The Benefits of Whole Genome Sequencing for Foodborne Outbreak Investigation from the Perspective of a National Reference Laboratory in a Smaller Country

 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Conventional Methods Applied for the Initial Outbreak Investigation
2.2. Retrospective WGS-Based Outbreak Analysis
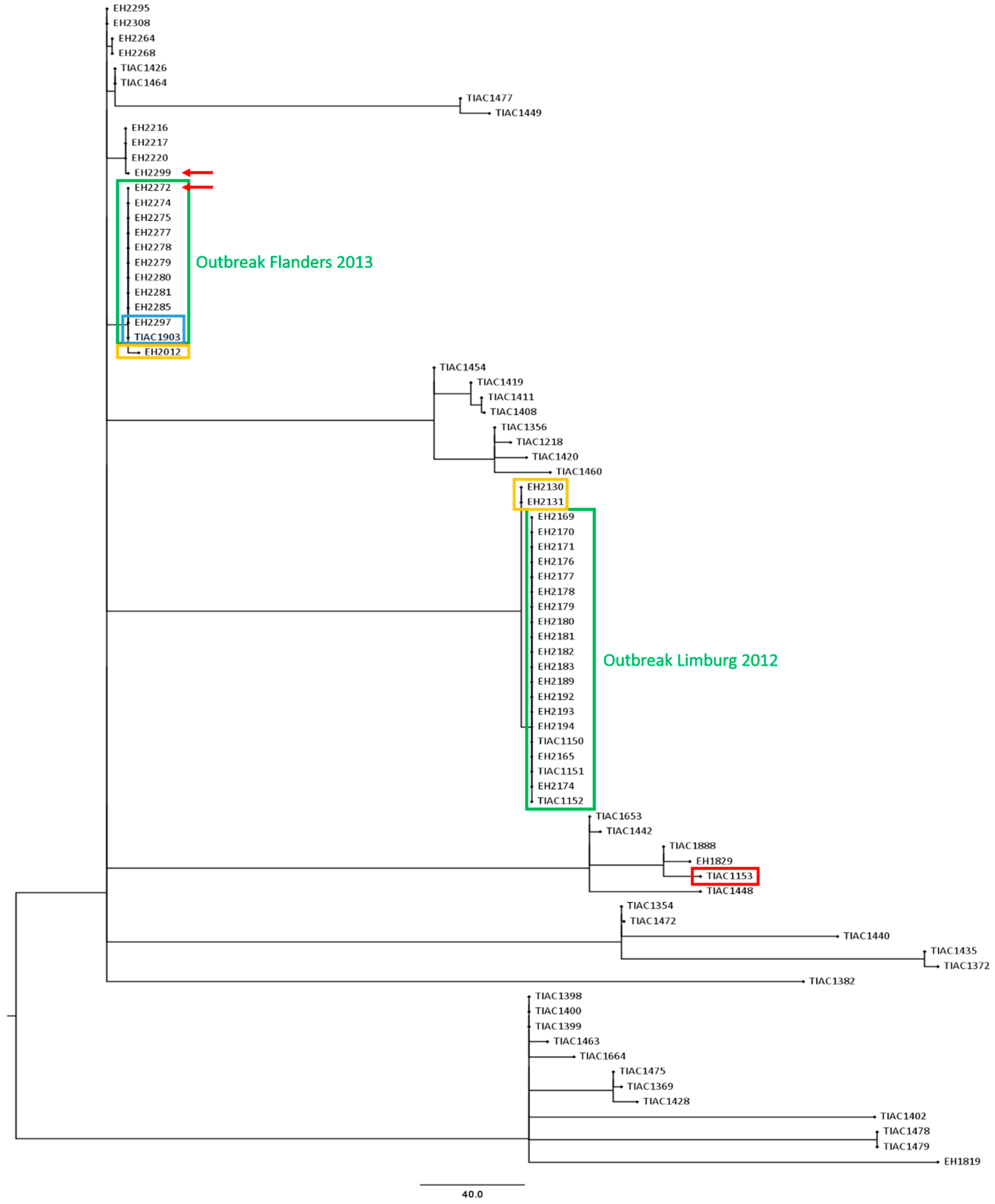
3. Results
3.1. Description of the Selected Outbreaks and Isolates
3.2. Comparing Isolate Characterization Using WGS and Conventional Methods
3.3. Comparison of the Phylogenetic Performance between WGS and Conventional Methods in Resolving Relatedness
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Foodborne Disease Outbreaks: Guidelines for Investigation and Control; World Health Organization: Geneva, Switzerland, 2008. [Google Scholar]
- Baker, C.A.; Rubinelli, P.M.; Park, S.H.; Carbonero, F.; Ricke, S.C. Shiga Toxin-Producing Escherichia coli in Food: Incidence, Ecology, and Detection Strategies. Food Control 2016, 59, 407–419. [Google Scholar] [CrossRef]
- Amézquita-López, B.A.; Soto-Beltrán, M.; Lee, B.G.; Yambao, J.C.; Quiñones, B. Isolation, Genotyping and Antimicrobial Resistance of Shiga Toxin-Producing Escherichia coli. J. Microbiol. Immunol. Infect. 2018, 51, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Holmes, A.; Allison, L.; Ward, M.; Dallman, T.J.; Clark, R.; Fawkes, A.; Murphy, L.; Hanson, M. Utility of Whole-Genome Sequencing of E. coli O157 for Outbreak Detection and Epidemiological Surveillance. J. Clin. Microbiol. 2015, 53, 3565–3573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooka, T.; Ogura, Y.; Asadulghani, M.; Ohnishi, M.; Nakayama, K.; Terajima, J.; Watanabe, H.; Hayashi, T. Inference of the Impact of Insertion Sequence (IS) Elements on Bacterial Genome Diversification through Analysis of Small-Size Structural Polymorphisms in Escherichia coli O157 Genomes. Genome Res. 2009, 19, 1809–1816. [Google Scholar] [CrossRef] [Green Version]
- Braeye, T.; Denayer, S.; De Rauw, K.; Forier, A.; Verluyten, J.; Fourie, L.; Dierick, K.; Botteldoorn, N.; Quoilin, S.; Cosse, P.; et al. Lessons Learned from a Textbook Outbreak: EHEC-O157. Arch. Public Heal. 2014, 72, 44. [Google Scholar] [CrossRef] [Green Version]
- De Rauw, K.; Denayer, S.; Piérard, D. Rapid Molecular Typing of Shiga Toxin-Producing E.coli (STEC) O157:H7 during an Outbreak in Belgium by IS629-Printing. In Proceedings of the 10th International Meeting on Microbial Epidemiological Markers (IMMEM X), Paris, France, 2–5 October 2013. [Google Scholar]
- Butcher, H.; Elson, R.; Chattaway, M.A.; Featherstone, C.A.; Willis, C.; Jorgensen, F.; Dallman, T.J.; Jenkins, C.; McLauchlin, J.; Beck, C.R.; et al. Whole Genome Sequencing Improved Case Ascertainment in an Outbreak of Shiga Toxin-Producing Escherichia coli O157 Associated with Raw Drinking Milk. Epidemiol. Infect. 2016, 144, 2812–2823. [Google Scholar] [CrossRef] [Green Version]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-Time Whole-Genome Sequencing for Routine Typing, Surveillance, and Outbreak Detection of Verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef] [Green Version]
- Rumore, J.; Tschetter, L.; Kearney, A.; Kandar, R.; McCormick, R.; Walker, M.; Peterson, C.L.; Reimer, A.; Nadon, C. Evaluation of Whole-Genome Sequencing for Outbreak Detection of Verotoxigenic Escherichia coli O157:H7 from the Canadian Perspective. BMC Genom. 2018, 19, 870. [Google Scholar] [CrossRef]
