
Solid-Phase Extraction Approaches for Improving Oligosaccharide and Small Peptide Identification with Liquid Chromatography-High-Resolution Mass Spectrometry: A Case Study on Proteolyzed Almond Extract

 ,
,  ,
,  and
and 
Abstract
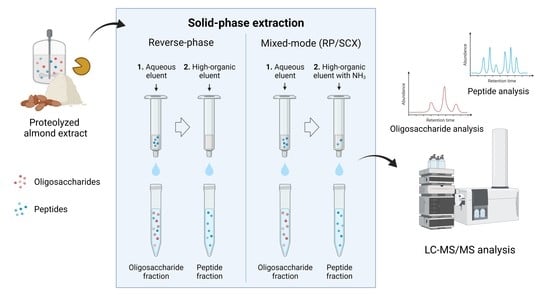
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Comparison of Procedures for Protein Removal
2.3. Comparison of Solid-Phase Extraction Approaches
2.3.1. Reverse-Phase Solid-Phase Extraction
2.3.2. Mixed-Mode Solid-Phase Extraction
2.4. Analysis of Peptide Standards
2.5. Measuring the Recovery of Peptides
2.6. Measuring the Recovery of Oligosaccharides
2.7. Characterization of Oligosaccharides in the Proteolyzed Almond Extract by LC-MS/MS
2.8. Characterization of Peptides in the Proteolyzed Almond Extract by LC-MS/MS
2.9. Peptide Data Analysis
2.10. Statistical Analysis
3. Results and Discussion
3.1. Efficacy of Different Solid-Phase Extraction Approaches in Binding Peptides
3.1.1. Reverse-Phase Solid-Phase Extraction
3.1.2. Mixed-Mode Solid-Phase Extraction
3.2. Evaluating Oligosaccharide and Peptide Sample Preparation Approaches Using the Proteolyzed Almond Extract
3.2.1. Comparison of Procedures for Protein Removal
3.2.2. Comparison of Solid-Phase Extraction Approaches for Improving Oligosaccharide Characterization
3.2.3. Comparison of Solid-Phase Extraction Approaches for Improving Peptide Characterization
Medium-Sized Peptides
Small Peptides
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chichlowski, M.; German, J.B.; Lebrilla, C.B.; Mills, D.A. The Influence of Milk Oligosaccharides on Microbiota of Infants: Opportunities for Formulas. Annu. Rev. Food Sci. Technol. 2011, 2, 331–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mussatto, S.I.; Mancilha, I.M. Non-Digestible Oligosaccharides: A Review. Carbohydr. Polym. 2007, 68, 587–597. [Google Scholar] [CrossRef]
- Human Milk Oligosaccharides Market Size, Share & Trends Analysis Report. Available online: https://www.grandviewresearch.com/industry-analysis/human-milk-oligosaccharides-market (accessed on 6 November 2021).
- Miralles, B.; Amigo, L.; Recio, I. Critical Review and Perspectives on Food-Derived Antihypertensive Peptides. J. Agric. Food Chem. 2018, 66, 9384–9390. [Google Scholar] [CrossRef] [PubMed]
- Guha, S.; Majumder, K. Structural-Features of Food-Derived Bioactive Peptides with Anti-Inflammatory Activity: A Brief Review. J. Food Biochem. 2019, 43, e12531. [Google Scholar] [CrossRef] [PubMed]
- Phazang, P.; Negi, N.P.; Raina, M.; Kumar, D. Plant Antimicrobial Peptides: Next-Generation Bioactive Molecules for Plant Protection. In Phyto-Microbiome in Stress Regulation; Kumar, M., Kumar, V., Prasad, R., Eds.; Environmental and Microbial Biotechnology; Springer: Singapore, 2020; pp. 281–293. [Google Scholar] [CrossRef]
