1. Introduction
Wheat (
Triticum aestivum L.) is one of the major constituents of the human diet because of its various end uses; it can be used to make a wide range of foods such as biscuits, bread and noodles [
1]. Starch is the predominant component in wheat grain, which is composed of two types of glucose polymers, amylose and amylopectin, and is present in different granule sizes. Protein is the second largest component in wheat grain, which is classified into two fractions according to its solubility in alcohol-water solutions: insoluble glutenins (about 40–50%) and soluble gliadins (about 50–60%). Glutenins are interchain disulfide (SS)-linked polymers with a wide molecular weight (Mw) distribution from 10
5 to 10
7 Da and impart the elasticity of dough. Two distinct groups of subunits can be distinguished: HMW-GS and LMW-GS. The majority of gliadins are monomeric proteins with Mw ranging from 3 to 8 × 10
4 Da and confer viscosity to the dough. Gliadins can be further classified into α-, ω- and γ-type subgroups. Heating is one of the most important processes in wheat flour processing, and the heat-induced physicochemical evolution of flour components is a complex process. It can improve food texture and hold moisture or functional components, which in turn affects the flavor, color and texture of the final product [
2]. Knowledge of the structure and physicochemistry changes of protein and starch during heat processing is important to understanding the mechanism of protein–starch interactions during the processing of flour as well as for the manufacture of wheat-based foods.
Protein and starch have complex hierarchical structures which are studied with multiple techniques. For example, the differences in the structure of gluten in different forms can be characterized by secondary, tertiary and spatial structure by Fourier transform infrared spectroscopy (FTIR), intrinsic fluorescence spectra and confocal laser scanning microscopy (CLSM). Meanwhile, FTIR, X-ray diffraction (XRD) and rapid viscosity analysis (RVA) can be used to determine the structure of starch from a nano-to-micrometers perspective [
3,
4]. During heat processing, starch granules are gelatinized and proteins are cross-linked, causing disruption of the multi-scale structure, with the extent of disruption depending on temperature and a series of heat processing conditions. Interactions between starch, proteins and other components during heat processing have been studied extensively in model systems [
5,
6]. Wang et al. [
7] extracted gliadin from wheat flour and found that gliadin polymers were formed at 90 °C and resulted in the loss of ethanol extractability. Wang et al. [
8] studied changes in chemical interactions during heat-induced wheat gluten gel formation, and found that crosslinking reactions among the wheat gluten molecules occurred when the temperature was higher than 60 °C. Liu et al. [
9] explored the disassembly mechanisms of starch granules during thermal food processing using purified wheat starch as a model material, and found the gelatinization and short-range molecular order of wheat starch were affected greatly by the heating temperature and water content.
The presence of multiple components in wheat-based food systems may affect the behavior of starch or protein during processing through the potential for complex formations [
10,
11]. The presence of multiple components in flour is assumed to affect the functionality of protein and starch in food systems. For example, the presence of proteins and other components in cereal-based food systems may affect the dynamic behaviors of starch or protein during heat processing through the potential for complex formation with them [
12]. However, there is little actual information on this and most research has been conducted on unitary studies of separated protein or starch. Therefore, study of the behavior of protein or starch which is isolated from the complete flour system cannot completely reflect the change and interaction of components in wheat-based foods during heat processing. For example, how protein and starch influence the behaviors of each other during the cooking of doughs and batters still remains unclear. Studying the mechanism of protein and starch behavior during wheat flour heating is an important step to predict and control the processing and functional properties of more complex cereal food systems. Therefore, in this study, a heating model system of wheat flour was used to study the molecular and physicochemical changes of starch and protein during heat processing.
2. Materials and Methods
2.1. Materials
Winter wheat (Triticum aestivum L.) cultivar Yangmai 22 was planted at the Center of Rice and Wheat Science and Technology demonstration in Jintan, Jiangsu province, China. The plant density was 1.6 × 106 ha−1. Fertilizers of N, P and K were applied as 180 kg N ha−1, 120 kg P2O5 ha−1 and 120 kg K2O ha−1, respectively. The ratio of basal to topdressing of N was 6:4, and topdressing fertilizer was applied at jointing stage.
At maturity, grains of Yangmai 22 were harvested and cleaned. After tempering to 14% moisture for 18 h, wheat grains were milled using a flour miller (ZS70-II, grain and oil foodstuff machine factory, Zhuozhou, China) with a 100μm mesh sieve. Flour was stored in refrigerator (4 °C) for further analysis.
