Oleic Acid Is not the Only Relevant Mono-Unsaturated Fatty Ester in Olive Oil


 ,
,  ,
,  and
and 
Abstract
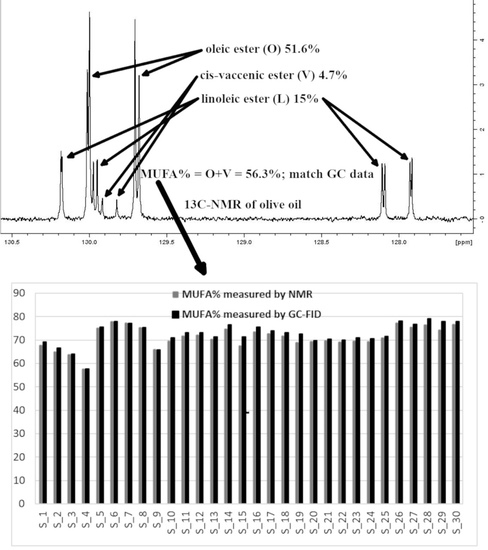
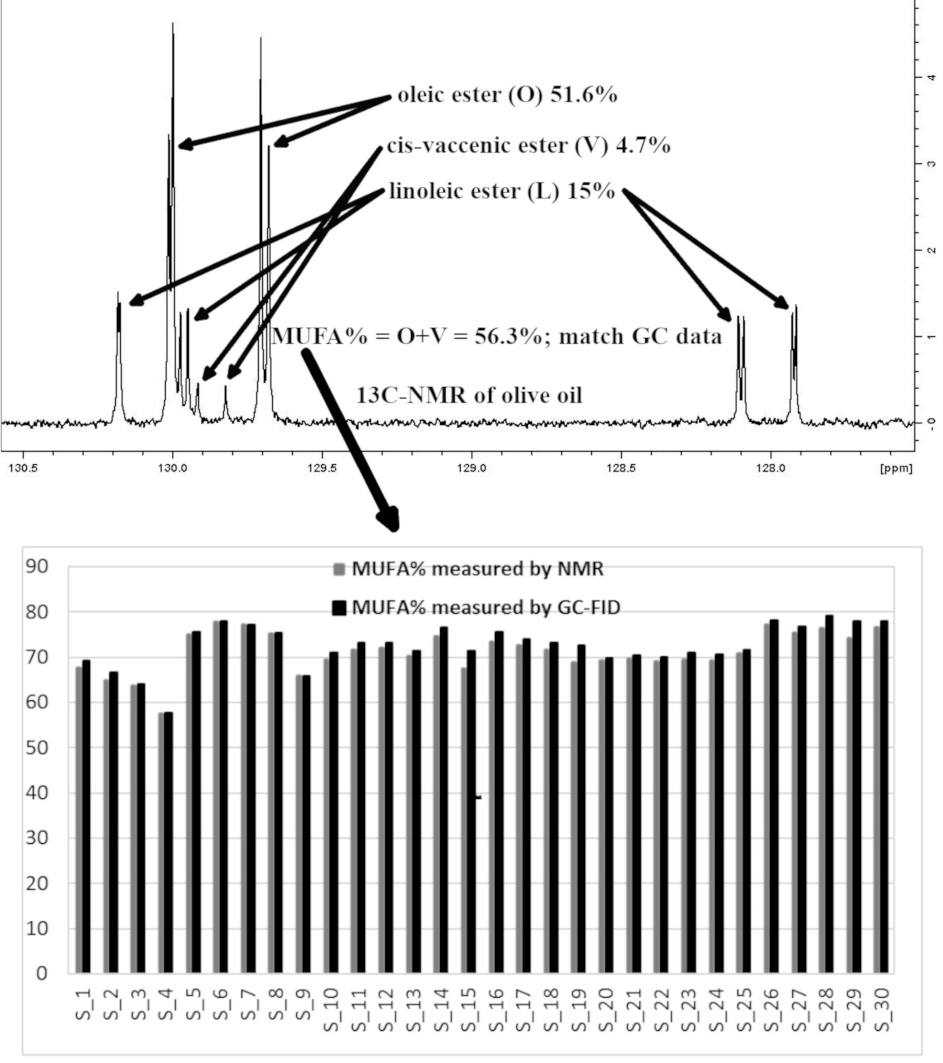
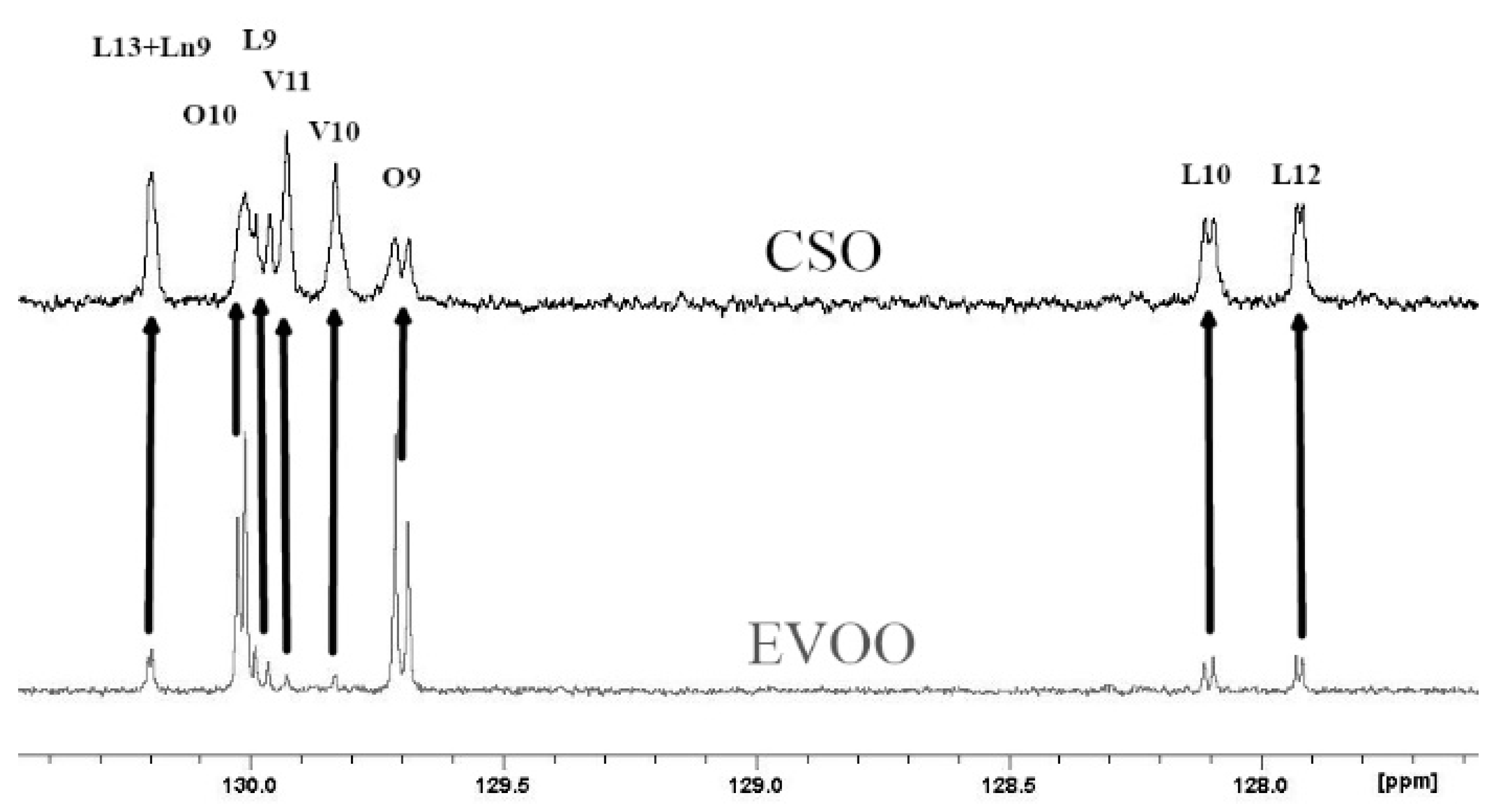
:
1. Introduction
2. Materials and Methods
2.1. Materials and Samples
2.2. GC-FID Analysis for the Comparative Tests
2.3. Sample Preparation for NMR
2.4. NMR Analysis
2.5. NMR Processing and Data Treatment
2.6. Mathematical Background of MARA-NMR and Updates
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Trichopolou, A.; Vasilopoulou, E. Mediterranean diet and longevity. Br. J. Nutr. 2000, 84, S205–S209. [Google Scholar] [CrossRef] [PubMed]
- Visioli, F.; Bernardini, E. Extra virgin olive oil’s polyphenols: Biological activities. Curr. Pharm. Des. 2011, 17, 786–804. [Google Scholar] [CrossRef] [PubMed]
- Frankel, E.N. Nutritional and biological properties of extra virgin olive oil. J. Agric. Food Chem. 2011, 59, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Jiménez, F.; Ruano, J.; Perez-Martinez, P.; Lopez-Segura, F.; Lopez-Miranda, J. The influence of olive oil on human health: Not a question of fat alone. Mol. Nutr. Food Res. 2007, 51, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Schwingshackl, L.; Christoph, M.; Hoffmann, G. Effects of Olive Oil on Markers of Inflammation and Endothelial Function—A Systematic Review and Meta-Analysis. Nutrients 2015, 7, 7651–7675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azadmard-Damirchi, S.; Dutta, P.C. Phytosterol Classes in Olive Oils and their Analysis by Common Chromatographic Method. In Olives and Olive Oil in Health and Disease Prevention; Victor, R., Preedy, V.R., Watson, R.R., Eds.; Academic Press: Cambridge, MA, USA, 2010; Chapter 27; pp. 249–257. [Google Scholar]
- Klikarova, J.; Rotondo, A.; Cacciola, F.; Ceslova, L.; Dugo, P.; Mondello, L.; Rigano, F. The Phenolic Fraction of Italian Extra Virgin Olive Oils: Elucidation through Combined Liquid Chromatography and NMR Approaches. Food Anal. Methods 2019, 12, 1759–1770. [Google Scholar] [CrossRef]
