
The Questionable Quality Profile of Food Supplements: The Case of Red Yeast Rice Marketed Products

 ,
,  ,
,  ,
,  ,
,  and
and
Abstract
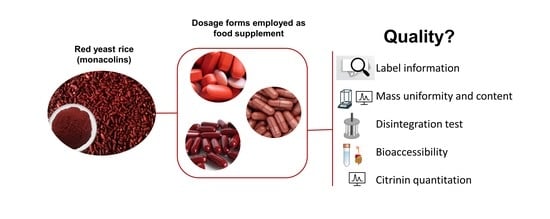
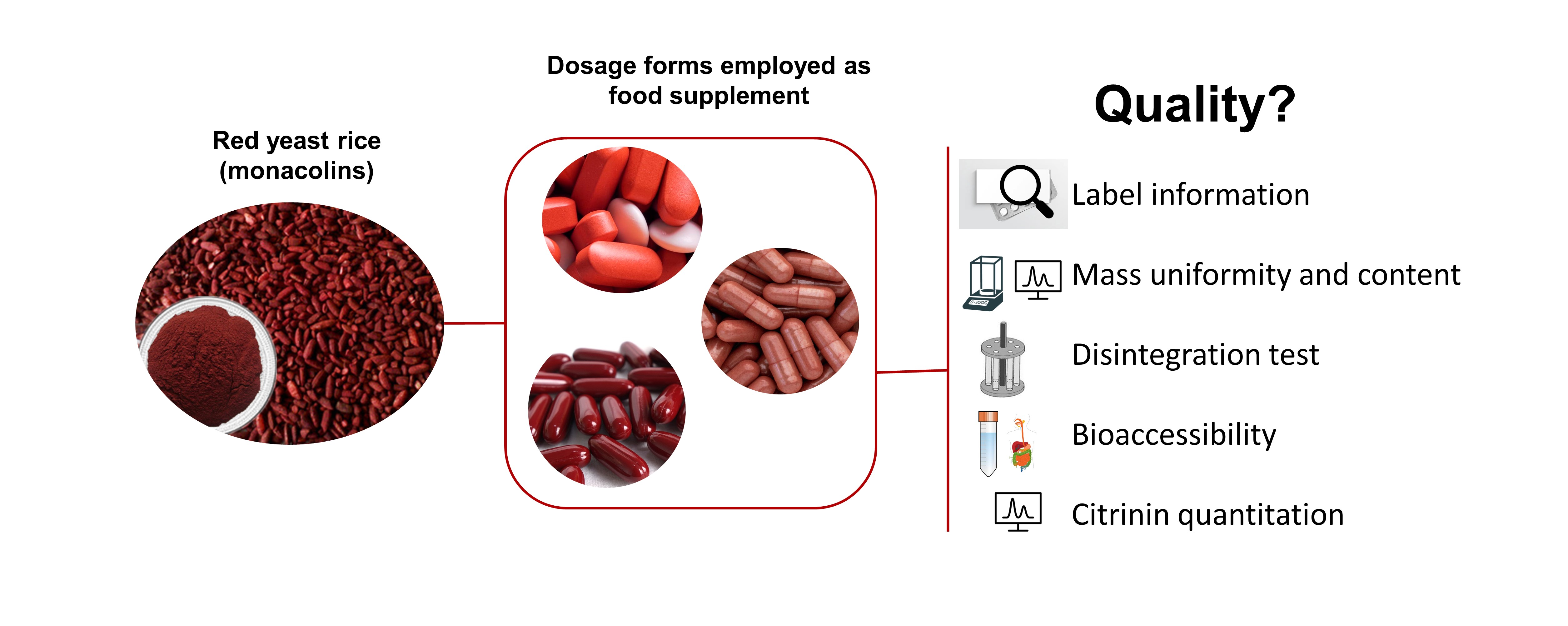
:
1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Food Supplements Properties
2.3. Mass Uniformity
2.4. Disintegration Test
2.5. In Vitro Bioaccessibility of Monacolin K
2.6. Citrinin Quantitative Determination
2.7. Statistical Analysis
3. Results and Discussion
3.1. Food Supplement Labelling
3.2. Mass Uniformity of Tablets and Capsules
3.3. Disintegration Test
3.4. Resistance to Crushing of Tablets
3.5. Relationship between FS Composition and Manufacturing
3.6. In Vitro Bioaccessibility of Monacolin K
3.7. CIT Quantitation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- (NMI) Natural Marketing Institute. Supplements/OTC/Rx Consumer Trends & Insights Report, 9th ed.; NMI: Newtown Square, PA, USA, 2021. [Google Scholar]
- Gusev, P.A.; Andrews, K.W.; Savarala, S.; Tey, P.-T.; Han, F.; Oh, L.; Pehrsson, P.R.; Dwyer, J.T.; Betz, J.M.; Kuszak, A.J.; et al. Disintegration and Dissolution Testing of Green Tea Dietary Supplements: Application and Evaluation of United States Pharmacopeial Standards. J. Pharm. Sci. 2020, 109, 1933–1942. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.G.; Fogacci, F.; Banach, M. Red Yeast Rice for Hypercholesterolemia. Methodist DeBakey Cardiovasc. J. 2019, 15, 192–199. [Google Scholar] [CrossRef]
- Burke, F.M. Red yeast rice for the treatment of dyslipidemia. Curr. Atheroscler. Rep. 2015, 17, 495. [Google Scholar] [CrossRef]
- Ma, J.; Li, Y.; Ye, Q.; Li, J.; Hua, Y.; Ju, D.; Zhang, D.; Cooper, R.; Chang, M. Constituents of Red Yeast Rice, a Traditional Chinese Food and Medicine. J. Agric. Food Chem. 2000, 48, 5220–5225. [Google Scholar] [CrossRef]
- Heber, D.; Yip, I.; Ashley, J.M.; Elashoff, D.A.; Elashoff, R.M.; Go, V.L.W. Cholesterol-lowering effects of a proprietary Chinese red-yeast-rice dietary supplement. Am. J. Clin. Nutr. 1999, 69, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-G.; Zhang, F.; Wang, Z.-T.; Hu, Z.-B. Identification and chemical profiling of monacolins in red yeast rice using high-performance liquid chromatography with photodiode array detector and mass spectrometry. J. Pharm. Biomed. Anal. 2004, 35, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-H.; Yang, J.-C.; Uang, Y.-S.; Lin, C.-J. Improved dissolution rate and oral bioavailability of lovastatin in red yeast rice products. Int. J. Pharm. 2013, 444, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Stancu, C.; Sima, A. Statins: Mechanism of action and effects. J. Cell. Mol. Med. 2001, 5, 378–387. [Google Scholar] [CrossRef]