- Brown, E.; Dessai, U.; Mcgarry, S.; Gerner-Smidt, P. Use of Whole-Genome Sequencing for Food Safety and Public Health in the United States. Foodborne Pathog. Dis. 2019, 16, 441–450. [Google Scholar] [CrossRef] [Green Version]
- Revez, J.; Espinosa, L.; Albiger, B.; Leitmeyer, K.C.; Struelens, M.J. Survey on the Use of Whole-Genome Sequencing for Infectious Diseases Surveillance: Rapid Expansion of European National Capacities, 2015–2016. Front. Public Health 2017, 5. [Google Scholar] [CrossRef] [Green Version]
- García Fierro, R.; Thomas-Lopez, D.; Deserio, D.; Liebana, E.; Rizzi, V.; Guerra, B. Outcome of EC/EFSA Questionnaire (2016) on Use of Whole Genome Sequencing (WGS) for Food- and Waterborne Pathogens Isolated from Animals, Food, Feed and Related Environmental Samples in EU/EFTA Countries. EFSA J. 2018, 15. [Google Scholar] [CrossRef]
- Apruzzese, I.; Song, E.; Bonah, E.; Sanidad, V.S.; Leekitcharoenphon, P.; Medardus, J.J.; Abdalla, N.; Hosseini, H.; Takeuchi, M. Investing in Food Safety for Developing Countries: Opportunities and Challenges in Applying Whole-Genome Sequencing for Food Safety Management. Foodborne Pathog. Dis. 2019, 16, 463–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Food and Agriculture Organization (FAO); World Health Organization (WHO). Applications of Whole Genome Sequencing in Food Safety Management; Food and Agriculture Organization: Rome, Italy, 2016. [Google Scholar]
- EFSA; ECDC. Technical Report on Multi-country Outbreak of Shiga Toxin-producing Escherichia coli Infection Associated with Haemolytic Uraemic Syndrome; European Food Safety Authority and European Centre for Disease Prevention and Control: Stockholm, Sweden, 2016; Volume 13. [Google Scholar] [CrossRef] [Green Version]
- FASFC. Advice 15-2012 of the Scientific Committee of the FASFC on the Prevention, Detection, Fast Tracing and Management of Outbreaks of Human Pathogenic Verotoxin Producing Escherichia coli in the Food Chain; Federal Agency for the Safety of the Food Chain: Brussels, Belgium, 2012; pp. 1–19. [Google Scholar]
- European Food Safety Authority (EFSA); European Center for Disease Prevention and Control (ECDC); Van Walle, I.; Guerra, B.; Borges, V.; André Carriço, J.; Cochrane, G.; Dallman, T.; Franz, E.; Karpíšková, R.; et al. EFSA and ECDC Technical Report on the Collection and Analysis of Whole Genome Sequencing Data from Food-Borne Pathogens and Other Relevant Microorganisms Isolated from Human, Animal, Food, Feed and Food/Feed Environmental Samples in the Joint ECDC-EFSA Mo; European Food Safety Authority: Parma, Italy, 2019; Volume 16. [Google Scholar] [CrossRef]
- De Rauw, K.; Crombé, F.; Piérard, D. National Reference Centre for Shiga Toxin/Verotoxin-Producing Escherichia coli (NRC-STEC/VTEC): Annual Report 2018; Laboratory of Microbiology and Infection Control: Brussels, Belgium, 2018. [Google Scholar]
- Denayer, S.; Verhaegen, B.; Van Hoorde, K. Voedselvergiftigingen in België: Jaaroverzicht 2019; Sciensano: Brussels, Belgium, 2019. [Google Scholar]
- International Organization for Standardization. ISO/TS 13136: 2012 Microbiology of Food and Animal Feed—Real-Time Polymerase Chain Reaction (PCR)-Based Method for the Detection of Food-Borne Pathogens—Horizontal Method for the Detection of Shiga Toxin-Producing Escherichia coli (STEC) and the Determin; International Organization for Standardization: Geneva, Switzerland, 2012. [Google Scholar]