2.2. Preparation of Flour Samples Heated with Different Temperature
In order to eliminate the influence of water, wheat flour (3.5 g) was weighed accurately into an aluminum case, and distilled water (25 g) was added to form wheat flour dispersions. Subsequently, the dispersions were homogenized for 30 s, then stirred continuously with a shear speed of 160 rpm/s. Meanwhile, the dispersions were incubated at 60, 70, 80, 90, 95 and 100 °C for 30 min, respectively, and the flour dispersions at room temperature (about 30 °C) were set as control. The dispersions treated with different temperatures were sampled and stored in liquid nitrogen. After lyophilizing, the samples were milled for further analysis.
2.3. Methods
2.3.1. Size Exclusion-Fast Protein Liquid Chromatography (SE-HPLC)
Freeze-dried samples (10 mg) were extracted with 1.0 mL of 0.05 M sodium phosphate buffer (pH 6.8, containing 2% SDS (
w/
v)) for 1 h at room temperature. After centrifugation (5000×
g, 4 °C, 5 min), 20 μL of the supernatant was filtered by 0.45 μm membrane, and then loaded on a Shodex Protein KW-804 column (Showa, Kyoto, Japan), which is suitable for the analysis of proteins with molecular weight ranging from several thousand to several million. The elution was achieved with a 0.2% sodium phosphate buffer (0.05 M, pH 6.8) at 30 °C with flow rate of 0.7 mL/min. Eluted protein was detected at 214 nm. The solution of reduced sample contained 1% DTT. The analyzed parameters were calculated as follows:
In the above formulas, SDS-P, SDS-M and SDS-I refer to SDS soluble polymers proteins, SDS soluble monomers proteins and SDS-insoluble proteins. Ap and Am are the peak areas of polymers and monomers extracted by 0.05 M sodium phosphate buffer, respectively. Ar was the peak area of reduced proteins which was extracted with reduced agent (1% DTT) [
13].
2.3.2. Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE)
SDS-PAGE analysis of proteins was performed in a vertical electrophoresis cell using 10% separating gel and 6% stacking gel. Each sample (50 mg) was dissolved in 1.5 mL of extraction buffer (Tris-HCl, 0.125 M, pH 6, containing 2% SDS (w/v), 10% glycerol (v/v) and 0.01% bromophenol blue (w/v)) and was left for 3 h at room temperature. For reduced proteins, the extraction buffer contained 5% (v/v) β-mercaptoethanol (β-ME). After centrifugation (10,000× g, 4 °C, 20 min), supernatant was heated for 5 min in boiling water and loaded after cooling.
2.3.3. Determination of Free Sulfhydryl (SH) Group
Free sulfhydryl content was determined according to the method of Lambrecht et al. [
14] with minor modification. A freeze-dried sample was mixed with 4 mL SDS-TGE buffer (pH 8.0, 2.5% SDS, 86 mM Tris-HCl, 92 mM glycine and 4.1 mM EDTA), and was homogenized for 30 s, then left for 30 min with homogenizing intermittently every 10 min 3 times. 40 μL Ellman’ reagent (DTNB in SDS-TGE buffer, 4 mg/mL) was added and the tubes were wrapped with aluminum foil, then left at room temperature for 30 min. After centrifugation (20,000×
g, 4 °C, 15 min), the absorbance of supernatant was detected at 412 nm.
2.3.4. Determination of Non-Covalent Bonds of Protein
Chemical reactions were determined according to the method described by Wang et al. [
15]. Selective buffers prepared in phosphate buffer (0.05 M, pH 7.0) were used to solubilize proteins as follows: 0.05 M NaCl (S1), 0.6 M NaCl (S2), 0.6 M NaCl + 1.5 M urea (S3) and 0.6 M NaCl + 8 M urea (S4). Flour samples (100 mg) was extracted with the above four agents (10 mL) at room temperature for 1 h. After being centrifuged (10,000×
g, 4 °C, 20 min), the soluble protein content in the supernatant was determined by micro-Kjeldahl method with conversion factor of 5.7. The difference of soluble protein between S1 and S2, S2 and S3, S3 and S4 reflected ionic bonds, hydrogen bonds and hydrophobic interactions, respectively.
2.3.5. Fourier Transform Infrared Spectroscopy (FTIR)
FTIR spectroscopy was used to measure infrared spectra in the region from 400–4000 cm
−1 using a Thermo Nicolet Nexus FTIR (Thermo Scientific, Waltham, MA, USA) with a single-reflection diamond attenuated total reflection (ATR) crystal and a mercury-cadmium-telluride (MCT) detector. Background spectrum of ATR was recorded at 64 scans and 4 cm
−1 resolution, and FTIR spectra of each sample was recorded under the same condition against the background. The amide I band (1600–1700 cm
−1) in spectrum was deconvoluted after baseline-correction and Gaussian smooth to quantify the secondary structure of protein using Omnic software (version 6.1a, Thermo Nicolet Corp., Madison, WI, USA) and Peakfit software (version 4.12, SPSS Inc., Chicago, IL, USA). The spectrum ranged from 980–1060 cm
−1 and was deconvoluted using Omnic software (version 6.1a, Thermo Nicolet Corp., Madison, WI, USA). The bands at 1047 and 1022 cm
−1 were detected; the ratio of the integrated area of absorption bands at 1047/1022 cm
−1 is generally used to quantify the internal changes of the starch molecule in the degree of short-range order [
16].