- Souza, T.S.P.; Dias, F.F.G.; Koblitz, M.G.B.; de Moura Bell, J.M.L.N. Aqueous and Enzymatic Extraction of Oil and Protein from Almond Cake: A Comparative Study. Processes 2019, 7, 472. [Google Scholar] [CrossRef] [Green Version]
- Koopman, R.; Crombach, N.; Gijsen, A.P.; Walrand, S.; Fauquant, J.; Kies, A.K.; Lemosquet, S.; Saris, W.H.; Boirie, Y.; van Loon, L.J. Ingestion of a Protein Hydrolysate Is Accompanied by an Accelerated in Vivo Digestion and Absorption Rate When Compared with Its Intact Protein. Am. J. Clin. Nutr. 2009, 90, 106–115. [Google Scholar] [CrossRef] [Green Version]
- Bahna, S.L. Hypoallergenic Formulas: Optimal Choices for Treatment versus Prevention. Ann. Allergy Asthma. Immunol. 2008, 101, 453–459. [Google Scholar] [CrossRef]
- Català-Clariana, S.; Benavente, F.; Giménez, E.; Barbosa, J.; Sanz-Nebot, V. Identification of Bioactive Peptides in Hypoallergenic Infant Milk Formulas by Capillary Electrophoresis–Mass Spectrometry. Anal. Chim. Acta 2010, 683, 119–125. [Google Scholar] [CrossRef]
- Wada, Y.; Lönnerdal, B. Bioactive Peptides Released by in Vitro Digestion of Standard and Hydrolyzed Infant Formulas. Peptides 2015, 73, 101–105. [Google Scholar] [CrossRef]
- Aluko, R.E. 15—Food Protein-Derived Peptides: Production, Isolation, and Purification. In Proteins in Food Processing, 2nd ed.; Yada, R.Y., Ed.; Woodhead Publishing Series in Food Science, Technology and Nutrition; Woodhead Publishing: Cambridge, UK, 2018; pp. 389–412. [Google Scholar] [CrossRef]
- Lafarga, T.; Hayes, M. Bioactive Protein Hydrolysates in the Functional Food Ingredient Industry: Overcoming Current Challenges. Food Rev. Int. 2017, 33, 217–246. [Google Scholar] [CrossRef]
- Piovesana, S.; Capriotti, A.L.; Cerrato, A.; Crescenzi, C.; La Barbera, G.; Laganà, A.; Montone, C.M.; Cavaliere, C. Graphitized Carbon Black Enrichment and UHPLC-MS/MS Allow to Meet the Challenge of Small Chain Peptidomics in Urine. Anal. Chem. 2019, 91, 11474–11481. [Google Scholar] [CrossRef] [PubMed]
- Piovesana, S.; Cerrato, A.; Antonelli, M.; Benedetti, B.; Capriotti, A.L.; Cavaliere, C.; Montone, C.M.; Laganà, A. A Clean-up Strategy for Identification of Circulating Endogenous Short Peptides in Human Plasma by Zwitterionic Hydrophilic Liquid Chromatography and Untargeted Peptidomics Identification. J. Chromatogr. A 2020, 1613, 460699. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Tamaya, K.; Seki, E.; Osajima, K.; Matsumoto, K.; Kawasaki, T. Val-Tyr as a Natural Antihypertensive Dipeptide can be Absorbed into the Human Circulatory Blood System. Clin. Exp. Pharmacol. Physiol. 2002, 29, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, J.Á.G.; Ramos, M.; Recio, I. Angiotensin Converting Enzyme-Inhibitory Activity of Peptides Isolated from Manchego Cheese. Stability under Simulated Gastrointestinal Digestion. Int. Dairy J. 2004, 14, 1075–1080. [Google Scholar] [CrossRef]