- International Organization for Standardization. ISO 16654:2001 Microbiology of Food and Animal Feeding Stuffs—Horizontal Method for the Detection of Escherichia coli O157; International Organization for Standardization: Geneva, Switzerland, 2001. [Google Scholar]
- Buvens, G.; De Gheldre, Y.; Dediste, A.; De Moreau, A.I.; Mascart, G.; Simon, A.; Allemeersch, D.; Scheutz, F.; Lauwers, S.; Piérard, D. Incidence and Virulence Determinants of Verocytotoxin-Producing Escherichia coli Infections in the Brussels-Capital Region, Belgium, in 2008–2010. J. Clin. Microbiol. 2012, 50, 1336–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paton, A.W.; Paton, J.C. Detection and Characterization of Shiga Toxigenic Escherichia Coli by Using Multiplex PCR Assays for Stx1, Stx2, EaeA, Enterohemorrhagic E. coli HlyA, Rfb(O111), and Rfb(O157). J. Clin. Microbiol. 1998, 36, 598–602. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, H.; Scheef, J.; Morabito, S.; Caprioli, A.; Wieler, L.H.; Karch, H. A New Shiga Toxin 2 Variant (Stx2f) from Escherichia coli Isolated from Pigeons. Appl. Environ. Microbiol. 2000, 66, 1205–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piérard, D.; Van Etterijck, R.; Breynaert, J.; Moriau, L.; Lauwers, S. Results of Screening for Verocytotoxin-Producing Escherichia coli in Faeces in Belgium. Eur. J. Clin. Microbiol. Infect. Dis. 1990, 9, 198–201. [Google Scholar] [CrossRef]
- Gannon, V.P.J.; D’Souza, S.; Graham, T.; King, R.K.; Rahn, K.; Read, S. Use of the Flagellar H7 Gene as a Target in Multiplex PCR Assays and Improved Specificity in Identification of Enterohemorrhagic Escherichia coli Strains. J. Clin. Microbiol. 1997, 35, 656–662. [Google Scholar] [CrossRef] [Green Version]
- De Rauw, K.; Jacobs, S.; Piérard, D. Twenty-Seven Years of Screening for Shiga Toxin-Producing Escherichia coli in a University Hospital. Brussels, Belgium, 1987–2014. PLoS ONE 2018, 13, e0199968. [Google Scholar] [CrossRef]
- Scheutz, F.; Teel, L.D.; Beutin, L.; Piérard, D.; Buvens, G.; Karch, H.; Mellmann, A.; Caprioli, A.; Tozzoli, R.; Morabito, S.; et al. Multicenter Evaluation of a Sequence-Based Protocol for Subtyping Shiga Toxins and Standardizing Stx Nomenclature. J. Clin. Microbiol. 2012, 50, 2951–2963. [Google Scholar] [CrossRef] [Green Version]
- Boisen, N.; Scheutz, F.; Rasko, D.A.; Redman, J.C.; Persson, S.; Simon, J.; Kotloff, K.L.; Levine, M.M.; Sow, S.; Tamboura, B.; et al. Genomic Characterization of Enteroaggregative Escherichia coli from Children in Mali. J. Infect. Dis. 2012, 205, 431–444. [Google Scholar] [CrossRef]
- Tenover, F.C.; Arbeit, R.D.; Goering, R.V.; Mickelsen, P.A.; Murray, B.E.; Persing, D.H.; Swaminathan, B. Interpreting Chromosomal DNA Restriction Patterns Produced by Pulsed- Field Gel Electrophoresis: Criteria for Bacterial Strain Typing. J. Clin. Microbiol. 1995, 33, 2233–2239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, T.J.; Gerner-Smidt, P.; Swaminathan, B. Interpretation of Pulsed-Field Gel Electrophoresis Patterns in Foodborne Disease Investigations and Surveillance. Foodborne Pathog. Dis. 2006, 3, 20–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nouws, S.; Bogaerts, B.; Verhaegen, B.; Denayer, S.; Piérard, D.; Marchal, K.; Roosens, N.H.; Vanneste, K.; De Keersmaecker, S.C.J. Impact of DNA Extraction on Whole Genome Sequencing Analysis for Characterization and Relatedness of Shiga Toxin-Producing Escherichia coli Isolates. Sci. Rep. Under Review.