- Rotondo, A.; Salvo, A.; Gallo, V.; Rastrelli, L.; Dugo, G. Quick unreferenced NMR quantification of Squalene in vegetable oils. Eur. J. Lipid Sci. Technol. 2019, 119, 1700151. [Google Scholar] [CrossRef] [Green Version]
- EU. Commission Implementing Regulation (EU) No 1348/2013 of 16 December 2013 amending Regulation (EEC) No 2568/91 on the characteristics of olive oil and olive-residue oil and on the relevant methods of analysis. Off. J. Eur. Union 2013, L 338, 31–67. [Google Scholar]
- EU. Commission Delegated Regulation (EU) No 2015/1830 of 8 July 2015 amending Regulation (EEC) No 2568/91 on the characteristics of olive oil and olive-residue oil and on the relevant methods of analysis. Off. J. Eur. Union 2015, L 266, 9–13. [Google Scholar]
- Mannina, L.; Sobolev, A.P.; Segre, A. NMR. Olive oil as seen by NMR and chemometrics. Spectroscopy 2003, 15, 6–14. [Google Scholar]
- Simmler, C.; Napolitano, J.G.; Mc Alpine, J.B.; Chen, S.-N.; Pauli, G.F. Universal quantitative NMR analysis of complex natural samples. Curr. Opin. Biotechnol. 2014, 25, 51–59. [Google Scholar] [CrossRef]
- Salvo, A.; Rotondo, A.; La Torre, G.L.; Cicero, N.; Dugo, G. Determination of 1,2/1,3-diglycerides in Sicilian extra-virgin olive oils by 1H-NMR over a one-year storage period. Nat. Prod. Res. 2017, 31, 822–828. [Google Scholar] [CrossRef]
- Bharti, S.K.; Roi, R. Quantitative 1H NMR spectroscopy. Trends Anal. Chem. 2012, 35, 5–26. [Google Scholar] [CrossRef]
- Monakhova, Y.B.; Tsikin, A.M.; Kuballa, T.; Lachenmeiera, D.W.; Mushtakovab, S.P. Independent component analysis (ICA) algorithms for improved spectral deconvolution of overlapped signals in 1H NMR analysis: Application to foods and related products. Magn. Reson. Chem. 2014, 52, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Aracama, A.; Goicoechea, E.; Guillén, M.D. Direct study of minor extra-virgin olive oil components without any sample modification. 1H NMR multisupression experiment: A powerful tool. Food Chem. 2017, 228, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Mannina, L.; Sobolev, A.P. High resolution NMR characterization of olive oils in terms of quality, authenticity and geographical origin. Magn. Reson. Chem. 2011, 49, S3–S11. [Google Scholar] [CrossRef] [PubMed]