2.3.6. Intrinsic Fluorescence Spectra
The intrinsic fluorescence spectra were determined by LS-55 luminescence spectrometer (Perkin Elmer Life Sciences, Shelton, 181 CT, USA) equipped with the Perkin Elmer FL Winlab software. The sample (25 mg) was dissolved in 5 mL 50 mM acetic acid solution at room temperature for 1 h and subsequently centrifuged (10,000× g, 15 min). The supernatant was diluted to 1 mg/mL with extraction solution. The excitation wavelength was set as 280 nm and the emission spectra were recorded from 300–420 nm with a 1 nm slit. The intrinsic fluorescence spectroscopy was used to depict the alterations in protein tertiary structure due to the sensitivity of the protein amino acid residues to the polarity of the microenvironment.
2.3.7. Pasting Properties
Pasting properties were analyzed with Rapid Viscosity Analyzer 130 (RVA-3D super type, Newport Scientific, Stockholm, Sweden) following the method described by Chang et al. [
17] with some modifications. A freeze-dried sample (1.5 g, on 14% dry basis) was suspended in deionized water (25 mL). The procedure of RVA included a heating step, a holding step and a cooling step. During heating, temperature increased from 30 °C to 40 °C for 1 min, and then linearly increased to 95 °C at a rate of 6 °C/min, then held at 95 °C for 5 min. During cooling, the temperature linearly decreased to 50 °C at a rate of 7.5 °C/min, and finally held at 50 °C for 3 min. The parameters of peak viscosity, trough viscosity, breakdown, final viscosity, pasting temperature and peak time were recorded.
2.3.8. Relative Crystallinity of Starch Determined by X-ray Diffraction (XRD)
Samples were sieved by 320 μm sieves after grinding by mortar prior to XRD. The X-ray diffractometer (TD-3500, Tongda, China) was operated at 40 KV and 40 mA with Cu Kα radiation (λ = 0.154 nm). The sample was packed tightly in a glass cell and scanned over the range of 5–40° 2θ angles at 2°/min at room temperature. The relative crystallinity of the starch was calculated using the following formation:
where Ac is the crystalline area on the X-ray diffractogram, and Aa is the amorphous area.
2.3.9. Texture Characteristics
Texture profile analysis was performed using a texture analyzer (TA. XT2i, Stable Micro Systems, Surrey, UK) using a P/0.5 probe at room temperature. The selected settings were 1.5 mm/s of pre-test speed, 1.0 mm/s of test speed and 1.0 mm/s of post-test speed with trigger force of 3 g. The force of probe puncture to 4 mm was expressed as bloom strength. The force of first peak is expressed as the rupture strength, which can reflect the parameters of the process or how much force is required to bite when chewing. Adhesiveness was determined by the area of the domain formed by force and time when the probe returned.
2.3.10. Confocal Laser Scanning Microscopy (CLSM)
A lyophilized sample was mixed with water to form dough organization, and was cut into 20 μm slices by freezing microtome (Leica CM3050S, Leica Bioystems Nussloch Gmbh, Germany). Slices were stained with Rhodamine B agent (0.001%,
w/
v) to observe the protein network in the dough by CLSM (ZEISS LSM800, Oberkochen, Germany). Parameters of protein network such as protein area (μm
2), junction density (×10
−3), total protein length (μm) and lacunarity (×10
−2) were analyzed using AngioTool version 0.5 [
18] according to Bernklau et al. [
19].
2.4. Statistics Analysis
All data were expressed as mean ± standard deviation (SD) of three replicates. Data was analyzed using one-way analysis of variance (ANOVA), and Duncan’s multiple range test was used to compare the means with a significance using an SPSS package (version 10.0 for Windows, SPSS Inc. Chicago, IL, USA). The probability value of p < 0.05 was considered significant.