- Schachter, M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: An update. Fundam. Clin. Pharmacol. 2005, 19, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Sirtori, C.R. The pharmacology of statins. Pharmacol. Res. 2014, 88, 3–11. [Google Scholar] [CrossRef]
- Avula, B.; Cohen, P.A.; Wang, Y.-H.; Sagi, S.; Feng, W.; Wang, M.; Zweigenbaum, J.; Shuangcheng, M.; Khan, I.A. Chemical profiling and quantification of monacolins and citrinin in red yeast rice commercial raw materials and dietary supplements using liquid chromatography-accurate QToF mass spectrometry: Chemometrics application. J. Pharm. Biomed. Anal. 2014, 100, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.Y.; Cooperman, T.; Obermeyer, W.; Becker, D.J. Marked Variability of Monacolin Levels in Commercial Red Yeast Rice Products: Buyer Beware! Arch. Intern. Med. 2010, 170, 1722–1727. [Google Scholar] [CrossRef]
- Klimek, M.; Wang, S.; Ogunkanmi, A. Safety and efficacy of red yeast rice (Monascus purpureus) as an alternative therapy for hyperlipidemia. Pharmacy and Therapeutics. 2009, 34, 313–327. [Google Scholar]
- Cohen, P.A.; Avula, B.; Khan, I.A. Variability in strength of red yeast rice supplements purchased from mainstream retailers. Eur. J. Prev. Cardiol. 2017, 24, 1431–1434. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Seeram, N.P.; Lee, R.; Thames, G.; Minutti, C.; Wang, H.-J.; Heber, D. Plasma Clearance of Lovastatin Versus Chinese Red Yeast Rice in Healthy Volunteers. J. Altern. Complement. Med. 2005, 11, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Flajs, D.; Peraica, M. Toxicological properties of citrinin. Arch. Ind. Hyg. Toxicol. 2009, 60, 457–464. [Google Scholar] [CrossRef]
- Silva, L.J.; Pereira, A.M.; Pena, A.; Lino, C.M. Citrinin in foods and supplements: A review of occurrence and analytical methodologies. Foods 2020, 10, 14. [Google Scholar] [CrossRef]
- Liao, C.-D.; Chen, Y.-C.; Lin, H.-Y.; Chiueh, L.-C.; Shih, D.Y.-C. Incidence of citrinin in red yeast rice and various commercial Monascus products in Taiwan from 2009 to 2012. Food Control 2014, 38, 178–183. [Google Scholar] [CrossRef]
- Ferre, F.S. Worldwide occurrence of mycotoxins in rice. Food Control 2016, 62, 291–298. [Google Scholar] [CrossRef]
- EU Commission. Commission Regulation (EU) No 212/2014 of 6 March 2014 Amending Regulation (EC) No 1881/2006 as Regards Maximum Levels of the Contaminant Citrinin in Food Supplements Based on Rice Fermented with Red Yeast Monascus purpureus; EU Commission: Brussels, Belgium, 2014. [Google Scholar]
- EU Commission. Commission Regulation (EU) 2019/1901 Amending Regulation (EC) No 1881/2006 as Regards Maximum Levels of Citrinin in Food Supplements Based on Rice Fermented with Red Yeast Monascus Purpureus; EU Commission: Brussels, Belgium, 2019. [Google Scholar]
