Exploring the Potential of Airyscan Microscopy for Live Cell Imaging



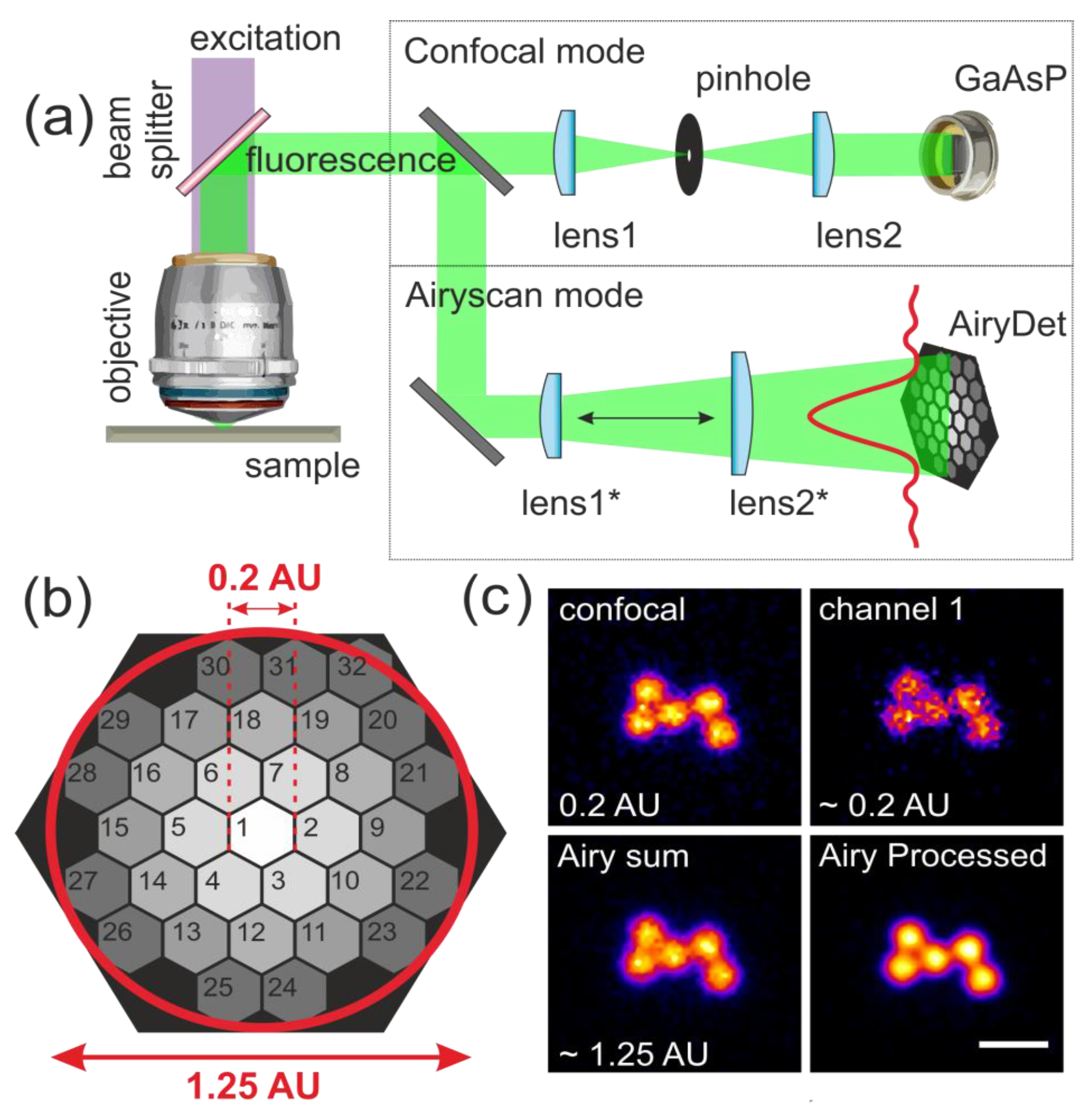{kind=link}
{kind=link}
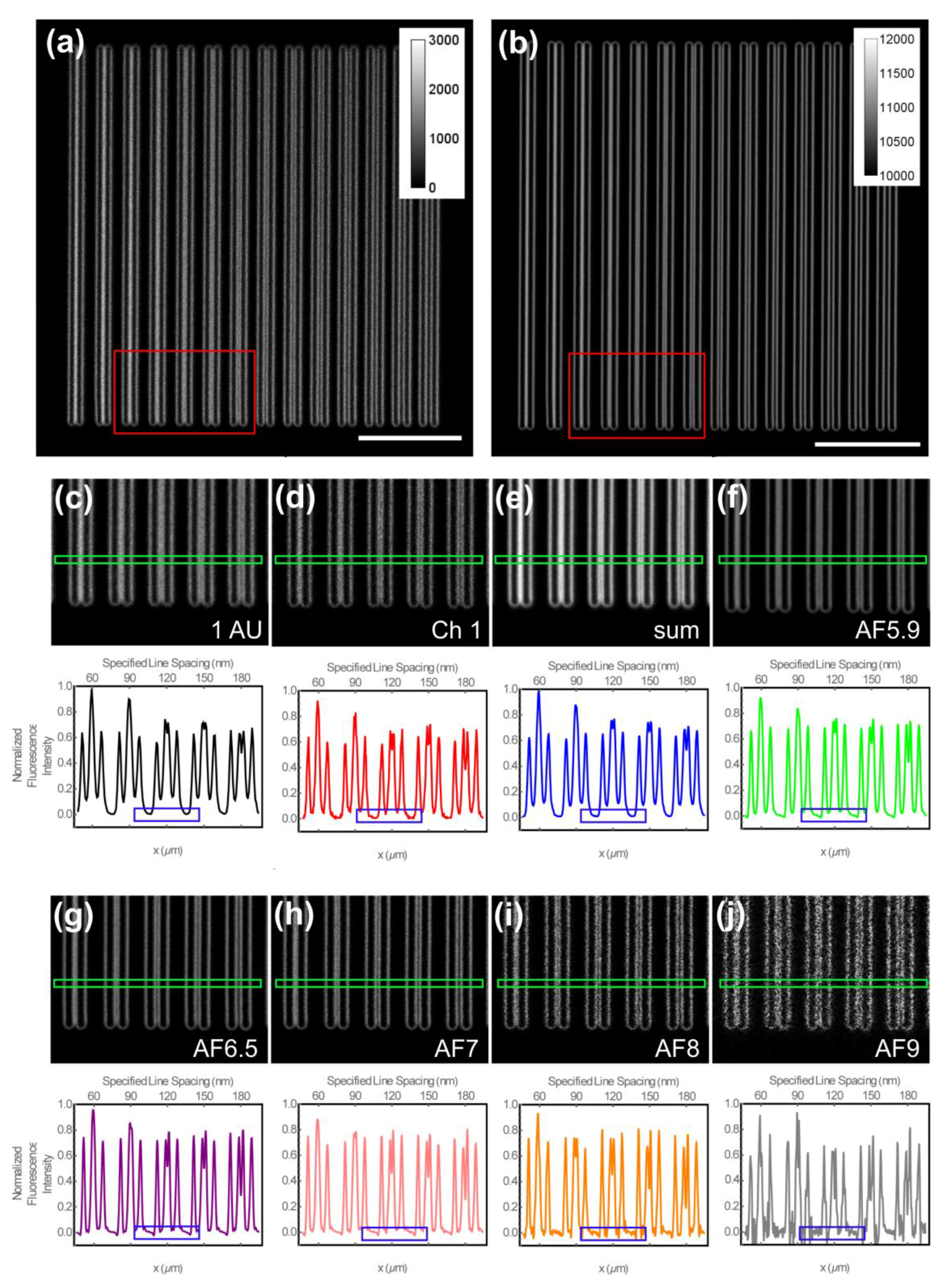{kind=link}
{kind=link}
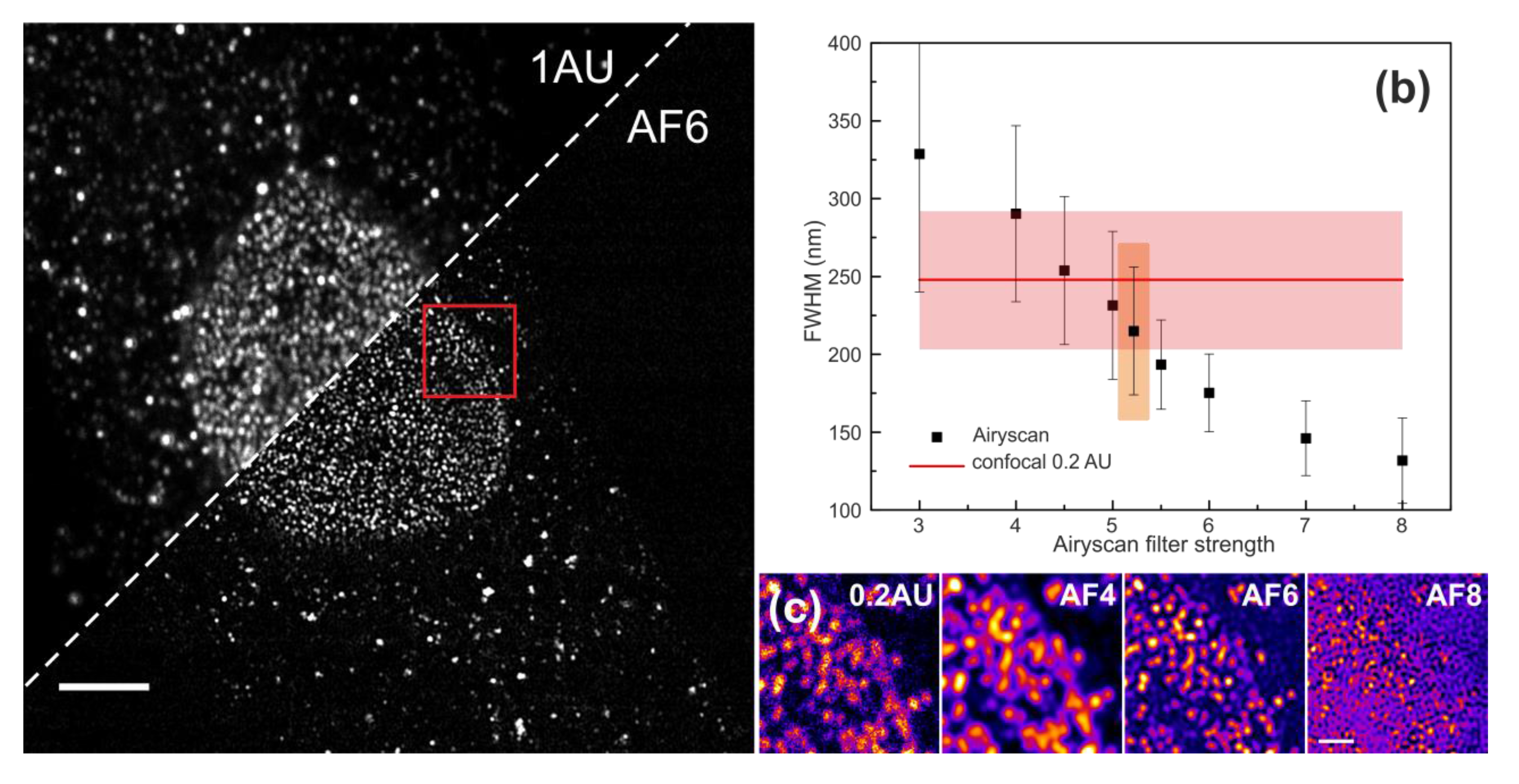{kind=link}
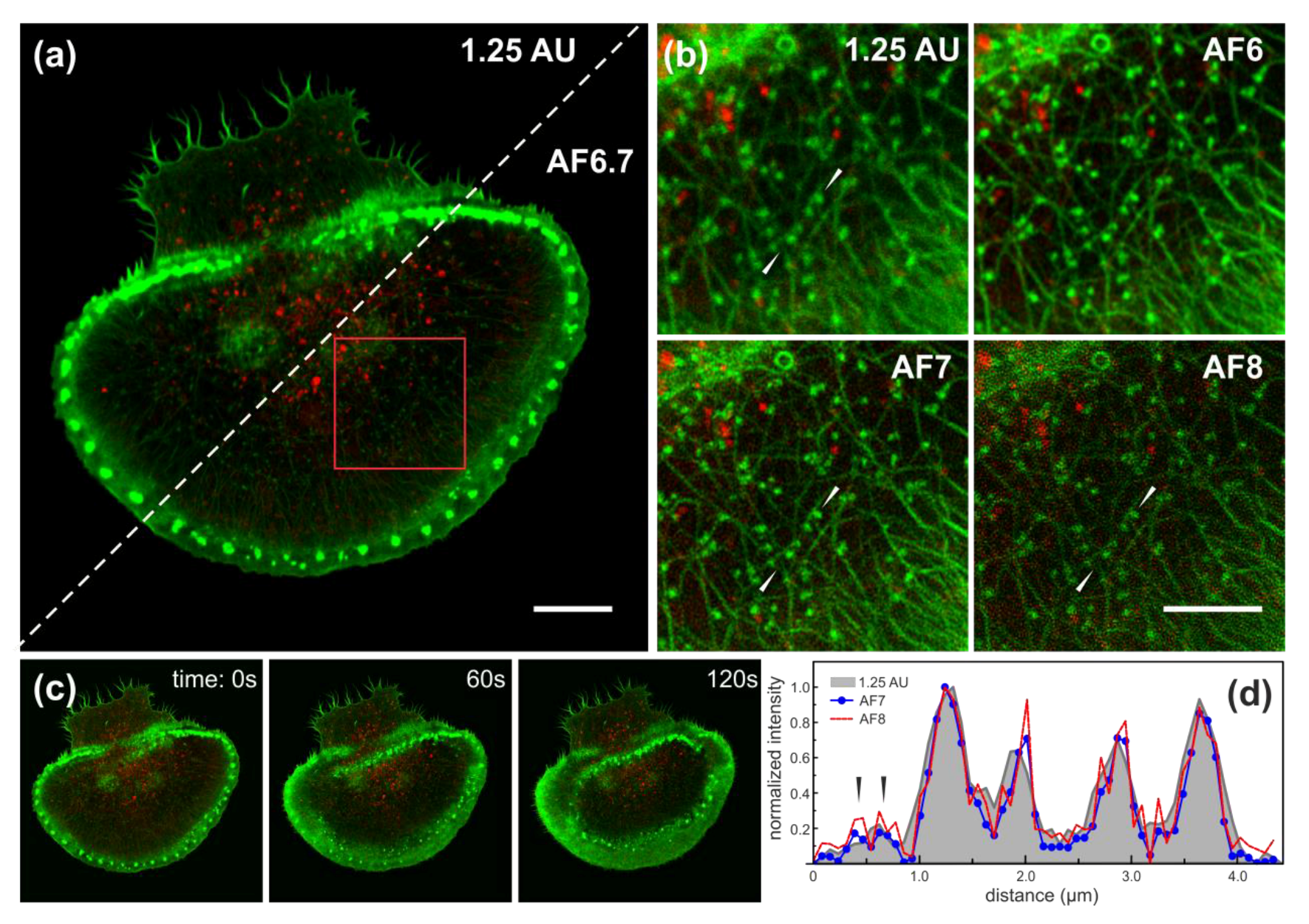{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell and Tissue Culture
2.1.1. HeLa Cells
2.1.2. Immunostaining of Nuclear Pore Samples
2.1.3. Rat Basophilic Leukaemia Cells
2.1.4. Plasmids
2.1.5. Generation of Stable RBL Cell Lines
2.2. Experimental Setup
2.2.1. GATTA-Beads
2.2.2. TetraSpeck Beads
2.2.3. Argolight SIM Slide
2.2.4. Nuclear Pore Complexes in HeLa Cells
2.2.5. Microscopy Cover Slide Preparation
2.2.6. Rat Basophilic Leukaemia Live-Cell Imaging
2.2.7. SNAP-Tag Labelling
2.3. Airyscan Processing
2.4. Data Analysis
2.4.1. Determination of the Spatial Resolution
2.4.2. Fitting of Fluorescent Bead Reference Samples and NPC Samples
2.4.3. Argo-SIM Slide Reference Samples
2.5. Determination of the SNR
3. Results
3.1. Evaluation of the Spatial Resolution and Signal-To-Noise Ratio Using Bead Reference Samples
3.2. Determination of the Spatial Resolution and Signal-To-Noise Ratio Using Argolight-SIM Slide
3.3. Airyscan Imaging of Nuclear Pore Complexes
3.4. Airyscan Imaging of Live Rat Basophilic Leukaemia Cells
4. Discussion and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Godin, A.G.; Lounis, B.; Cognet, L. Super-resolution microscopy approaches for live cell imaging. Biophys. J. 2014, 107, 1777–1784. [Google Scholar] [CrossRef] [PubMed]