4. Discussion
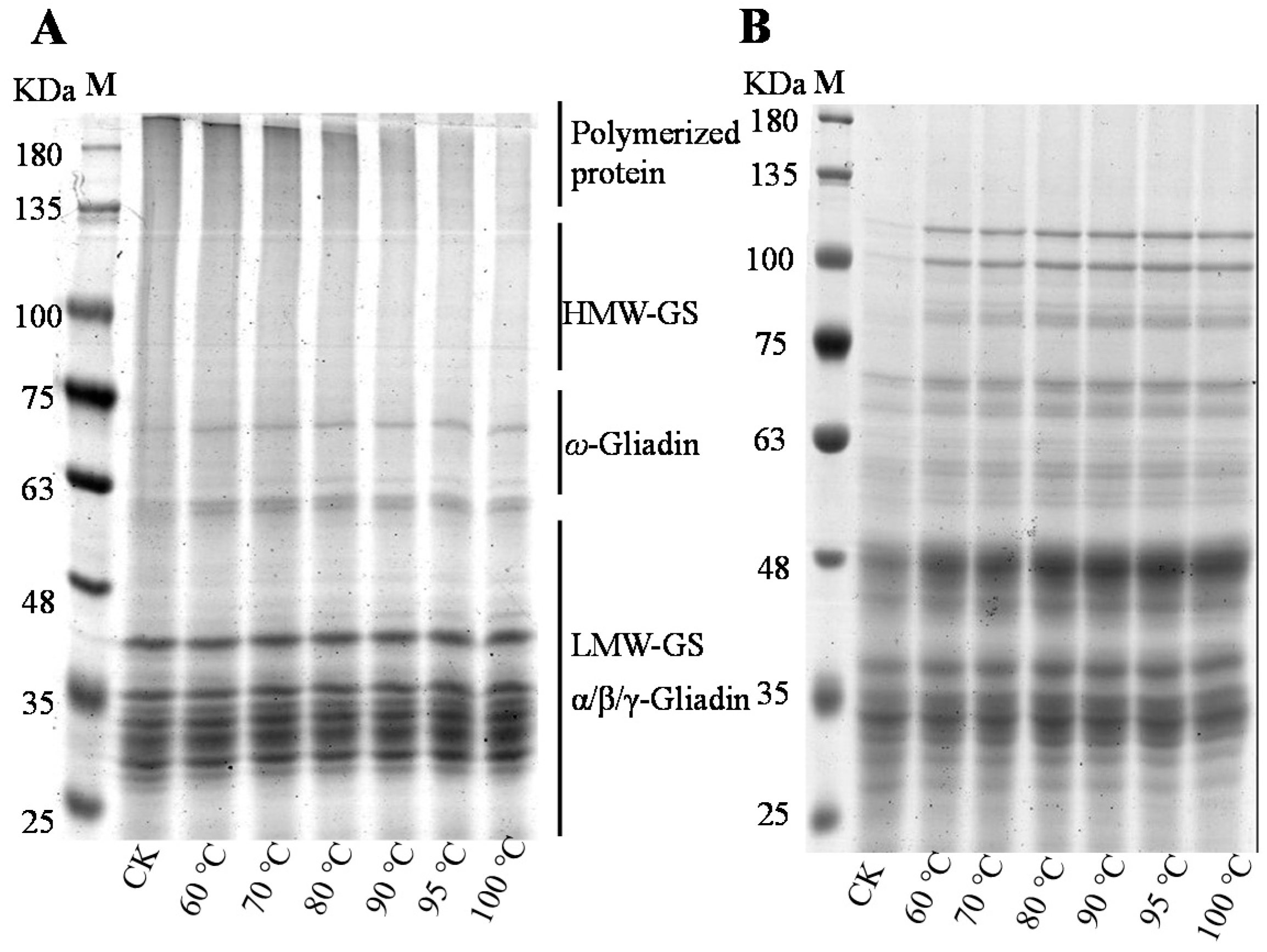
In this study, the molecular and physicochemical changes of protein and starch in wheat flour suspension during heating were monitored by a combination of analytical methods. The advantage of using wheat flour as a model system to study dynamic changes of protein or starch compared to pure starch or protein during the heating process or simulated food processing is that it can reveal the influence of other ingredients (fat, vitamins, minerals, etc.) in the native flour. Therefore, this study is related to foods made from dough (e.g., baked products, noodles, pasta) and batters (e.g., pancakes, coatings for fried foods). The results showed that the band density of SDS-soluble polymers diminished gradually with the increase of temperature in non-reduced SDS-PAGE profiles, especially when the temperature exceeded 80 °C, which indicated that protein aggregated at higher temperatures. The band density of HMW-GS and LMW-GS in reduced SDS-PAGE increased at higher temperatures, indicating that more HMW-GS and LMW-GS participated in protein aggregation. The difference between non-reduced and reduced SDS-PAGE showed that polymerized protein could be reduced by β-Me, which showed that the SS bond was the main intermolecular covalent bond in aggregated protein. The decrease of free SH levels coincided with the increase of SS bonds, which further demonstrated that new SS bonds should be formed by the oxidation of SH [
20]. In addition, the absorption peaks of at 2850 cm