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.; Dvorkin, M.; Kulikov, A.; Lesin, V.; Nikolenko, S.; Pham, S.; Prjibelski, A.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inouye, M.; Dashnow, H.; Raven, L.A.; Schultz, M.B.; Pope, B.J.; Tomita, T.; Zobel, J.; Holt, K.E. SRST2: Rapid Genomic Surveillance for Public Health and Hospital Microbiology Labs. Genome Med. 2014, 6, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Zankari, E.; Hasman, H.; Vestergaard, M.; Rasmussen, S.; Cosentino, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of Acquired Antimicrobial Resistance Genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Kleinheinz, K.A.; Joensen, K.G.; Larsen, M.V. Applying the ResFinder and VirulenceFinder Web-Services for Easy Identification of Acquired Antibiotic Resistance and E. coli Virulence Genes in Bacteriophage and Prophage Nucleotide Sequences. Bacteriophage 2014, 4, e27943. [Google Scholar] [CrossRef] [Green Version]
- Carattoli, A.; Zankari, E.; Garciá-Fernández, A.; Larsen, M.V.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. In Silico Detection and Typing of Plasmids Using Plasmidfinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [Green Version]
- Joensen, K.G.; Tetzschner, A.M.M.; Iguchi, A.; Aarestrup, F.M.; Scheutz, F. Rapid and Easy in Silico Serotyping of Escherichia coli Isolates by Use of Whole-Genome Sequencing Data. J. Clin. Microbiol. 2015, 53, 2410–2426. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Godzik, A. Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Alikhan, N.; Mohamed, K.; The Agama Study Group; Achtman, M. The User’s Guide to Comparative Genomics with EnteroBase. Three Case Studies: Micro-Clades within Salmonella enterica Serovar Agama, Ancient and Modern Populations of Yersinia pestis, and Core Genomic Diversity of All Escherichia. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Alikhan, N.F.; Sergeant, M.J.; Luhmann, N.; Vaz, C.; Francisco, A.P.; Carriço, J.A.; Achtman, M. Grapetree: Visualization of Core Genomic Relationships among 100,000 Bacterial Pathogens. Genome Res. 2018, 28, 1395–1404. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A. FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 11 March 2020).
- NCBI SRA. Available online: https://www.ncbi.nlm.nih.gov/sra (accessed on 14 July 2020).
- Bogaerts, B.; Nouws, S.; Verhaegen, B.; Denayer, S.; Braekel, J.V.; Winand, R.; Fu, Q.; Piérard, D.; Marchal, K.; Roosens, N.H.C.; et al. Validation of a Bioinformatics Workflow for Characterization of Shiga Toxin-Producing Escherichia coli, Applied to a High-Quality Reference Collection, Demonstrates the Feasibility of Switching to Whole-Genome Sequencing for Routine Pathogen Typing. In Preparation.
- Piérard, D.; De Rauw, K. National Reference Centre for Shiga Toxin/Verotoxin-Producing Escherichia coli (STEC/VTEC) Annual Report 2015; Laboratory of Microbiology and Infection Control: Brussels, Belgium, 2015. [Google Scholar]
- Edgar, R.; Bibi, E. MdfA, an Escherichia coli Multidrug Resistance Protein with an Extraordinarily Broad Spectrum of Drug Recognition. J. Bacteriol. 1997, 179, 2274–2280. [Google Scholar] [CrossRef] [Green Version]
- Dallman, T.J.; Byrne, L.; Ashton, P.M.; Cowley, L.A.; Perry, N.T.; Adak, G.; Petrovska, L.; Ellis, R.J.; Elson, R.; Underwood, A.; et al. Whole-Genome Sequencing for National Surveillance of Shiga Toxin-Producing Escherichia coli O157. Clin. Infect. Dis. 2015, 61, 305–312. [Google Scholar] [CrossRef] [Green Version]
- Turabelidze, G.; Lawrence, S.J.; Gao, H.; Sodergren, E.; Weinstock, G.M.; Abubucker, S.; Wylie, T.; Mitreva, M.; Shaikh, N.; Gautom, R.; et al. Precise Dissection of an Escherichia coli O157:H7 Outbreak by Single Nucleotide Polymorphism Analysis. J. Clin. Microbiol. 2013, 51, 3950–3954. [Google Scholar] [CrossRef] [Green Version]
- Jackson, B.R.; Tarr, C.; Strain, E.; Jackson, K.A.; Conrad, A.; Carleton, H.; Katz, L.S.; Stroika, S.; Gould, L.H.; Mody, R.K.; et al. Implementation of Nationwide Real-Time Whole-Genome Sequencing to Enhance Listeriosis Outbreak Detection and Investigation. Clin. Infect. Dis. 2016, 63, 380–386. [Google Scholar] [CrossRef] [Green Version]