- Ohsawa, K.; Satsu, H.; Ohki, K.; Enjoh, M.; Takano, T.; Shimizu, M. Producibility and Digestibility of Antihypertensive β-Casein Tripeptides, Val-Pro-Pro and Ile-Pro-Pro, in the Gastrointestinal Tract: Analyses Using an in Vitro Model of Mammalian Gastrointestinal Digestion. J. Agric. Food Chem. 2008, 56, 854–858. [Google Scholar] [CrossRef]
- Sato, K. Structure, Content, and Bioactivity of Food-Derived Peptides in the Body. J. Agric. Food Chem. 2018, 66, 3082–3085. [Google Scholar] [CrossRef]
- Kailemia, M.J.; Ruhaak, L.R.; Lebrilla, C.B.; Amster, I.J. Oligosaccharide Analysis by Mass Spectrometry: A Review of Recent Developments. Anal. Chem. 2014, 86, 196–212. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Huang, G.; Liao, W.; Gong, S.; Xiao, J.; Bai, J.; Hsiao, W.L.W.; Li, N.; Wu, J.L. Discovery of the Bioactive Peptides Secreted by Bifidobacterium Using Integrated MCX Coupled with LC–MS and Feature-Based Molecular Networking. Food Chem. 2021, 347, 129008. [Google Scholar] [CrossRef]
- Gilar, M.; Yu, Y.Q.; Ahn, J.; Fournier, J.; Gebler, J.C. Mixed-Mode Chromatography for Fractionation of Peptides, Phosphopeptides, and Sialylated Glycopeptides. J. Chromatogr. A 2008, 1191, 162–170. [Google Scholar] [CrossRef]
- Li, X.; Guo, Z.; Sheng, Q.; Xue, X.; Liang, X. Sequential Elution of Multiply and Singly Phosphorylated Peptides with Polar-Copolymerized Mixed-Mode RP18/SCX Material. Analyst 2012, 137, 2774–2776. [Google Scholar] [CrossRef]
- Huang, Y.P.; Robinson, R.C.; Barile, D. Food Glycomics: Dealing with Unexpected Degradation of Oligosaccharides during Sample Preparation and Analysis. J. Food Drug Anal. 2022; in press. [Google Scholar]
- de Almeida, N.M.; Dias, F.F.G.; Rodrigues, M.I.; de Moura Bell, J.M.L.N. Effects of Processing Conditions on the Simultaneous Extraction and Distribution of Oil and Protein from Almond Flour. Processes 2019, 7, 844. [Google Scholar] [CrossRef] [Green Version]
- Sischo, W.M.; Short, D.M.; Geissler, M.; Bunyatratchata, A.; Barile, D. Comparative Composition, Diversity, and Abundance of Oligosaccharides in Early Lactation Milk from Commercial Dairy and Beef Cows. Int. J. Dairy Sci. 2017, 100, 3883–3892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, H.S.; Wang, F.L.; Ondetti, M.A.; Sabo, E.F.; Cushman, D.W. Binding of Peptide Substrates and Inhibitors of Angiotensin-Converting Enzyme. Importance of the COOH-Terminal Dipeptide Sequence. J. Biol. Chem. 1980, 255, 401–407. [Google Scholar] [CrossRef]
- Lan, V.T.T.; Ito, K.; Ohno, M.; Motoyama, T.; Ito, S.; Kawarasaki, Y. Analyzing a Dipeptide Library to Identify Human Dipeptidyl Peptidase IV Inhibitor. Food Chem. 2015, 175, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Méndez, A.; Bosch, E.; Rosés, M.; Neue, U.D. Comparison of the Acidity of Residual Silanol Groups in Several Liquid Chromatography Columns. J. Chromatogr. A 2003, 986, 33–44. [Google Scholar] [CrossRef]