−1 indicated the interaction of the mercaptan group and the intermolecular hydroxyl group, and the band intensity increased with the increase of temperature, which indicated that it was easier to form SS bonds from mercaptan groups of sulfur-containing amino acids under a higher temperature, which further demonstrated that higher temperatures facilitated the formation of SS bonds by thiol end functional groups in sulfur-containing amino acids [
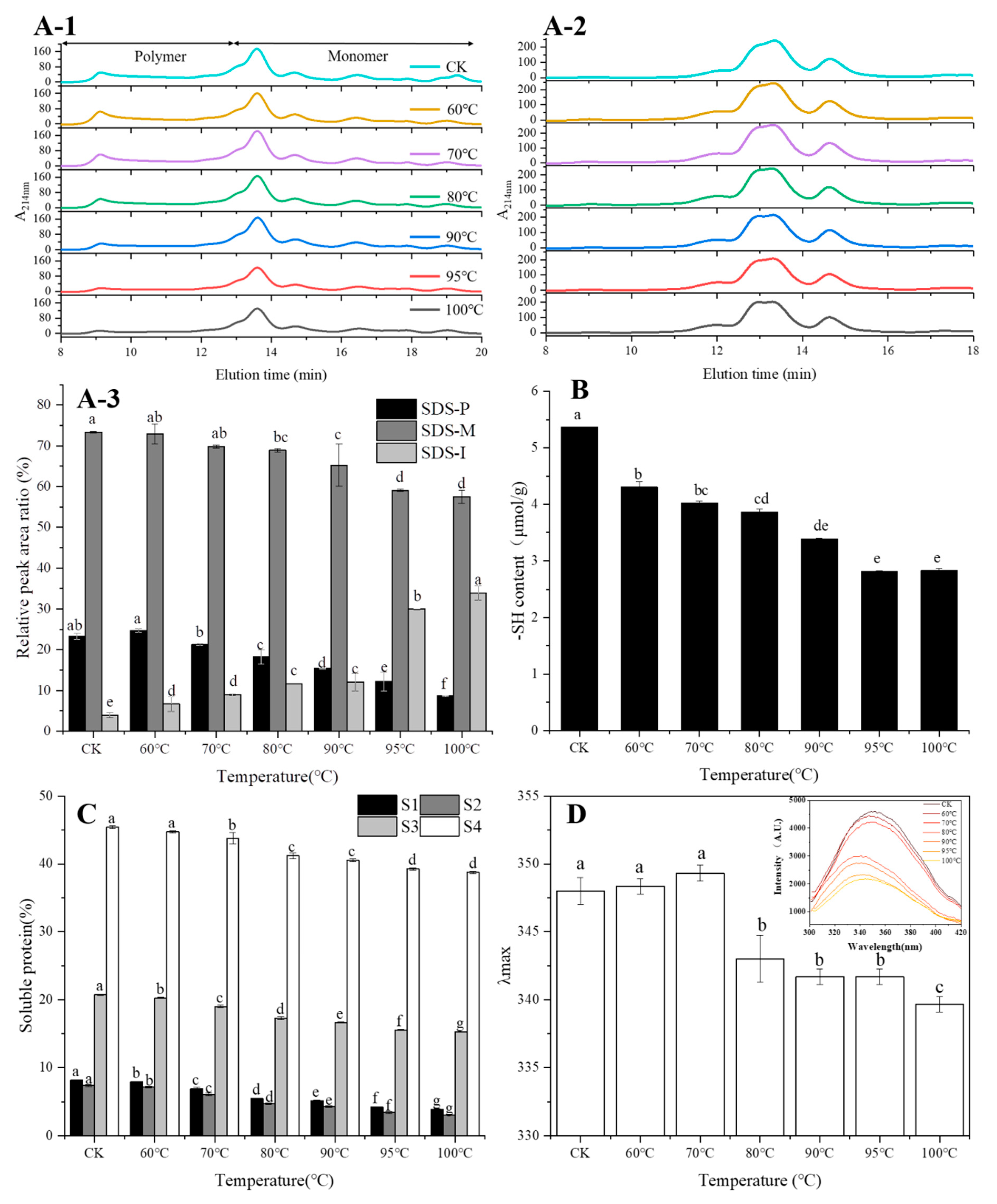
21]. SEC-HPLC is an important method for protein size fractionation and extractability. In this study, monomeric protein vanished with the DTT addition while the peak of polymeric protein was retained (
Figure 2A-2), indicating that SDS-soluble monomeric proteins participated in the formation of SDS-soluble protein polymers through the intermolecular SS bond [
7]. Meanwhile, SDS-insoluble protein levels increased with the increase of temperature and increased greatly when the temperature exceeded 70 °C, indicating the formation of protein aggregates at higher temperatures, and this was in line with the results of SDS-PAGE and free SH content. Apart from SS bonds, interactions of non-covalent bonds such as hydrogen bonds, ionic bonds and hydrophobic interactions also played essential roles in maintaining the three-dimensional spatial structure of the protein network. According to the results of protein solubility in different agents (
Figure 2C), the ionic bond and hydrogen bond participated in the formation of the protein chain, but they were easily destroyed during the heating of the wheat flour. The destroyed ionic and hydrogen bonds caused a lot of exposure of hydrophobic groups and formed a hydrophobic interaction, which contributed the most in maintaining protein spatial structure.