- Moura, A.; Criscuolo, A.; Pouseele, H.; Maury, M.M.; Leclercq, A.; Tarr, C.; Björkman, J.T.; Dallman, T.; Reimer, A.; Enouf, V.; et al. Whole Genome-Based Population Biology and Epidemiological Surveillance of Listeria monocytogenes. Nat. Microbiol. 2016, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Moore, G.; Cookson, B.; Gorden, N.C.; Jackson, R.; Kearns, A.; Singleton, J.; Smyth, D.; Wilson, A.P.R. Whole-Genome Sequencing in Hierarchy with Pulsed-Field Gel Electrophoresis: The Utility of This Approach to Establish Possible Sources of MRSA Cross-Transmission. J. Hosp. Infect. 2015, 90, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Allard, M.W.; Strain, E.; Melka, D.; Bunning, K.; Musser, S.M.; Brown, E.W.; Timme, R. Practical Value of Food Pathogen Traceability through Building a Whole-Genome Sequencing Network and Database. J. Clin. Microbiol. 2016, 54, 1975–1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greig, D.R.; Jenkins, C.; Gharbia, S.; Dallman, T.J. Comparison of Single-Nucleotide Variants Identified by Illumina and Oxford Nanopore Technologies in the Context of a Potential Outbreak of Shiga Toxin-Producing Escherichia coli. Gigascience 2019, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.D.; Knox, N.C.; Ronholm, J.; Pagotto, F.; Reimer, A. Metagenomics: The next Culture-Independent Game Changer. Front. Microbiol. 2017, 8, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Saltykova, A.; Buytaers, F.E.; Denayer, S.; Verhaegen, B.; Piérard, D.; Roosens, N.H.C.; Marchal, K.; De Keersmaecker, S.C.J. Strain-Level Metagenomic Data Analysis of Enriched in Vitro and in Silico Spiked Food Samples: Paving the Way towards Culture-Free Foodborne Outbreak Investigation Using STEC as a Case Study. In Preparation.
- DebRoy, C.; Fratamico, P.M.; Yan, X.; Baranzoni, G.M.; Liu, Y.; Needleman, D.S.; Tebbs, R.; O’Connell, C.D.; Allred, A.; Swimley, M.; et al. Comparison of O-Antigen Gene Clusters of All O-Serogroups of Escherichia coli and Proposal for Adopting a New Nomenclature for O-Typing. PLoS ONE 2016, 11, e0147434. [Google Scholar] [CrossRef] [Green Version]
- Koutsoumanis, K.; Allende, A.; Alvarez-Ordóñez, A.; Bover-Cid, S.; Chemaly, M.; Davies, R.; De Cesare, A.; Herman, L.; Hilbert, F.; Lindqvist, R.; et al. Pathogenicity Assessment of Shiga Toxin-Producing Escherichia coli (STEC) and the Public Health Risk Posed by Contamination of Food with STEC. EFSA J. 2020, 18, 1–105. [Google Scholar] [CrossRef]
- EFSA Panel on Biological Hazards (BIOHAZ). Scientific Opinion on VTEC-Seropathotype and Scientific Criteria Regarding Pathogenicity Assessment. EFSA J. 2013, 11, 106. [Google Scholar] [CrossRef]
- Melton-Celsa, A.R. Shiga Toxin (Stx) Classification, Structure, and Function. Microbiol. Spectr. 2014, 2, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Scheutz, F. Taxonomy Meets Public Health: The Case of Shiga Toxin-Producing Escherichia coli. Microbiol. Spectr. 2014, 2, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielaszewska, M.; Aldick, T.; Bauwens, A.; Karch, H. Hemolysin of Enterohemorrhagic Escherichia coli: Structure, Transport, Biological Activity and Putative Role in Virulence. Int. J. Med. Microbiol. 2014, 304, 521–529. [Google Scholar] [CrossRef]
- Gould, L.H.; Bopp, C.; Strockbine, N.; Atkinson, R.; Baselski, V.; Body, B.; Carey, R.; Crandall, C.; Hurd, S.; Kaplan, R.; et al. Update: Recommendations for Diagnosis of Shiga Toxin-Producing Escherichia coli Infections by Clinical Laboratories. Clin. Microbiol. Newsl. 2012, 34, 75–83. [Google Scholar] [CrossRef]
- Gyles, C.L. Shiga Toxin-Producing Escherichia coli: An Overview. J. Anim. Sci. 2007, 85, E45–E62. [Google Scholar] [CrossRef] [PubMed]
- Bettelheim, K.A. The Non-O157 Shiga-Toxigenic (Verocytotoxigenic) Escherichia coli; under-Rated Pathogens. Crit. Rev. Microbiol. 2007, 33, 67–87. [Google Scholar] [CrossRef] [PubMed]
- Gould, L.H.; Mody, R.K.; Ong, K.L.; Clogher, P.; Cronquist, A.B.; Garman, K.N.; Lathrop, S.; Medus, C.; Spina, N.L.; Webb, T.H.; et al. Increased Recognition of Non-O157 Shiga Toxin-Producing Escherichia coli Infections in the United States during 2000–2010: Epidemiologic Features and Comparison with E. Coli O157 Infections. Foodborne Pathog. Dis. 2013, 10, 453–460. [Google Scholar] [CrossRef]
- Lathrop, S.; Edge, K.; Bareta, J. Shiga Toxin-Producing Escherichia coli, New Mexico, USA, 2004–2007. Emerg. Infect. Dis. 2009, 15, 1289–1291. [Google Scholar] [CrossRef]
- Tseng, M.; Sha, Q.; Rudrik, J.T.; Collins, J.; Henderson, T.; Funk, J.A.; Manning, S.D. Increasing Incidence of Non-O157 Shiga Toxin-Producing Escherichia coli (STEC) in Michigan and Association with Clinical Illness. Epidemiol. Infect. 2016, 144, 1394–1405. [Google Scholar] [CrossRef] [Green Version]