- Sulpizi, M.; Gaigeot, M.P.; Sprik, M. The Silica–Water Interface: How the Silanols Determine the Surface Acidity and Modulate the Water Properties. J. Chem. Theory Comput. 2012, 8, 1037–1047. [Google Scholar] [CrossRef]
- Benavente, F.; Medina-Casanellas, S.; Barbosa, J.; Sanz-Nebot, V. Investigation of Commercial Sorbents for the Analysis of Opioid Peptides in Human Plasma by On-Line SPE-CE. J. Sep. Sci. 2010, 33, 1294–1304. [Google Scholar] [CrossRef]
- Arakawa, T.; Ejima, D.; Akuta, T. Protein Aggregation under High Concentration/Density State during Chromatographic and Ultrafiltration Processes. Int. J. Biol. Macromol. 2017, 95, 1153–1158. [Google Scholar] [CrossRef]
- Groleau, P.E.; Lapointe, J.F.; Gauthier, S.F.; Pouliot, Y. Effect of Aggregating Peptides on the Fractionation of β-LG Tryptic Hydrolysate by Nanofiltration Membrane. J. Membr. Sci. 2004, 234, 121–129. [Google Scholar] [CrossRef]
- Guo, X.; Kristal, B.S. The Use of Underloaded C18 Solid-Phase Extraction Plates Increases Reproducibility of Analysis of Tryptic Peptides from Unfractionated Human Plasma. Anal. Biochem. 2012, 426, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Stone, T.A.; Cole, G.B.; Ravamehr-Lake, D.; Nguyen, H.Q.; Khan, F.; Sharpe, S.; Deber, C.M. Positive Charge Patterning and Hydrophobicity of Membrane-Active Antimicrobial Peptides as Determinants of Activity, Toxicity, and Pharmacokinetic Stability. J. Med. Chem. 2019, 62, 6276–6286. [Google Scholar] [CrossRef] [PubMed]
- Piovesana, S.; Capriotti, A.L.; Cavaliere, C.; La Barbera, G.; Montone, C.M.; Zenezini Chiozzi, R.; Laganà, A. Recent Trends and Analytical Challenges in Plant Bioactive Peptide Separation, Identification and Validation. Anal. Bioanal. Chem. 2018, 410, 3425–3444. [Google Scholar] [CrossRef] [PubMed]
- Giacometti, J.; Buretić-Tomljanović, A. Peptidomics as a Tool for Characterizing Bioactive Milk Peptides. Food Chem. 2017, 230, 91–98. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Solid-Phase Extraction | Glycyl Tyrosine | Leucine Enkephalin (YGGFL) | Methionine Enkephalin (YGGFM) | Angiotensin II (DRVYIHPF) | ||||
|---|---|---|---|---|---|---|---|---|
| aq | org | aq | org | aq | org | aq | org | |
| no modifier | ||||||||
| C18 100 mg | ✔ 1 | ✔ | ✔ | ✔ (low) | ||||
| C18 500 mg | ✔ | ✔ | ✔ | |||||
| HLB 60 mg | ✔ | ✔ | ✔ | ✔ | ||||
| FA2as modifier | ||||||||
| C18 100 mg | ✔ | ✔ | ✔ | ✔ | ||||
| C18 500 mg | ✔ | ✔ | ✔ | ✔ | ||||
| HLB 60 mg | ✔ | ✔ | ✔ | ✔ | ✔ | |||
| TFA as modifier | ||||||||
| C18 100 mg | ✔ | ✔ | ✔ | ✔ | ✔ | |||
| C18 500 mg | ✔ | ✔ | ✔ | ✔ | ||||
| HLB 60 mg | ✔ | ✔ | ✔ | ✔ | ✔ | |||
| FA as modifier for aq; NH3 as modifier for org | ||||||||
| X-C 30 mg | ✔ | ✔ | ✔ | ✔ | ||||
| Solid-Phase Extraction | Composition of High-Organic Eluent | Angiotensin I | Neurotensin |
|---|---|---|---|
| NH3 as modifier | |||
| X-C 30 mg | 80% ACN, 1% NH3 | ✔ 2 | |
| MCX 30 mg | 80% ACN, 1% NH3 | ✔ | |
| MCAX 100 mg | 80% ACN, 1% NH3 | ✔ | |
| NH4COOH as modifier | |||
