QSAR Prediction Model to Search for Compounds with Selective Cytotoxicity Against Oral Cell Cancer




Abstract
:1. Introduction
2. Materials and Methods
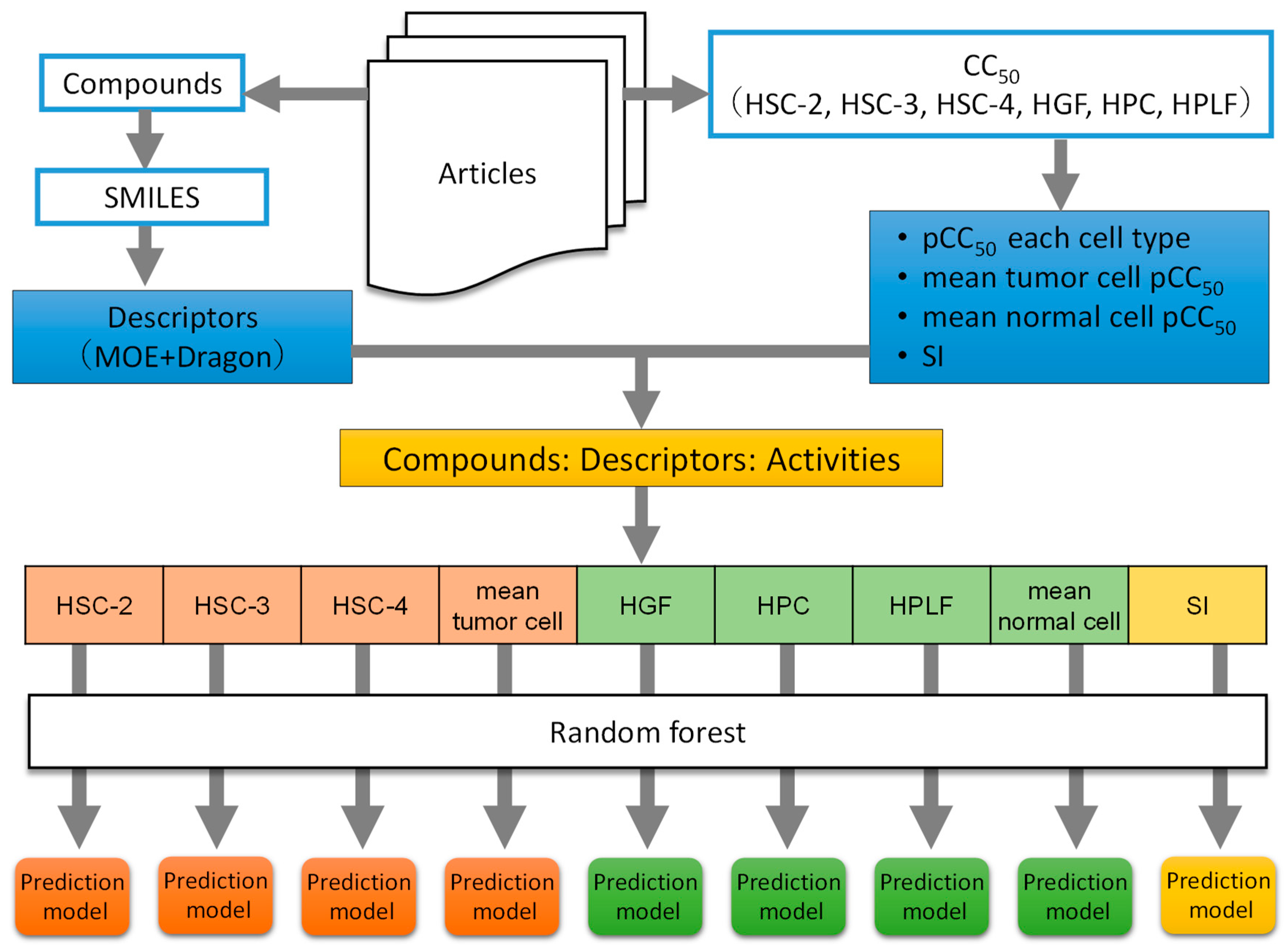
2.1. Data Collection and Preparation
2.2. Chemical Structure Data Acquisition and Descriptor Calculation
2.3. Preparation of Data Table
2.4. Construction of Prediction Models by RF
3. Results
3.1. Data Collection
3.2. Construction of Prediction Models by RF
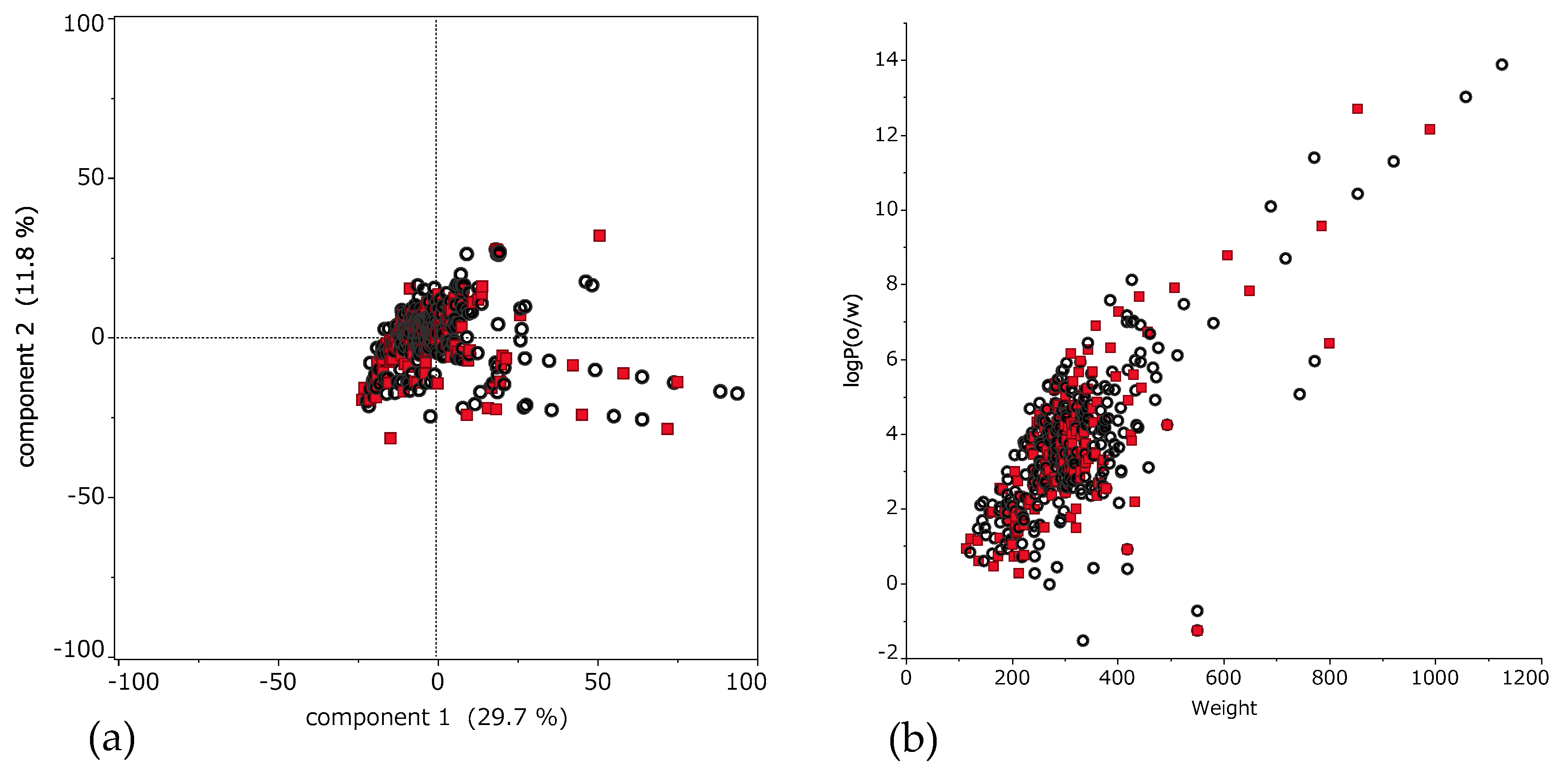
3.3. Large Contribution Descriptor for Prediction Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HGF | Human gingival fibroblast |
| HPC | Human pulp cell |
| HPLF | Human periodontal fibroblast |
| Log P | Octanol-water partitioning coefficient |
| MOE | Molecular Operating Environment |
| OM | Oral mucositis |
| OOB | Out-of-bag |
| OSCC | Oral squamous cell carcinoma |
| pCC50 | −logCC50, a negative common logarithm |
| QASR | Quantitative structure-activity relationship |
| R2 | Coefficient of determination |
| RF | Random forest |
| RMSE | Root-mean-square error |
| SAR | Structure-activity relationship |
References
- Sonis, S.T. Mucositis: The impact, biology and therapeutic opportunities of oral mucositis. Oral Oncol. 2009, 45, 1015–1020. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, F.; Yoshida, A.; Nakajima, A.; Wada-Takahashi, S.; Takahashi, S.S.; Lee, M.C. Alteration of the redox state with reactive oxygen species for 5-fluorouracil-induced oral mucositis in hamsters. PLoS ONE 2013, 8, e82834. [Google Scholar] [CrossRef] [PubMed]