- Agger, M.; Scheutz, F.; Villumsen, S.; Mølbak, K.; Petersen, A.M. Antibiotic Treatment of Verocytotoxin-Producing Escherichia coli (VTEC) Infection: A Systematic Review and a Proposal. J. Antimicrob. Chemother. 2015, 70, 2440–2446. [Google Scholar] [CrossRef] [Green Version]
- Freedman, S.B.; Xie, J.; Neufeld, M.S.; Hamilton, W.L.; Hartling, L.; Tarr, P.I. Shiga Toxin-Producing Escherichia coli Infection, Antibiotics, and Risk of Developing Hemolytic Uremic Syndrome: A Meta-Analysis. Clin. Infect. Dis. 2016, 62, 1251–1258. [Google Scholar] [CrossRef] [Green Version]
- Holmes, A.; Dallman, T.J.; Shabaan, S.; Hanson, M.; Allison, L. Validation of Whole-Genome Sequencing for Identification and Characterization of Shiga Toxin-Producing Escherichia coli to Produce Standardized Data to Enable Data Sharing. J. Clin. Microbiol. 2018, 56, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentle, A.; Jenkins, C.; Day, M.; Greig, D.; Painset, A.; Do Nascimento, V. Antimicrobial Resistance in Non-O157 Shiga Toxin-Producing E. coli. Access Microbiol. 2019. [Google Scholar] [CrossRef]
- Lindsey, R.L.; Pouseele, H.; Chen, J.C.; Strockbine, N.A.; Carleton, H.A. Implementation of Whole Genome Sequencing (WGS) for Identification and Characterization of Shiga Toxin-Producing Escherichia coli (STEC) in the United States. Front. Microbiol. 2016, 7, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossen, J.W.A.; Friedrich, A.W.; Moran-Gilad, J. Practical Issues in Implementing Whole-Genome-Sequencing in Routine Diagnostic Microbiology. Clin. Microbiol. Infect. 2018, 24, 355–360. [Google Scholar] [CrossRef] [Green Version]
- Pasquali, F.; Do Valle, I.; Palma, F.; Remondini, D.; Manfreda, G.; Castellani, G.; Hendriksen, R.S.; De Cesare, A. Application of Different DNA Extraction Procedures, Library Preparation Protocols and Sequencing Platforms: Impact on Sequencing Results. Heliyon 2019, 5, e02745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, L.; Steglich, M.; Fuchs, S.; Werner, G.; Nübel, U. Comparison of Six Commercial Kits to Extract Bacterial Chromosome and Plasmid DNA for MiSeq Sequencing. Sci. Rep. 2016, 6, 10–14. [Google Scholar] [CrossRef]
- Grützke, J.; Malorny, B.; Hammerl, J.A.; Busch, A.; Tausch, S.H.; Tomaso, H.; Deneke, C. Fishing in the Soup—Pathogen Detection in Food Safety Using Metabarcoding and Metagenomic Sequencing. Front. Microbiol. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Seth-Smith, H.M.B.; Bonfiglio, F.; Cuénod, A.; Reist, J.; Egli, A.; Wüthrich, D. Evaluation of Rapid Library Preparation Protocols for Whole Genome Sequencing Based Outbreak Investigation. Front. Public Heal. 2019, 7, 241. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.F.; Rankin, S.C.; Schurer, J.M.; Cole, S.; Conti, L.; Rabinowitz, P.; Gray, G.; Kahn, L.; Machalaba, C.; Mazet, J.; et al. Checklist for One Health Epidemiological Reporting of Evidence (COHERE). One Health 2017, 4, 14–21. [Google Scholar] [CrossRef] [Green Version]
- European Center for Disease Prevention and Control; Struelens, M.; Albiger, B.; Catchpole, M.; Ciancio, B.; Coulombier, D.; Espinosa, L.; Johansson, K.; Ködmön, C.; Palm, D.; et al. Expert Opinion on Whole Genome Sequencing for Public Health Surveillance; European Center for Disease Prevention and Control: Stockholm, Sweden, 2016. [Google Scholar] [CrossRef]
- Timme, R.E.; Leon, M.S.; Allard, M.W. Utilizing the Public GenomeTrakr Database for Foodborne Pathogen Traceback. In Methods in Molecular Biology; Springer: Berlin, Germany, 2019; Volume 1918, pp. 201–212. [Google Scholar] [CrossRef]
- Wielinga, P.R.; Hendriksen, R.S.; Aarestrup, F.M.; Lund, O.; Smits, S.L.; Koopmans, M.P.G.; Schlundt, J. Global Microbial Identifier. Appl. Genom. Foodborne Pathog. 2017. [Google Scholar] [CrossRef]
- Boqvist, S.; Söderqvist, K.; Vågsholm, I. Food Safety Challenges and One Health within Europe. Acta Vet. Scand. 2018, 60, 1–13. [Google Scholar] [CrossRef]