| X-C 30 mg | 50% ACN, 250 mM NH4COOH | ✔ | |
| MCX 30 mg | 50% ACN, 250 mM NH4COOH | ||
| MCAX 100 mg | 50% ACN, 250 mM NH4COOH | ✔ | ✔ |
| X-C 30 mg | 40% ACN, 375 mM NH4COOH | ✔ | ✔ |
| MCX 30 mg | 40% ACN, 375 mM NH4COOH | ✔ | ✔ |
| MCAX 100 mg | 40% ACN, 375 mM NH4COOH | ✔ | ✔ |
| Peptide Sequence | Retention Time (min) | C18 100 mg | C18 500 mg | C8 100 mg | HLB 60 mg | X-C 30 mg |
|---|---|---|---|---|---|---|
| Gln-Gln | 2.00 | ✔ 2 | ||||
| Gly-Gln | 2.00 | ✔ | ||||
| Ala-Pro | 2.21 | ✔ | ||||
| Gly-Val | 2.28 | ✔ | ||||
| Lxx-Glu 3 | 3.24 | ✔ | ||||
| Ser-Tyr | 3.58 | ✔ | ✔ | |||
| Gly-Tyr | 3.65 | ✔ | ✔ | |||
| Val-Pro | 3.84 | ✔ | ✔ | |||
| Thr-Tyr | 3.93 | ✔ | ||||
| Ser-Lxx | 4.80 | ✔ | ||||
| Gly-Lxx | 5.13 | ✔ | ✔ | |||
| Ala-Lxx | 5.23 | ✔ | ||||
| Thr-Lxx | 6.17 | ✔ | ✔ | |||
| Val-Tyr | 6.28 | ✔ | ✔ | ✔ | ||
| Lxx-Val | 6.83 | ✔ | ✔ | |||
| Ser-Phe | 6.91 | ✔ | ||||
| Gly-Phe | 7.12 | ✔ | ✔ | ✔ | ✔ | |
| Ala-Phe | 7.27 | ✔ | ||||
| Lxx-Pro | 7.38 | ✔ | ✔ | ✔ | ||
| Val-Lxx | 7.66 | ✔ | ✔ | |||
| Phe-Pro | 9.45 | ✔ | ✔ | ✔ | ✔ | ✔ |
| Lxx-Phe | 10.42 | ✔ | ✔ | ✔ | ||
| Trp-Pro | 11.08 | ✔ | ✔ | ✔ | ✔ | ✔ |
| Tyr-Trp | 11.72 | ✔ | ||||
| Val-Met | 11.74 | ✔ | ✔ | ✔ | ✔ | ✔ |
| Lxx-Phe | 11.88 | ✔ | ✔ | ✔ | ✔ | ✔ |
| Lxx-Trp | 12.75 | ✔ | ✔ | ✔ | ✔ | ✔ |
| Lxx-Trp | 13.39 | ✔ | ✔ | ✔ | ✔ | ✔ |
| Phe-Phe | 13.40 | ✔ | ✔ | ✔ | ✔ | ✔ |
| Phe-Trp | 15.06 | ✔ | ✔ | ✔ | ✔ | ✔ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.-P.; Robinson, R.C.; Dias, F.F.G.; de Moura Bell, J.M.L.N.; Barile, D. Solid-Phase Extraction Approaches for Improving Oligosaccharide and Small Peptide Identification with Liquid Chromatography-High-Resolution Mass Spectrometry: A Case Study on Proteolyzed Almond Extract. Foods 2022, 11, 340. https://doi.org/10.3390/foods11030340
Huang Y-P, Robinson RC, Dias FFG, de Moura Bell JMLN, Barile D. Solid-Phase Extraction Approaches for Improving Oligosaccharide and Small Peptide Identification with Liquid Chromatography-High-Resolution Mass Spectrometry: A Case Study on Proteolyzed Almond Extract. Foods. 2022; 11(3):340. https://doi.org/10.3390/foods11030340
Chicago/Turabian StyleHuang, Yu-Ping, Randall C. Robinson, Fernanda Furlan Goncalves Dias, Juliana Maria Leite Nobrega de Moura Bell, and Daniela Barile. 2022. "Solid-Phase Extraction Approaches for Improving Oligosaccharide and Small Peptide Identification with Liquid Chromatography-High-Resolution Mass Spectrometry: A Case Study on Proteolyzed Almond Extract" Foods 11, no. 3: 340. https://doi.org/10.3390/foods11030340
APA StyleHuang, Y. -P., Robinson, R. C., Dias, F. F. G., de Moura Bell, J. M. L. N., & Barile, D. (2022). Solid-Phase Extraction Approaches for Improving Oligosaccharide and Small Peptide Identification with Liquid Chromatography-High-Resolution Mass Spectrometry: A Case Study on Proteolyzed Almond Extract. Foods, 11(3), 340. https://doi.org/10.3390/foods11030340