- Leung, B.O.; Chou, K.C. Review of super—Resolution fluorescence microscopy for biology. Appl. Spectrosc. 2011, 65, 967–980. [Google Scholar] [CrossRef] [PubMed]
- Hell, S.W. Far-Field Optical Nanoscopy. Science 2007, 316, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Babcock, H.; Zhuang, X. Breaking the Diffraction Barrier: Super-Resolution Imaging of Cells. Cell 2010, 143, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Stephens, D.J.; Allan, V.J. Light Microscopy Techniques for Live Cell Imaging. Science 2003, 300, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Fritzsche, M.; Erlenkamper, C.; Moeendarbary, E.; Charras, G.T.; Kruse, K. Actin kinetics shapes cortical network structure and mechanics. Sci. Adv. 2016, 2, e1501337. [Google Scholar] [CrossRef] [PubMed]
- Clausen, M.P.; Colin-York, H.; Schneider, F.; Eggeling, C.; Fritzsche, M. Dissecting the actin cortex density and membrane-cortex distance in living cells by super-resolution microscopy. J. Phys. D 2017, 50, 64002. [Google Scholar] [CrossRef] [PubMed]
- Schermelleh, L.; Heintzmann, R.; Leonhardt, H. A guide to super-resolution fluorescence microscopy. J. Cell Biol. 2010, 190, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Shao, L.; Chen, B.-C.; Zhang, X.; Zhang, M.; Moses, B.; Milkie, D.E.; Beach, J.R.; Hammer, J.A.; Pasham, M.; et al. Extended-resolution structured illumination imaging of endocytic and cytoskeletal dynamics. Science 2015, 349, aab3500. [Google Scholar] [CrossRef] [PubMed]