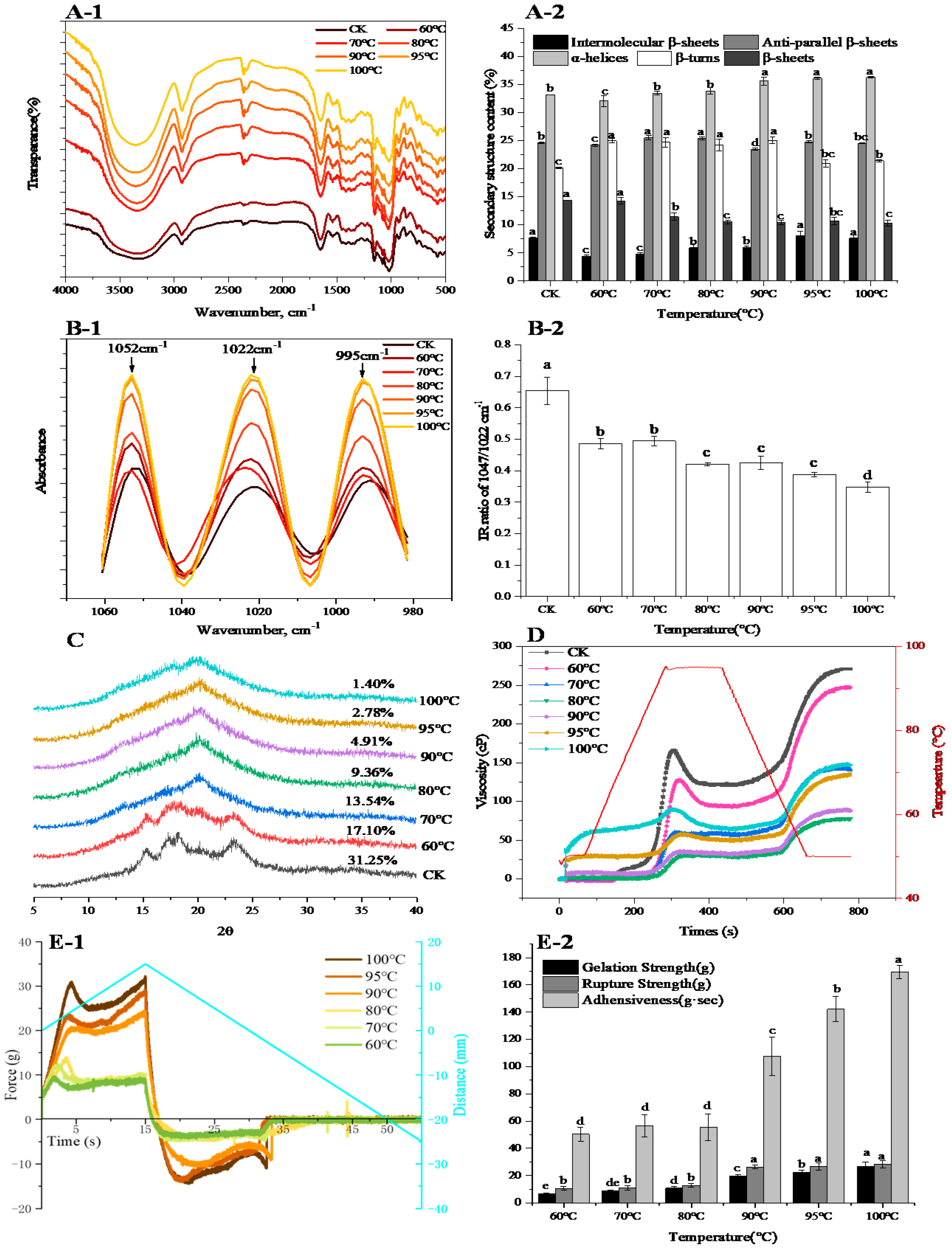
In addition, heat treatment induced the fluctuation of a protein secondary structure, α-helix and intermolecular β-sheets content increased, while β-sheets decreased with the increasing temperature. An especially significant decrease was observed at 70 °C, which was consistent with the results of Francesco et al. [
22]. The increase in α-helix meant the formation of ordered and compact protein spatial structure [
23] by enhancing protein folding [
24]. The increase in intermolecular β-sheets further demonstrated that protein polymerized during heating of wheat flour. In order to obtain further information concerning protein structural changes, an intrinsic fluorescence spectroscopy was used to depict the alterations in protein tertiary structure [
25]. The intrinsic fluorescence intensity reduced largely when the temperature exceeded 70 °C and reached its minimum at 100 °C. This was mainly attributed to a shift in the Trp microenvironment toward a more hydrophilic microenvironment, indicating that the extent of protein polymerization was enhanced. Besides, red-shifts in λmax of 2 nm were observed from CK to 70 °C, indicating that the Trp residues were exposed to a solvent, and the peptide strands became extended, which is an indicator of protein unfolding [
26]. The λmax significantly blue-shifted when the temperature exceeded 70 °C and reached its minimum at 100 °C, further suggesting that Trp was exposed in the hydrophilic microenvironment.
During heating, starch granules also changed greatly: the long-range order and short-range order of the starch structure were determined. XRD was used to analyze the regular arrangement of the long-range ordered double helix structure of starch [
27]. The treatment of control displayed a typical A-type pattern with four major signal peaks and showed the highest crystallinity. When the temperature increased to 60 °C, the intensity of the four reflection peaks decreased slightly and relative crystallinity decreased as compared with control indicating the slight degradation of the crystalline structure [