- Salvo, A.; Rotondo, A.; Mangano, V.; Grimaldi, V.; Stillitano, I.; D’Ursi, A.M.; Dugo, G.; Rastrelli, L. High-resolution magic angle spinning nuclear magnetic resonance (HR-MAS-NMR) as quick and direct insight of almonds. Nat. Prod. Res. 2019, 34, 71–77. [Google Scholar] [CrossRef]
- Rotondo, A.; Mannina, L.; Salvo, A. Multiple Assignment Recovered Analysis (MARA) NMR for a Direct Food Labeling, the Case Study of Olive Oils. Food Anal. Methods 2019, 12, 1238–1245. [Google Scholar] [CrossRef]
- Christie, W.W. Preparation of Ester Derivatives of Fatty Acids for Chromatographic Analysis. Adv. Lipid Methodol. 1993, 2, 69–111. [Google Scholar]
- Naccari, C.; Rando, R.; Salvo, A.; Donato, D.; Bartolomeo, G.; Mangano, V.; Lo Turco, V.; Dugo, G. Study on the composition and quality of several sicilian EVOOs (harvesting year 2015). Rivista Italiana Delle Sostanze Grasse 2017, 94, 231–237. [Google Scholar]
- Zhang, H.; Wang, Z.; Liu, O. Development and validation of a GC–FID method for quantitative analysis of oleic acid and related fatty acids. J. Pharm. Anal. 2015, 5, 223–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scano, P.; Casu, M.; Lai, A.; Saba, G.; Dessi, M.A.; Deiana, M.; Corongiu, F.P.; Bandino, G. Recognition and quantitation of cis-vaccenic and eicosenoic fatty acids in olive oils by 13C nuclear magnetic resonance spectroscopy. Lipids 1999, 34, 757–759. [Google Scholar] [CrossRef] [PubMed]
- Barison, A.; Pereira da Silva, C.W.; Ramos Campos, F.; Simonelli, F.; Lenz, C.A.; Ferreira, A.G. A simple methodology for the determination of fatty acid composition in edible oils through 1H NMR spectroscopy. Magn. Reson. Chem. 2010, 48, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Knothe, G.; Kenar, J.A. Determination of the fatty acid profile by 1H-NMR spectroscopy. Eur. J. Lipid Sci. Technol. 2004, 106, 88–96. [Google Scholar] [CrossRef]