- Wegel, E.; Göhler, A.; Lagerholm, B.C.; Wainman, A.; Uphoff, S.; Kaufmann, R.; Dobbie, I.M. Imaging cellular structures in super-resolution with SIM, STED and Localisation Microscopy: A practical comparison. Sci. Rep. 2016, 6, 27290. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Babcock, H.P.; Zhuang, X. Dual-objective STORM reveals three-dimensional filament organization in the actin cytoskeleton. Nat. Methods 2012, 9, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [PubMed]
- Gould, T.J.; Gunewardene, M.S.; Gudheti, M.V.; Verkhusha, V.V.; Yin, S.-R.; Gosse, J.A.; Hess, S.T. Nanoscale imaging of molecular positions and anisotropies. Nat. Methods 2008, 5, 1027–1030. [Google Scholar] [CrossRef] [PubMed]
- Vogelsang, J.; Cordes, T.; Forthmann, C.; Steinhauer, C.; Tinnefeld, P. Controlling the fluorescence of ordinary oxazine dyes for single-molecule switching and superresolution microscopy. Proc. Natl. Acad. Sci. USA 2009, 106, 8107–8112. [Google Scholar] [CrossRef] [PubMed]
- Colin-York, H.; Eggeling, C.; Fritzsche, M. Dissection of mechanical force in living cells by super-resolved traction force microscopy. Nat. Protoc. 2017, 12, 783–796. [Google Scholar] [CrossRef] [PubMed]
- Bergermann, F.; Alber, L.; Sahl, S.J.; Engelhardt, J.; Hell, S.W. 2000-fold parallelized dual-color STED fluorescence nanoscopy. Opt. Express 2015, 23, 211. [Google Scholar] [CrossRef] [PubMed]
- Hein, B.; Willig, K.I.; Hell, S.W. Stimulated emission depletion (STED) nanoscopy of a fluorescent protein-labeled organelle inside a living cell. Proc. Natl. Acad. Sci. USA 2008, 105, 14271–14276. [Google Scholar] [CrossRef] [PubMed]