- Sugita, Y.; Takao, K.; Uesawa, Y.; Sakagami, H. Search for New Type of Anticancer Drugs with High Tumor Specificity and Less Keratinocyte Toxicity. Anticancer Res. 2017, 37, 5919–5924. [Google Scholar] [PubMed]
- Sakagami, H.; Okudaira, N.; Masuda, Y.; Amano, O.; Yokose, S.; Kanda, Y.; Suguro, M.; Natori, T.; Oizumi, H.; Oizumi, T. Induction of apoptosis in human oral keratinocyte by doxorubicin. Anticancer Res. 2017, 37, 1023–1029. [Google Scholar]
- SciFinder®. Available online: https://www.cas.org/products/scifinder (accessed on 5 February 2019).
- Wolfgang, S.; Dina, R. QSAR/QSPR. In Applied Chemoinformatics; Thomas, E., Johann, G., Eds.; Wiley-VCH: Weinheim, Germany; Berlin, Germany, 2018; pp. 9–13. [Google Scholar]
- Guohui, S.; Tengjiao, F.; Xiaodong, S.; Yuxing, H.; Xin, C.; Lijiao, Z.; Ting, R.; Yue, Z.; Rugang, Z.; Yongzhen, P. In Silico Prediction of O6-Methylguanine-DNA Methyltransferase Inhibitory Potency of Base Analogs with QSAR and Machine Learning Methods. Molecules 2018, 23, 2892. [Google Scholar]
- Tengjiao, F.; Guohui, S.; Lijiao, Z.; Xin, C.; Rugang, Z. QSAR and Classification Study on Prediction of Acute Oral Toxicity of N-Nitroso Compounds. Int. J. Mol. Sci. 2018, 19, 3015. [Google Scholar]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random forest: A classification and regression tool for compound classification and QSAR modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar] [CrossRef]