- Minekus, M.; Alminger, M.; Alvito, P.; Ballance, S.; Bohn, T.; Bourlieu, C.; Carriere, F.; Boutrou, R.; Corredig, M.; Dupont, D. A standardised static in vitro digestion method suitable for food—An international consensus. Food Funct. 2014, 5, 1113–1124. [Google Scholar] [CrossRef]
- Castaldo, L.; Izzo, L.; Narváez, A.; Rodríguez-Carrasco, Y.; Grosso, M.; Ritieni, A. Colon Bioaccessibility under In Vitro Gastrointestinal Digestion of Different Coffee Brews Chemically Profiled through UHPLC-Q-Orbitrap HRMS. Foods 2021, 10, 179. [Google Scholar] [CrossRef]
- Nigović, B.; Sertić, M.; Mornar, A. Simultaneous determination of lovastatin and citrinin in red yeast rice supplements by micellar electrokinetic capillary chromatography. Food Chem. 2013, 138, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Narvaez, A.; Izzo, L.; Rodriguez-Carrasco, Y.; Ritieni, A. Citrinin dietary exposure assessment approach through human biomonitoring high-resolution mass spectrometry-based data. J. Agric. Food Chem. 2021, 69, 6330–6338. [Google Scholar] [CrossRef]
- EU Commission. Commission Decision 657/EC Implementing Council Directive 96/23/EC Concerning the Performances of Analytical Methods and the Interpretation of Results; EU Commission: Brussels, Belgium, 2002; p. L221. [Google Scholar]
- Pellett, J.D.; Dwaraknath, S.; Nauka, E.; Dalziel, G. Chapter 18—Accelerated Predictive Stability (APS) Applications: Packaging Strategies for Controlling Dissolution Performance. In Accelerated Predictive Stability; Qiu, F., Scrivens, G., Eds.; Academic Press: Boston, MA, USA, 2018; pp. 383–401. [Google Scholar] [CrossRef]
- Adeleye, O.A. Relationship between compression pressure, mechanical strength and release properties of tablets. Polim. Med. 2019, 49, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Vodáčková, P.; Vraníková, B.; Svačinová, P.; Franc, A.; Elbl, J.; Muselík, J.; Kubalák, R.; Solný, T. Evaluation and Comparison of Three Types of Spray Dried Coprocessed Excipient Avicel® for Direct Compression. BioMed Res. Int. 2018, 2018, 2739428. [Google Scholar] [CrossRef]
- Ordu, J.A.; Abidde, T.O.; Okafo, S.E. Evaluation of the binding properties of gum obtained from dried leaves of Cochoros olitorious on metronidazole tablets formulation. Pharma Innov. J. 2018, 7, 688–694. [Google Scholar]
- Lieberman, H.A.; Lachman, L.; Schwartz, J.B. Pharmaceutical Dosage Forms: Tablets Vol. 2, 2nd ed.; Marcel Dekker, Inc.: New York, NY, USA, 1990; pp. 1–71. [Google Scholar]
- Carlin, B. Pharmaceutical Dosage Forms—Tablets: Rational Design and Formulation, 3rd ed.; Direct Compression and the Role of Filler-Binders; Augsburger, L.L., Hoag, S.W., Eds.; CRC Press: Boca Raton, FL, USA, 2008; pp. 173–216. [Google Scholar] [CrossRef]
- Kása, P.; Bajdik, J.; Zsigmond, Z.; Pintye-Hódi, K. Study of the compaction behaviour and compressibility of binary mixtures of some pharmaceutical excipients during direct compression. Chem. Eng. Process. Process Intensif. 2009, 48, 859–863. [Google Scholar] [CrossRef]