28]. When the temperature exceeded 60 °C, the reflection peaks of the samples changed from A-type patterns into V-type, the typical A-type pattern disappeared, and there was a low and wide dispersion peak, which can be interpreted as the destruction of starch crystallization due to the movement of amylopectin double helices [
29]. From the results of the XRD, it could be seen that under high temperature treatment the starch was completely gelatinized, and the long-range ordered structure of starch had been completely destroyed. Therefore, the short-range order of the starch structure was determined by infrared spectrum. The infrared spectrum is very sensitive to the short-range ordered structure of starch granules and can distinguish the short-range ordered structure [
30]. The absorbance of 1047 and 1022 cm
−1 detected by FTIR corresponded to the crystalline and amorphous structure of starch, respectively [
31]. In our study, the IR ratio of 1047/1022 cm
−1 decreased with the increase of temperature indicating that the starch granule structure was destroyed, and more amorphous structure was formed with increasing the temperature.
Different heat treatments had different effects on the peak viscosity. The viscosity decreased as the temperature of the heating treatment increased from control to 80 °C. Amylose acted as a diluent or an inhibitor of swelling and even formed insoluble complexes with wheat lipid which restricted swelling during starch gelatinization [
5]. In addition, heat-induced protein aggregates acted as an inhibitor of starch swelling [
6], and the results were in line with previous research [
12,
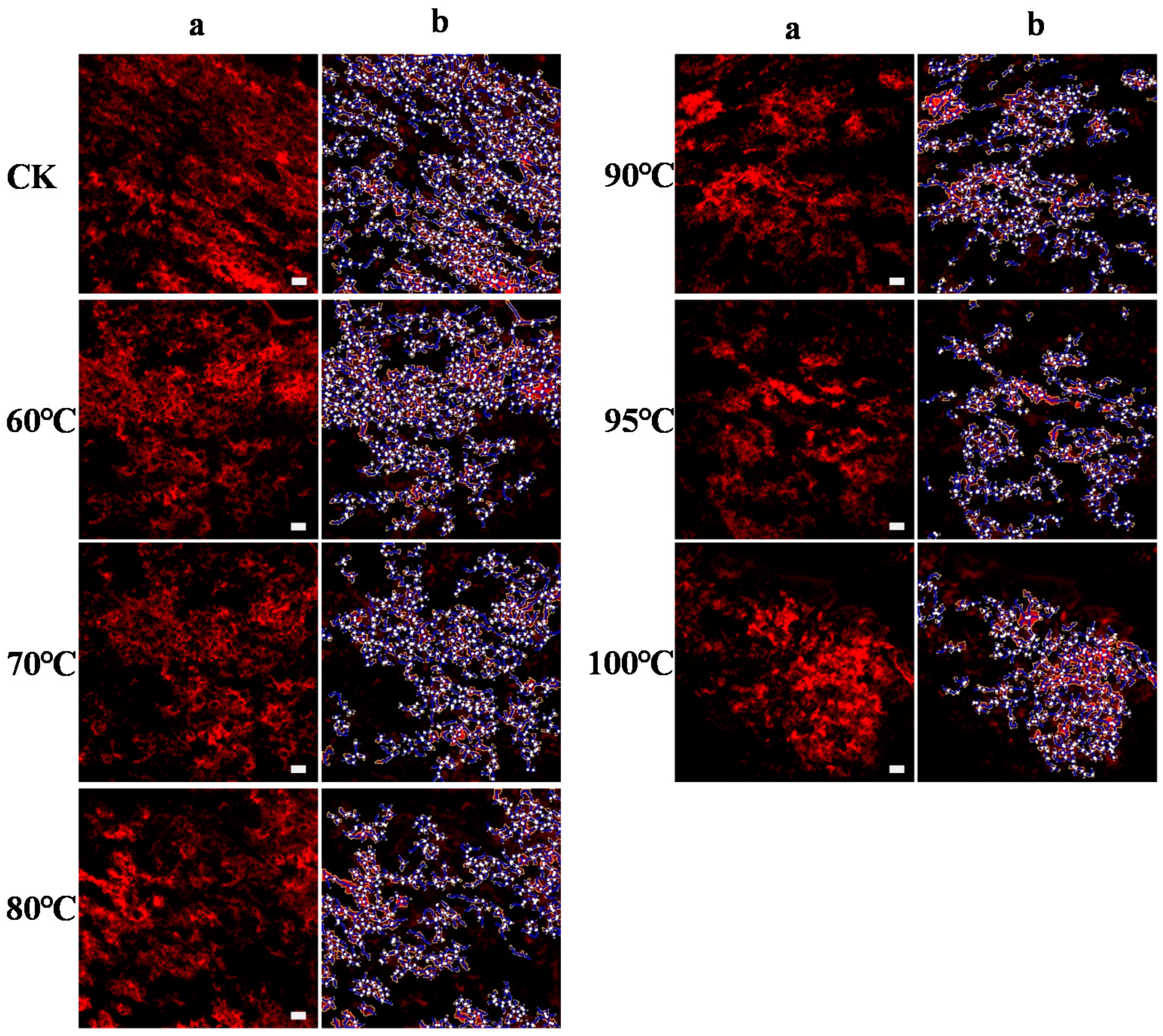
32]. Meanwhile, the denser and more compact morphology of the gluten network at higher temperatures observed by CLSM (
Figure 4) demonstrated the role of protein aggregates as barriers, and the faded band of soluble polymerized protein in non-reduced SDS-PAGE (