- Boudour-Benrachou, N.; Plard, J.; Pinatel, C.; Artaud, J.; Dupuy, N. Fatty acid compositions of olive oils from six cultivars from East and South-Western Algeria. Adv. Food Technol. Nutr. Sci. Open J. 2017, 3, 1–5. [Google Scholar] [CrossRef]
- Retief, L.; McKenzie, J.M.; Koch, K.R. A novel approach to the rapid assignment of 13C NMR spectra of major components of vegetable oils such as avocado, mango kernel andmacadamia nut oils. Magn. Reson. Chem. 2009, 47, 771–781. [Google Scholar] [CrossRef]
- Chira, N.A.; Nicolescu, A.; Stan, R.; Rosca, S. Fatty Acid Composition of Vegetable Oils Determined from 13C-NMR Spectra. Rev. Chim. 2016, 67, 1257–1263. [Google Scholar]
- Vlahov, G.; Schiavone, C.; Simone, N. Quantitative 13C NMR method using the DEPT pulse sequence for the determination of the geographical origin (DOP) of olive oils. Magn. Reson. Chem. 2001, 39, 689–695. [Google Scholar] [CrossRef]
- Zamora, R.; Alba, V.; Hidalgo, F.J. Use of High-Resolution 13C Nuclear Magnetic Resonance Spectroscopy for the Screening of Virgin Olive Oils. J. Am. Oil Chem. Soc. 2001, 78, 89–94. [Google Scholar] [CrossRef]
- Ng, S.; Koh, H.F. Detection of cis-Vaccenic Acid in Palm Oil by 13C NMR Spectroscopy. Lipids 1988, 23, 140–143. [Google Scholar] [CrossRef]
- Kuznetsova, E.I.; Pchelkin, V.P.; Tsydendambaev, V.D.; Vereshchagin, A.G. Distribution of unusual fatty acids in the mesocarp triacylglycerols of maturing sea buckthorn fruits. Russ. J. Plant Physiol. 2010, 57, 852–858. [Google Scholar] [CrossRef]
- Vlahov, G.; Chepkwony, P.K.; Ndalut, P.K. 13C NMR Characterization of Triacylglycerols of Moringa oleifera Seed Oil: An “Oleic-Vaccenic Acid” Oil. J. Agric. Food Chem. 2002, 50, 970–975. [Google Scholar] [CrossRef] [PubMed]
- Barding, G.A.; Béni, S.; Fukao, T.; Bailey-Serres, J.; Larive, C.K. Comparison of GC-MS and NMR for Metabolite Profiling of Rice Subjected to Submergence Stress. J. Proteome Res. 2012, 12, 898–909. [Google Scholar] [CrossRef] [PubMed]