- Carroll, L.M.; Wiedmann, M.; Mukherjee, M.; Nicholas, D.C.; Mingle, L.A.; Dumas, N.B.; Cole, J.A.; Kovac, J. Characterization of Emetic and Diarrheal Bacillus Cereus Strains from a 2016 Foodborne Outbreak Using Whole-Genome Sequencing: Addressing the Microbiological, Epidemiological, and Bioinformatic Challenges. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrario, C.; Lugli, G.A.; Ossiprandi, M.C.; Turroni, F.; Milani, C.; Duranti, S.; Mancabelli, L.; Mangifesta, M.; Alessandri, G.; van Sinderen, D.; et al. Next Generation Sequencing-Based Multigene Panel for High Throughput Detection of Food-Borne Pathogens. Int. J. Food Microbiol. 2017, 256, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Mukhopadhyay, K.; Thomassin, P.J. An Economic Analysis of Salmonella Detection in Fresh Produce, Poultry, and Eggs Using Whole Genome Sequencing Technology in Canada. Food Res. Int. 2019, 116, 802–809. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Isolate Reference | Isolation Date (dd/mm/yyyy) | Origin | Conventional Methods | WGS | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Serotype | Virulence Genes | AMR | Relation to Outbreak | Serotype | Virulence Genotype | AMR Genotype | Relation to Outbreak | ||||||||||||||||||||||||||||||
| stx1 | stx2 | eae | ehxA | aggR | aaiC | stx1 | stx2 | eae | ehxA | aggR | aaiC | katP | toxB | astA | esp | gad | nle | tir | iss | etpD | iha | tccP | blaTEM-1B | aph(3’)-Ia | str | sul2 | tetA | dfrA8 | mdfA | ||||||||
| EH2165 | 06/06/2012 | Human | O157:H7 | a | a | + | + | - | - | S | O | O157:H7 | a | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O |
| EH2169 | 07/06/2012 | Human | O157:H7 | a | a | + | + | - | - | S | O | O157:H7 | a | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O |
| EH2170 | 12/06/2012 | Human | O157:H7 | a | a | + | + | - | - | S | O | O157:H7 | a | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O |
| EH2171 | 13/06/2012 | Human | O157:H7 | a | a | + | + | - | - | S | O | O157:H7 | a | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O |
| EH2174 | 12/06/2012 | Human | O157:H7 | a | a | + | + | - | - | S | O | O157:H7 | a | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O |
| EH2176 | 08/06/2012 | Human | O157:H7 | a | a | + | + | - | - | S | O | O157:H7 | a | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O |
| EH2177 | 08/06/2012 | Human | O157:H7 | a | a | + | + | - | - | S | O | O157:H7 | a | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O |
| EH2178 | 13/06/2012 | Human | O157:H7 | a | a | + | + | - | - | S | O | O157:H7 | a | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O |
| EH2179 | 14/06/2012 | Human | O157:H7 | a | a | + | + | - | - | S | O | O157:H7 | a | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O |
| EH2180 | 14/06/2012 | Human | O157:H7 | a | a | + | + | - | - | S | O | O157:H7 | a | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O |
| EH2181 | 14/06/2012 | Human | O157:H7 | a | a | + | + | - | - | S | O | O157:H7 | a | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O |
| EH2182 | 14/06/2012 | Human | O157:H7 | a | a | + | + | - | - | S | O | O157:H7 | a | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O |
| EH2183 | 19/06/2012 | Human | O157:H7 | a | a | + | + | - | - | S | O | O157:H7 | a | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O |
| EH2189 | 22/06/2012 | Human | O157:H7 | a | a | + | + | - | - | S | O | O157:H7 | a | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O |
| EH2192 | 26/06/2012 | Human | O157:H7 | a | a | + | + | - | - | S | O | O157:H7 | a | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O |
| EH2193 | 26/06/2012 | Human | O157:H7 | a | a | + | + | - | - | S | O | O157:H7 | a | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O |
| EH2194 | 02/07/2012 | Human | O157:H7 | a | a | + | + | - | - | S | O | O157:H7 | a | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O |
| TIAC1150 | 27/06/2012 | Filet americain | O157:H7 | a * | a * | + | + * | - | - | S * | O | O157:H7 | a | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O |
| TIAC1151 | 19/06/2012 | Filet americain | O157:H7 | a * | a * | + | + * | - | - | S * | O | O157:H7 | a | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O |
| TIAC1152 | 19/06/2012 | Filet americain | O157:H7 | a * | a * | + | + * | - | - | S * | O | O157:H7 | a | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O |
| TIAC1153 | 18/06/2012 | Carcass swab | O157:H7 | a * | c * | + | + * | - | - | AMP, KAN, STR, SUL, SXT, TET * | N | O157:H7 | a | c | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | + | + | A, B | + | + | + | + | N |