Figure 1) and higher levels of SDS-I and blue-shifted emission wavelength at higher temperatures also confirmed that. Therefore, interactions between denatured protein and starch delayed the starch swelling and caused the decrease of viscosity. Interestingly, although the overall crystallinity of starch was completely disrupted at higher temperatures, the existence of a starch short-range ordered structure made starch expand, which caused the heated sample to develop the typical viscosity pattern. While viscosity increased when the temperature of heat treatment exceeded 80 °C, this was related to the presence of cold water-swelling starch. Cold water-swelling starch, sometimes called pregelatinized starch, had complete degradation of the starch structure, and similar results were also found in rice [
33]. These results indicated that starch, especially amylose, and protein in wheat flour prevented the starch granules from swelling which led to a decrease in peak viscosity at higher temperatures.
Flour gelation can be obtained by cooling flour pasta after gelatinization. During starch retrogradation, the rheological properties and crystallinity of the gelation change significantly, and this is the main factor that affects food texture. Meanwhile, starch retrogradation is based on the degree of starch gelatinization. Accordingly, flour gelation is the result of starch gelatinization and protein aggregation, and it is the macroscopic manifestation of micro level changes of protein and starch during the heating of wheat flour. In our study, gelation strength and rupture strength increased with the increase of treatment temperature, especially when the temperature exceeded 80 °C. This was related to the acceleration of the rearrangement of double helices of starch molecules with the elevating temperature, and hence the gelatinization of starch was accelerated and the gelation strength was increased. Besides, the interaction of starch and protein also contributed to the changes of gelation strength at different temperatures. The heat-induced protein molecule was in a “melting sphere” state between the natural state and denaturation state [
2,
34], which intensely interacted with starch molecules at higher temperatures, and thus enhanced their gelation strength.









{kind=link}
{kind=link}
{kind=link}
{kind=link}