- Romano, R.; Giordano, A.; Le Grottaglie, L.; Manzo, N.; Paduano, A.; Sacchi, R.; Santini, A. Volatile compounds in intermittent frying by gas chromatography and nuclear magnetic resonance. Eur. J. Lipid Sci. Technol. 2013, 115, 764–773. [Google Scholar] [CrossRef]
- Shibaharaa, A.; Yamamoto, K.; Nakayama, T.; Kajimotoa, G. cis-Vaccenic Acid in Pulp Lipids of Commonly Available Fruits. J. Am. Oil Chem. Soc. 1987, 64, 397–398. [Google Scholar] [CrossRef]
- Circi, S.; Capitani, D.; Randazzo, A.; Ingallina, C.; Mannina, L.; Sobolev, A. Panel test and chemical analyses of commercial olive oils: A comparative study. Chem. Biol. Technol. Agric. 2017, 4, 1–10. [Google Scholar] [CrossRef]
- Naviglio, D.; Romano, R.; Pizzolongo, F.; Santini, A.; Vito, A.D.; Schiavo, L.; Nota, G.; Musso, S.S. Rapid determination of esterified glycerol and glycerides in triglyceride fats and oils by means of periodate method after transesterification. Food Chem. 2007, 102, 399–405. [Google Scholar] [CrossRef]





{kind=link}
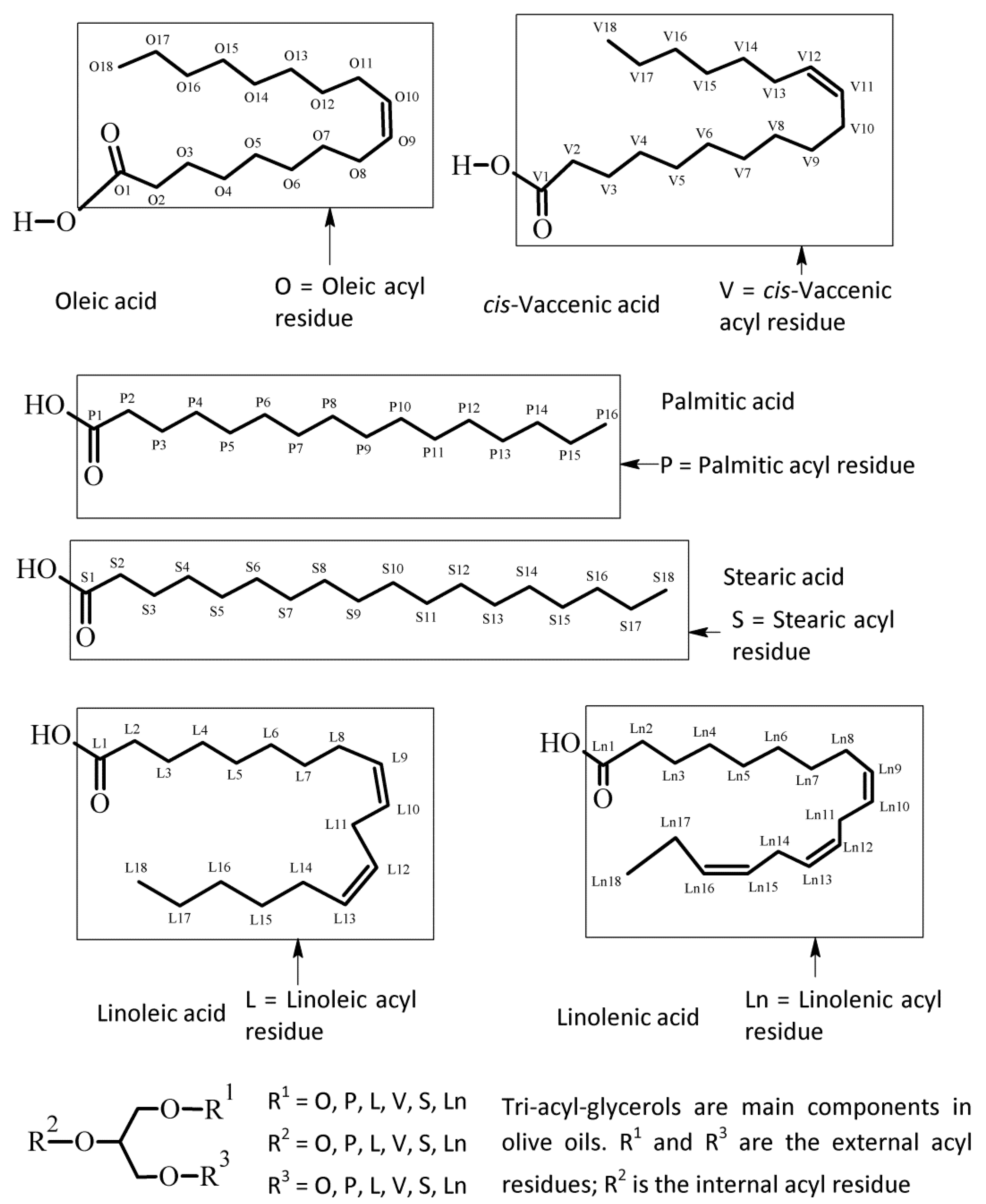{kind=link}
{kind=link}
{kind=link}
{kind=link}
| tri-acyl-glycerol percent | TG% |
| 1,2 di-acyl-glycerol percent | 1,2-DG% |
| 1,3 di-acyl-glycerol percent | 1,3-DG% |
| squalene molecular% | SQmol% |
| linolenate esters % | Ln% |
| linoleates esters % | L% |
| oleic esters % | O% |
| palmitoleic esters % | PO% |
| cis-vaccenic esters % | V% |
| palmitate esters % | P% |
| stearate esters | S% |
| linolenate esters % in internal glyceril position | Lni% |
| linoleates esters % in internal glyceril position | Li% |
| oleic esters % in internal glyceril position | Oi% |
| palmitoleic esters % in internal glyceril position | POi% |
| cis-vaccenic esters % in internal glyceril position | Vi% |
| palmitate esters % in internal glyceril position | Pi% |
| sterarate esters % in internal glyceril position | Si% |
| β-sitosterol + avenasterol + camposterol in molecular ppm | VSTR |
