby, C.; Galletly, N.P.; Stamp, G.W.; Chu, A.C.; Anand, U.; Anand, P.; Benham, C.D.; Naylor, A.; French, P.M.W. A hyperspectral fluorescence lifetime probe for skin cancer diagnosis. Rev. Sci. Instrum. 2007, 78, 123101. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeh, S.-C.A.; Patterson, M.S.; Hayward, J.E.; Fang, Q. Time-Resolved Fluorescence in Photodynamic Therapy. Photonics 2014, 1, 530-564. https://doi.org/10.3390/photonics1040530
Yeh S-CA, Patterson MS, Hayward JE, Fang Q. Time-Resolved Fluorescence in Photodynamic Therapy. Photonics. 2014; 1(4):530-564. https://doi.org/10.3390/photonics1040530
Chicago/Turabian StyleYeh, Shu-Chi Allison, Michael S. Patterson, Joseph E. Hayward, and Qiyin Fang. 2014. "Time-Resolved Fluorescence in Photodynamic Therapy" Photonics 1, no. 4: 530-564. https://doi.org/10.3390/photonics1040530
APA StyleYeh, S.-C. A., Patterson, M. S., Hayward, J. E., & Fang, Q. (2014). Time-Resolved Fluorescence in Photodynamic Therapy. Photonics, 1(4), 530-564. https://doi.org/10.3390/photonics1040530