- Patel, S.; Kaushal, A.M.; Bansal, A.K. Compression Physics in the Formulation Development of Tablets. Crit. Rev. Ther. Drug Carr. Syst. 2006, 23, 1–66. [Google Scholar] [CrossRef] [PubMed]
- Tho, I.; Bauer-Brandl, A. Quality by design (QbD) approaches for the compression step of tableting. Expert Opin. Drug Deliv. 2011, 8, 1631–1644. [Google Scholar] [CrossRef]
- Thoorens, G.; Krier, F.; Leclercq, B.; Carlin, B.; Evrard, B. Microcrystalline cellulose, a direct compression binder in a quality by design environment—A review. Int. J. Pharm. 2014, 473, 64–72. [Google Scholar] [CrossRef]
- Horio, T.; Yasuda, M.; Matsusaka, S. Effect of particle shape on powder flowability of microcrystalline cellulose as determined using the vibration shear tube method. Int. J. Pharm. 2014, 473, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Doelker, E.; Mordier, D.; Iten, H.; Humbert-Droz, P. Comparative Tableting Properties of Sixteen Microcrystalline Celluloses. Drug Dev. Ind. Pharm. 1987, 13, 1847–1875. [Google Scholar] [CrossRef]
- Ferrari, F.; Bertoni, M.; Bonferoni, M.C.; Rossi, S.; Caramella, C.; Nyström, C. Investigation on bonding and disintegration properties of pharmaceutical materials. Int. J. Pharm. 1996, 136, 71–79. [Google Scholar] [CrossRef]
- Mostafa, H.F.; Ibrahim, M.A.; Sakr, A. Development and optimization of dextromethorphan hydrobromide oral disintegrating tablets: Effect of formulation and process variables. Pharm. Dev. Technol. 2013, 18, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Bolhuis, G.K.; Chowhan, Z.T. Materials for Direct Compaction; Alderborn, G., Nyström, C., Eds.; Marcel Dekker, Inc.: New York, NY, USA, 1996. [Google Scholar]
- Lahdenpää, E.; Niskanen, M.; Yliruusi, J. Crushing strength, disintegration time and weight variation of tablets compressed from three Avicel® PH grades and their mixtures. Eur. J. Pharm. Biopharm. 1997, 43, 315–322. [Google Scholar] [CrossRef]
- Bala, R.; Khanna, S.; Pawar, P. Formulation and optimization of fast dissolving intraoral drug delivery system for clobazam using response surface methodology. J. Adv. Pharm. Technol. Res. 2013, 4, 151–159. [Google Scholar] [CrossRef]
- Van Veen, B.; Bolhuis, G.K.; Wu, Y.S.; Zuurman, K.; Frijlink, H.W. Compaction mechanism and tablet strength of unlubricated and lubricated (silicified) microcrystalline cellulose. Eur. J. Pharm. Biopharm. 2005, 59, 133–138. [Google Scholar] [CrossRef]
- Kraboun, K.; Phanumong, P.; Tochampa, W.; Jittrepotch, N.; Rojsuntornkitti, K.; Chatdamrong, W.; Kongbangkerd, T. Impact of in vitro digestion phases on antioxidant properties of monascal waxy corn from 2-step fermentation. J. Microbiol. Biotechnol. Food Sci. 2021, 2021, 454–456. [Google Scholar]