| cyclo arthenol and other cyclosterols in molecular ppm | CYSR |
| Sample | TG% | 1,2-DG% | 1,3-DG% | SQmol% | Ln% | L% | O% | PO% | V% | P% | S% |
|---|---|---|---|---|---|---|---|---|---|---|---|
| S_1 | 96.7 ± 0.1 | 1.19 ± 0.06 | 2.1 ± 0.1 | 1.7 ± 0.1 | 0.59 ± 0.03 | 10.2 ± 0.1 | 63.9 ± 0.5 | 1.3 ± 0.3 | 2.8 ± 0.3 | 19.1 ± 0.3 | 2.2 ± 0.2 |
| S_2 | 97.4 ± 0.1 | 1.30 ± 0.07 | 1.34 ± 0.09 | 2.2 ± 0.1 | 0.57 ± 0.03 | 12.5 ± 0.1 | 61.8 ± 0.4 | 0.9 ± 0.2 | 2.5 ± 0.2 | 19.7 ± 0.3 | 1.9 ± 0.2 |
| S_3 | 97.7 ± 0.1 | 1.65 ± 0.09 | 0.68 ± 0.05 | 1.3 ± 0.1 | 0.51 ± 0.03 | 12.6 ± 0.1 | 59.4 ± 0.4 | 0.8 ± 0.2 | 3.7 ± 0.3 | 21.1 ± 0.3 | 2.0 ± 0.2 |
| S_4 | 97.4 ± 0.1 | 1.39 ± 0.07 | 1.21 ± 0.08 | 0.8 ± 0.0 | 0.63 ± 0.03 | 15.4 ± 0.1 | 51.9 ± 0.4 | 1.1 ± 0.2 | 4.9 ± 0.4 | 23.7 ± 0.3 | 2.4 ± 0.2 |
| S_5 | 97.2 ± 0.1 | 2.1 ± 0.1 | 0.70 ± 0.05 | 2.9 ± 0.2 | 0.67 ± 0.04 | 5.7 ± 0.1 | 72.8 ± 0.5 | 0.7 ± 0.1 | 1.8 ± 0.2 | 16.0 ± 0.2 | 2.4 ± 0.2 |
| S_6 | 97.3 ± 0.1 | 1.70 ± 0.09 | 0.97 ± 0.07 | 2.7 ± 0.2 | 0.65 ± 0.03 | 2.6 ± 0.0 | 73.9 ± 0.5 | 0.7 ± 0.1 | 3.5 ± 0.3 | 16.7 ± 0.2 | 1.9 ± 0.2 |
| S_7 | 97.3 ± 0.1 | 2.0 ± 0.1 | 0.77 ± 0.05 | 2.5 ± 0.2 | 0.61 ± 0.03 | 3.1 ± 0.0 | 74.2 ± 0.5 | 0.8 ± 0.2 | 2.7 ± 0.2 | 16.1 ± 0.2 | 2.5 ± 0.2 |
| S_8 | 97.7 ± 0.1 | 1.59 ± 0.08 | 0.74 ± 0.05 | 2.2 ± 0.1 | 0.61 ± 0.03 | 6.4 ± 0.1 | 70.8 ± 0.5 | 0.9 ± 0.2 | 3.6 ± 0.3 | 15.5 ± 0.2 | 2.1 ± 0.2 |
| S_9 | 96.8 ± 0.1 | 1.78 ± 0.09 | 1.4 ± 0.1 | 1.3 ± 0.1 | 0.44 ± 0.02 | 10.2 ± 0.1 | 60.0 ± 0.4 | 1.1 ± 0.2 | 5.0 ± 0.4 | 21.7 ± 0.3 | 1.6 ± 0.1 |
| S_10 | 96.9 ± 0.1 | 1.30 ± 0.07 | 1.8 ± 0.1 | 2.3 ± 0.1 | 0.68 ± 0.04 | 7.6 ± 0.1 | 65.2 ± 0.5 | 1.1 ± 0.2 | 3.4 ± 0.3 | 20.0 ± 0.3 | 2.1 ± 0.2 |
| S_11 | 97.4 ± 0.1 | 1.21 ± 0.06 | 1.4 ± 0.1 | 2.9 ± 0.2 | 0.61 ± 0.03 | 5.9 ± 0.1 | 67.2 ± 0.5 | 1.2 ± 0.3 | 3.6 ± 0.3 | 19.2 ± 0.3 | 2.4 ± 0.2 |
| S_12 | 97.6 ± 0.1 | 1.32 ± 0.07 | 1.04 ± 0.07 | 3.1 ± 0.2 | 0.64 ± 0.03 | 7.7 ± 0.1 | 65.4 ± 0.5 | 2.5 ± 0.5 | 4.6 ± 0.4 | 17.4 ± 0.3 | 1.9 ± 0.2 |