| Isolate Reference | Isolation Date (dd/mm/yyyy) | Origin | Conventional Methods | WGS | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Serotype | Virulence Genes | AMR | Relation to Outbreak | Serotype | Virulence Genotype | AMR Genotype | Relation to Outbreak | ||||||||||||||||||||||||||||||
| stx1 | stx2 | eae | ehxA | aggR | aaiC | stx1 | stx2 | eae | ehxA | aggR | aaiC | katP | toxB | astA | esp | gad | nle | tir | iss | etpD | iha | tccP | blaTEM-1B | aadA1 | str | tetA | dfrA1 | sul | mdfA | ||||||||
| EH2264 | 15/05/2013 | Human | O157:H7 | - | a | + | + | - | - | S | N | O157:H7 | - | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | N |
| EH2268 | 28/05/2013 | Human | O157:H7 | - | a | + | + | - | - | S | N | O157:H7 | - | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | N |
| EH2272 | 08/06/2013 | Human | O157:H7 | - | a | + | + | - | - | S | ? | O157:H7 | - | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O * |
| EH2274 | 13/06/2013 | Human | O157:H7 | - | a | + | + | - | - | S | O* | O157:H7 | - | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O * |
| EH2275 | 13/06/2013 | Human | O157:H7 | - | a | + | + | - | - | S | O * | O157:H7 | - | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O * |
| EH2277 | 18/06/2013 | Human | O157:H7 | - | a | + | + | - | - | S | O * | O157:H7 | - | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O * |
| EH2278 | 18/06/2013 | Human | O157:H7 | - | a | + | + | - | - | S | O * | O157:H7 | - | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O * |
| EH2279 | 18/06/2013 | Human | O157:H7 | - | a | + | + | - | - | S | O * | O157:H7 | - | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O * |
| EH2280 | 18/06/2013 | Human | O157:H7 | - | a | + | + | - | - | S | O * | O157:H7 | - | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O * |
| EH2281 | 20/06/2013 | Human | O157:H7 | - | a | + | + | - | - | S | O * | O157:H7 | - | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O * |
| EH2285 | 28/06/2013 | Human | O157:H7 | - | a | + | + | - | - | AMP, STR, TET, SXT | O * | O157:H7 | - | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | + | + | A, B | + | + | 1, 2 | + | O * |
| EH2295 | 12/07/2013 | Human | O157:H7 | - | a | + | + | - | - | S | N | O157:H7 | - | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | N |
| EH2297 | 16/07/2013 | Human | O157:H7 | - | a | + | + | - | - | S | O | O157:H7 | - | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O |
| EH2299 | 18/07/2013 | Human | O157:H7 | - | a | + | + | - | - | S | ? | O157:H7 | - | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | N |
| EH2308 | 31/07/2013 | Human | O157:H7 | a | a | + | + | - | - | S | N | O157:H7 | a | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | N |
| TIAC1903 | 01/07/2013 | Filet americain | O157:H7 | - | a * | + | + * | - * | - * | S * | O | O157:H7 | - | a | + | + | - | - | + | + | + | A, B, F, J, P | + | A, B, C | + | + | + | + | + | - | - | - | - | - | - | + | O |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nouws, S.; Bogaerts, B.; Verhaegen, B.; Denayer, S.; Crombé, F.; De Rauw, K.; Piérard, D.; Marchal, K.; Vanneste, K.; Roosens, N.H.C.; et al. The Benefits of Whole Genome Sequencing for Foodborne Outbreak Investigation from the Perspective of a National Reference Laboratory in a Smaller Country. Foods 2020, 9, 1030. https://doi.org/10.3390/foods9081030
Nouws S, Bogaerts B, Verhaegen B, Denayer S, Crombé F, De Rauw K, Piérard D, Marchal K, Vanneste K, Roosens NHC, et al. The Benefits of Whole Genome Sequencing for Foodborne Outbreak Investigation from the Perspective of a National Reference Laboratory in a Smaller Country. Foods. 2020; 9(8):1030. https://doi.org/10.3390/foods9081030
Chicago/Turabian StyleNouws, Stéphanie, Bert Bogaerts, Bavo Verhaegen, Sarah Denayer, Florence Crombé, Klara De Rauw, Denis Piérard, Kathleen Marchal, Kevin Vanneste, Nancy H. C. Roosens, and et al. 2020. "The Benefits of Whole Genome Sequencing for Foodborne Outbreak Investigation from the Perspective of a National Reference Laboratory in a Smaller Country" Foods 9, no. 8: 1030. https://doi.org/10.3390/foods9081030
APA StyleNouws, S., Bogaerts, B., Verhaegen, B., Denayer, S., Crombé, F., De Rauw, K., Piérard, D., Marchal, K., Vanneste, K., Roosens, N. H. C., & De Keersmaecker, S. C. J. (2020). The Benefits of Whole Genome Sequencing for Foodborne Outbreak Investigation from the Perspective of a National Reference Laboratory in a Smaller Country. Foods, 9(8), 1030. https://doi.org/10.3390/foods9081030