- Fritzsche, M.; Li, D.; Colin-York, H.; Chang, V.T.; Moeendarbary, E.; Felce, J.H.; Sezgin, E.; Charras, G.; Betzig, E.; Eggeling, C. Self-organizing actin patterns shape membrane architecture but not cell mechanics. Nat. Commun. 2017, 8, 14347. [Google Scholar] [CrossRef] [PubMed]
- Conchello, J.-A.; Lichtman, J.W. Optical sectioning microscopy. Nat. Methods 2005, 2, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Cox, G.; Sheppard, C.J.R. Practical Limits of Resolution in Confocal and Non-Linear Microscopy. Microsc. Res. Tech. 2004, 63, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Cremer, C.; Masters, B.R. Resolution enhancement techniques in microscopy. Eur. Phys. J. H 2013, 38, 281–344. [Google Scholar] [CrossRef]
- Sarder, P.; Nehorai, A. Deconvolution methods for 3-D fluorescence microscopy images. IEEE Signal. Process. Mag. 2006, 23, 32–45. [Google Scholar] [CrossRef]
- De Luca, G.M.R.; Breedijk, R.M.P.; Brandt, R.A.J.; Zeelenberg, C.H.C.; de Jong, B.E.; Timmermans, W.; Azar, L.N.; Hoebe, R.A.; Stallinga, S.; Manders, E.M.M. Re-scan confocal microscopy: Scanning twice for better resolution. Biomed. Opt. Express 2013, 4, 2644. [Google Scholar] [CrossRef] [PubMed]
- Heintzmann, R.; Sarafis, V.; Munroe, P.; Nailon, J.; Hanley, Q.S.; Jovin, T.M. Resolution enhancement by subtraction of confocal signals taken at different pinhole sizes. Micron 2003, 34, 293–300. [Google Scholar] [CrossRef]