| S_13 | 97.7 ± 0.1 | 1.47 ± 0.08 | 0.88 ± 0.06 | 2.8 ± 0.2 | 0.79 ± 0.04 | 8.3 ± 0.1 | 67.1 ± 0.5 | 1.0 ± 0.2 | 2.4 ± 0.2 | 18.3 ± 0.3 | 2.0 ± 0.2 |
| S_14 | 97.8 ± 0.1 | 1.40 ± 0.07 | 0.80 ± 0.06 | 3.4 ± 0.2 | 0.64 ± 0.03 | 4.5 ± 0.0 | 71.3 ± 0.5 | 1.2 ± 0.2 | 2.5 ± 0.2 | 17.7 ± 0.3 | 2.3 ± 0.2 |
| S_15 | 97.8 ± 0.1 | 1.14 ± 0.06 | 1.07 ± 0.07 | 4.0 ± 0.3 | 0.60 ± 0.03 | 8.3 ± 0.1 | 64.0 ± 0.5 | 0.7 ± 0.1 | 3.1 ± 0.3 | 20.7 ± 0.3 | 2.7 ± 0.2 |
| S_16 | 97.8 ± 0.1 | 1.48 ± 0.08 | 0.72 ± 0.05 | 3.1 ± 0.2 | 0.58 ± 0.03 | 6.2 ± 0.1 | 70.2 ± 0.5 | 0.6 ± 0.1 | 3.0 ± 0.3 | 17.5 ± 0.3 | 2.0 ± 0.2 |
| S_17 | 97.4 ± 0.1 | 1.53 ± 0.08 | 1.08 ± 0.07 | 2.1 ± 0.1 | 0.61 ± 0.03 | 6.4 ± 0.1 | 68.4 ± 0.5 | 1.2 ± 0.3 | 3.3 ± 0.3 | 17.9 ± 0.3 | 2.2 ± 0.2 |
| S_18 | 97.4 ± 0.1 | 1.22 ± 0.06 | 1.4 ± 0.1 | 2.3 ± 0.1 | 0.58 ± 0.03 | 6.7 ± 0.1 | 68.5 ± 0.5 | 0.8 ± 0.2 | 2.5 ± 0.2 | 19.1 ± 0.3 | 1.7 ± 0.1 |
| S_19 | 97.6 ± 0.1 | 1.43 ± 0.07 | 0.98 ± 0.07 | 3.5 ± 0.2 | 0.79 ± 0.04 | 7.5 ± 0.1 | 65.6 ± 0.5 | 0.7 ± 0.2 | 2.8 ± 0.3 | 20.7 ± 0.3 | 1.9 ± 0.2 |
| S_20 | 97.2 ± 0.1 | 1.59 ± 0.08 | 1.17 ± 0.08 | 2.0 ± 0.1 | 0.63 ± 0.03 | 9.3 ± 0.1 | 64.8 ± 0.5 | 1.1 ± 0.2 | 3.7 ± 0.3 | 19.0 ± 0.3 | 1.5 ± 0.1 |
| S_21 | 97.2 ± 0.1 | 1.70 ± 0.09 | 1.10 ± 0.08 | 3.0 ± 0.2 | 0.70 ± 0.04 | 7.6 ± 0.1 | 66.2 ± 0.5 | 0.6 ± 0.1 | 3.1 ± 0.3 | 19.9 ± 0.3 | 1.9 ± 0.2 |
| S_22 | 97.7 ± 0.1 | 1.49 ± 0.08 | 0.78 ± 0.05 | 3.6 ± 0.2 | 0.73 ± 0.04 | 7.5 ± 0.1 | 64.8 ± 0.5 | 1.3 ± 0.3 | 3.4 ± 0.3 | 20.7 ± 0.3 | 1.7 ± 0.1 |
| S_23 | 98.0 ± 0.1 | 1.63 ± 0.09 | 0.37 ± 0.03 | 3.4 ± 0.2 | 0.76 ± 0.04 | 6.9 ± 0.1 | 65.4 ± 0.5 | 1.4 ± 0.3 | 2.8 ± 0.3 | 20.1 ± 0.3 | 2.5 ± 0.2 |
| S_24 | 97.6 ± 0.1 | 1.58 ± 0.08 | 0.85 ± 0.06 | 3.4 ± 0.2 | 0.73 ± 0.04 | 7.9 ± 0.1 | 65.9 ± 0.5 | 1.1 ± 0.2 | 2.5 ± 0.2 | 20.0 ± 0.3 | 2.0 ± 0.2 |
| S_25 | 98.0 ± 0.1 | 1.21 ± 0.06 | 0.78 ± 0.05 | 2.4 ± 0.2 | 0.58 ± 0.03 | 7.2 ± 0.1 | 66.9 ± 0.5 | 0.6 ± 0.1 | 3.6 ± 0.3 | 19.2 ± 0.3 | 2.0 ± 0.2 |
| S_26 | 97.3 ± 0.1 | 1.27 ± 0.07 | 1.4 ± 0.1 | 2.0 ± 0.1 | 0.68 ± 0.04 | 4.4 ± 0.0 | 73.3 ± 0.5 | 1.3 ± 0.3 | 3.0 ± 0.3 | 15.6 ± 0.2 | 1.8 ± 0.1 |