- EFSA Panel on Food Additives and Nutrient Sources added to Food (ANS); Younes, M.; Aggett, P.; Aguilar, F.; Crebelli, R.; Dusemund, B.; Filipič, M.; Frutos, M.J.; Galtier, P.; Gott, D.; et al. Scientific opinion on the safety of monacolins in red yeast rice. EFSA J. 2018, 16, e05368. [Google Scholar]
- Li, Y.; Zhou, Y.-C.; Yang, M.-H.; Ou-Yang, Z. Natural occurrence of citrinin in widely consumed traditional Chinese food red yeast rice, medicinal plants and their related products. Food Chem. 2012, 132, 1040–1045. [Google Scholar] [CrossRef]
- Lachenmeier, D.W.; Monakhova, Y.B.; Kuballa, T.; Löbell-Behrends, S.; Maixner, S.; Kohl-Himmelseher, M.; Steffen, C. NMR evaluation of total statin content and HMG-CoA reductase inhibition in red yeast rice (Monascus spp.) food supplements. Chin. Med. 2012, 7, 8. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Sample | Dosage Form | Single Unit Weight-Label (mg) | Single Unit Weight Average-Measured (mg) | Declared-Measured Weight Deviation (%) | Compliance to Ph. Eur. (Mass Uniformity) |
|---|---|---|---|---|---|
| #1 | Film-coated tablet | 400 | 421 | 5.2 | Pass |
| #2 | Film-coated tablet | 1000 | 1022 | 2.2 | Pass |
| #3 | Film-coated tablet | 1100 | 1141 | 3.8 | Pass |
| #4 | Film-coated tablet | 1340 | 1352 | 0.9 | Pass |
| #5 | Film-coated tablet | 983 | 978 | −0.5 | Pass |
| #6 | Uncoated tablet | 1000 | 999 | −0.1 | Pass |
| #7 | Uncoated tablet | 550 | 551 | 0.1 | Pass |
| #8 | Uncoated tablet | NR | 898 | - | Pass |
| #9 | Uncoated tablet | 330 | 329 | −0.3 | Pass |
| #10 | Hard-shell capsule | 450 | 463 | 6.1 | Pass |
| #11 | Hard-shell capsule | 500 | 498 | 1.8 | Pass |
| #12 | Hard-shell capsule | 450 | 463 | 3 | Pass |
| #13 | Softgels | 1600 | 1657 | 3.6 | Pass |
| #14 | Softgels | 1777 | 1892 | 6.6 | Pass |
| Sample | Dosage Form | Thickness | Diameter | Resistance to Crushing | Compliance to Disintegration Specifications | Compliance to Revised Disintegration Specifications |
|---|---|---|---|---|---|---|
| (mm ± SD) | (mm ± SD) | (N ± SD) | (Ph. Eur. 11) | (in House) * | ||
| #1 | Film-coated tablet | 5.58 ± 0.09 | 10.19 ± 0.01 | 84 ± 17 | Pass | - |
| #2 | Film-coated tablet | 7.82 ± 0.02 | 19.16 ± 0.01 | 289 ± 7 | Fail | Fail |
| #3 | Film-coated tablet | 7.16 ± 0.01 | 20.7 ± 0.0 | 238 ± 5 | Fail | Fail |
| #4 | Film-coated tablet | - | - | 280 ± 7 | Fail | Fail |
| #5 | Film-coated tablet | 6.76 ± 0.03 | 19.16 ± 0.02 | 211 ± 11 | Pass | - |
| #6 | Uncoated tablet | 7.13 ± 0.04 | 20.65 ± 0.19 | 76 ± 8 | Pass | - |
| #7 | Uncoated tablet | 7.00 ± 0.03 | 10.17 ± 0.01 | 47 ± 3 | Fail | Pass |
| #8 | Uncoated tablet | - | - | - | Pass | - |
| #9 | Uncoated tablet | 4.10 ± 0.04 | 10.12 | 125 ± 14 | Pass | - |
| #10 | Hard-shell capsule | - | - | NA | Pass | - |
| #11 | Hard-shell capsule | - | - | NA | Pass | - |
| #12 | Hard-shell capsule | - | - | NA | Pass | - |