- Korobchevskaya, K.; Peres, C.; Li, Z.; Antipov, A.; Sheppard, C.J.R.; Diaspro, A.; Bianchini, P. Intensity weighted subtraction microscopy approach for image contrast and resolution enhancement. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, C.J.R.; Mehta, S.B.; Heintzmann, R. Superresolution by image scanning microscopy using pixel reassignment. Opt. Lett. 2013, 38, 2889–2892. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, R.C.; Woods, R.E. Digital Image Processing; Pearson Higher Ed: Cambridge, UK, 2011; pp. 231–346. [Google Scholar]
- Sánchez-Ortiga, E.; Sheppard, C.J.R.; Saavedra, G.; Martínez-Corral, M.; Doblas, A.; Calatayud, A. Subtractive imaging in confocal scanning microscopy using a CCD camera as a detector. Opt. Lett. 2012, 37, 1280–1282. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, R.; Weisshart, K. Airyscanning. G.I.T. Imaging Microsc. 2014, 3, 20–21. [Google Scholar]
- Bertero, M.; Brianzi, P.; Pike, E.R. Super-resolution in confocal scanning microscopy. Inverse Probl. 1999, 3, 195–212. [Google Scholar] [CrossRef]
- Sheppard, C.J.R. Superresolution in confocal Imaging. Optik 1988, 80, 53–54. [Google Scholar]
- Huff, J. The Airyscan detector from ZEISS: Confocal imaging with improved signal-to-noise ratio and super-resolution. Nat. Methods 2015, 12. [Google Scholar] [CrossRef]
- Sivaguru, M.; Urban, M.A.; Fried, G.; Wesseln, C.J.; Mander, L.; Punyasena, S.W. Comparative performance of airyscan and structured illumination superresolution microscopy in the study of the surface texture and 3D shape of pollen. Microsc. Res. Tech. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ball, G.; Demmerle, J.; Kaufmann, R.; Davis, I.; Dobbie, I.M.; Schermelleh, L. SIMcheck: A Toolbox for Successful Super-resolution Structured Illumination Microscopy. Sci. Rep. 2015, 5, 15915. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.C.; Fornerod, M.; Pickersgill, H.; Goldberg, M.; Allen, T.D.; Mattaj, I.W. The nucleoporin Nup153 is required for nuclear pore basket formation, nuclear pore complex anchoring and import of a subset of nuclear proteins. EMBO J. 2001, 20, 5703–5714. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.; Lučić, V.; Förster, F.; Baumeister, W.; Medalia, O. Snapshots of nuclear pore complexes in action captured by cryo-electron tomography. Nature 2007, 449, 611–615. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korobchevskaya, K.; Lagerholm, B.C.; Colin-York, H.; Fritzsche, M. Exploring the Potential of Airyscan Microscopy for Live Cell Imaging. Photonics 2017, 4, 41. https://doi.org/10.3390/photonics4030041
Korobchevskaya K, Lagerholm BC, Colin-York H, Fritzsche M. Exploring the Potential of Airyscan Microscopy for Live Cell Imaging. Photonics. 2017; 4(3):41. https://doi.org/10.3390/photonics4030041
Chicago/Turabian StyleKorobchevskaya, Kseniya, B. Christoffer Lagerholm, Huw Colin-York, and Marco Fritzsche. 2017. "Exploring the Potential of Airyscan Microscopy for Live Cell Imaging" Photonics 4, no. 3: 41. https://doi.org/10.3390/photonics4030041
APA StyleKorobchevskaya, K., Lagerholm, B. C., Colin-York, H., & Fritzsche, M. (2017). Exploring the Potential of Airyscan Microscopy for Live Cell Imaging. Photonics, 4(3), 41. https://doi.org/10.3390/photonics4030041