| S_27 | 97.4 ± 0.1 | 1.48 ± 0.08 | 1.13 ± 0.08 | 2.5 ± 0.2 | 0.64 ± 0.03 | 4.4 ± 0.0 | 72.2 ± 0.5 | 0.9 ± 0.2 | 2.6 ± 0.2 | 16.9 ± 0.2 | 2.3 ± 0.2 |
| S_28 | 97.8 ± 0.1 | 1.31 ± 0.07 | 0.85 ± 0.06 | 2.5 ± 0.2 | 0.64 ± 0.03 | 7.1 ± 0.1 | 73.9 ± 0.5 | 1.3 ± 0.3 | 1.5 ± 0.1 | 13.7 ± 0.2 | 1.8 ± 0.1 |
| S_29 | 96.9 ± 0.1 | 1.45 ± 0.08 | 1.6 ± 0.1 | 2.7 ± 0.2 | 0.65 ± 0.04 | 5.2 ± 0.0 | 70.5 ± 0.5 | 1.4 ± 0.3 | 2.6 ± 0.2 | 17.1 ± 0.2 | 2.4 ± 0.2 |
| S_30 | 97.9 ± 0.1 | 1.22 ± 0.06 | 0.89 ± 0.06 | 1.9 ± 0.1 | 0.64 ± 0.03 | 4.8 ± 0.0 | 73.1 ± 0.5 | 0.9 ± 0.2 | 2.9 ± 0.3 | 15.4 ± 0.2 | 2.3 ± 0.2 |
| S_31 | 96.9 ± 0.1 | 2.5 ± 0.1 | 0.50 ± 0.03 | 2.6 ± 0.2 | 0.56 ± 0.03 | 6.3 ± 0.1 | 64.5 ± 0.5 | 0.5 ± 0.1 | 5.2 ± 0.5 | 21.5 ± 0.3 | 1.4 ± 0.1 |
| S_32 | 96.0 ± 0.1 | 3.0 ± 0.2 | 0.99 ± 0.07 | 3.1 ± 0.2 | 0.69 ± 0.04 | 9.4 ± 0.1 | 61.8 ± 0.4 | 1.0 ± 0.2 | 4.4 ± 0.4 | 21.1 ± 0.3 | 1.8 ± 0.1 |
| S_33 | 95.8 ± 0.1 | 2.3 ± 0.1 | 1.9 ± 0.1 | 2.2 ± 0.1 | 0.66 ± 0.04 | 8.7 ± 0.1 | 65.6 ± 0.5 | 1.0 ± 0.2 | 3.9 ± 0.4 | 18.3 ± 0.3 | 1.9 ± 0.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rotondo, A.; La Torre, G.L.; Dugo, G.; Cicero, N.; Santini, A.; Salvo, A. Oleic Acid Is not the Only Relevant Mono-Unsaturated Fatty Ester in Olive Oil. Foods 2020, 9, 384. https://doi.org/10.3390/foods9040384
Rotondo A, La Torre GL, Dugo G, Cicero N, Santini A, Salvo A. Oleic Acid Is not the Only Relevant Mono-Unsaturated Fatty Ester in Olive Oil. Foods. 2020; 9(4):384. https://doi.org/10.3390/foods9040384
Chicago/Turabian StyleRotondo, Archimede, Giovanna Loredana La Torre, Giacomo Dugo, Nicola Cicero, Antonello Santini, and Andrea Salvo. 2020. "Oleic Acid Is not the Only Relevant Mono-Unsaturated Fatty Ester in Olive Oil" Foods 9, no. 4: 384. https://doi.org/10.3390/foods9040384
APA StyleRotondo, A., La Torre, G. L., Dugo, G., Cicero, N., Santini, A., & Salvo, A. (2020). Oleic Acid Is not the Only Relevant Mono-Unsaturated Fatty Ester in Olive Oil. Foods, 9(4), 384. https://doi.org/10.3390/foods9040384