| #13 | Softgels | - | - | NA | - | - |
| #14 | Softgels | - | - | NA | Pass | - |
| Sample | mg of Monacolin K Declared in Labels Per Unit | mg of Monacolina K Per Unit ± SD | Bioaccessibility | |
|---|---|---|---|---|
| Gastric Phase | Intestinal Phase | |||
| mg of Monacolina K Per Unit ± SD | mg of Monacolina K Per Unit ± SD | |||
| #1 | 10 | 9.15 ± 0.38 | 8.78 ± 0.41 | 9.06 ± 0.34 |
| #2 | 1.45 | 1.47 ± 0.13 | 1.41 ± 0.11 | 1.43 ± 0.09 |
| #3 | 10 | 9.76 ± 0.26 | 9.47 ± 0.17 | 9.66 ± 0.22 |
| #4 | 5 | 4.94 ± 0.24 | 4.84 ± 0.29 | 4.89 ± 0.12 |
| #5 | 2.8 | 2.77 ± 0.09 | 2.66 ± 0.11 | 2.74 ± 0.08 |
| #6 | 10 | 10.04 ± 0.52 | 9.64 ± 0,27 | 9.94 ± 0.27 |
| #7 | 10 | 8.61 ± 0.47 | 8.27 ± 0.42 | 8.5 ± 0.36 |
| #8 | 10 | 7.92 ± 0.37 | 7.60 ± 0.29 | 7.84 ± 0.24 |
| #9 | 10 | 10.14 ± 0.23 | 9.73 ± 0.29 | 10.02 ± 0.19 |
| #10 | 5 | 4.68 ± 0.32 | 4.09 ± 0.24 | 4.63 ± 0.30 |
| #11 | 10 | 9.38 ± 0.42 | 8.71 ± 0.33 | 9.39 ± 0.23 |
| #12 | 10 | 9.96 ± 0.26 | 9.16 ± 0.23 | 9.86 ± 0.23 |
| #13 | 10 | 9.92 ± 0.37 | 9.12 ± 0.29 | 9.89 ± 0.31 |
| #14 | 5 | 4.68 ± 0.31 | 4.11 ± 0.33 | 4.62 ± 0.29 |
| Analyte | Retention Time | Elemental Composition | Adduct Ion | Theoretical Mass | Measured Mass | Accuracy |
|---|---|---|---|---|---|---|
| (min) | (m/z) | (m/z) | (Δ ppm) | |||
| CIT | 4.97 | C13H14O5 | [M + H]+ | 251.0914 | 251.0912 | −0.79 |
| Recovery (%) | Precision (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| [RSDr, (RSDR)] | ||||||||||
| Analyte | Linearity (r2) | SSE (%) | 100 ng/mL | 50 ng/mL | 10 ng/mL | 100 ng/mL | 50 ng/mL | 10 ng/mL | LOD (ng/mL) | LOQ (ng/mL) |
| CIT | 0.998 | 85 | 85 | 85 | 86 | 78(12) | 84(8) | 84(18) | 1.56 | 6.25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitiello, A.; Izzo, L.; Castaldo, L.; d’Angelo, I.; Ungaro, F.; Miro, A.; Ritieni, A.; Quaglia, F. The Questionable Quality Profile of Food Supplements: The Case of Red Yeast Rice Marketed Products. Foods 2023, 12, 2142. https://doi.org/10.3390/foods12112142
Vitiello A, Izzo L, Castaldo L, d’Angelo I, Ungaro F, Miro A, Ritieni A, Quaglia F. The Questionable Quality Profile of Food Supplements: The Case of Red Yeast Rice Marketed Products. Foods. 2023; 12(11):2142. https://doi.org/10.3390/foods12112142
Chicago/Turabian StyleVitiello, Antonella, Luana Izzo, Luigi Castaldo, Ivana d’Angelo, Francesca Ungaro, Agnese Miro, Alberto Ritieni, and Fabiana Quaglia. 2023. "The Questionable Quality Profile of Food Supplements: The Case of Red Yeast Rice Marketed Products" Foods 12, no. 11: 2142. https://doi.org/10.3390/foods12112142
APA StyleVitiello, A., Izzo, L., Castaldo, L., d’Angelo, I., Ungaro, F., Miro, A., Ritieni, A., & Quaglia, F. (2023). The Questionable Quality Profile of Food Supplements: The Case of Red Yeast Rice Marketed Products. Foods, 12(11), 2142. https://doi.org/10.3390/foods12112142