- Shirataki, Y.; Wakae, M.; Yamamoto, Y.; Hashimoto, K.; Satoh, K.; Ishihara, M.; Kikuchi, H.; Nishikawa, H.; Minagawa, K.; Motohashi, N.; et al. Cytotoxicity and radical modulating activity of isoflavones and isoflavanones from sophora species. Anticancer Res. 2004, 24, 1481–1488. [Google Scholar]
- Ishihara, M.; Sakagami, H. Re-evaluation of cytotoxicity and iron chelation activity of three β-diketones by semiempirical molecular orbital method. In Vivo 2005, 19, 119–124. [Google Scholar]
- Momoi, K.; Sugita, Y.; Ishihara, M.; Satoh, K.; Kikuchi, H.; Hashimoto, K.; Yokoe, I.; Nishikawa, H.; Fujisawa, S.; Sakagami, H. Cytotoxic activity of styrylchromones against human tumor cell lines. In Vivo 2005, 19, 157–164. [Google Scholar] [PubMed]
- Sakagami, H.; Ishihara, M.; Hoshino, Y.; Ishikawa, J.; Mikami, Y.; Fukai, T. Cytotoxicity of nocobactins NA-a, NA-b and their ferric complexes assessed by semiempirical molecular orbital method. In Vivo 2005, 19, 277–282. [Google Scholar] [PubMed]
- Ishihara, M.; Sakagami, H.; Liu, W.K. Quantitative structure-cytotoxicity relationship analysis of betulinic acid and its derivatives by semi-empirical molecular-orbital method. Anticancer Res. 2005, 25, 3951–3956. [Google Scholar] [PubMed]
- Inoue, K.; Kulsum, U.; Chowdhury, SA.; Fujisawa, S.; Ishihara, M.; Yokoe, I.; Sakagami, H. Tumor-specific cytotoxicity and apoptosis-inducing activity of berberines. Anticancer Res. 2005, 25, 4053–4060. [Google Scholar] [PubMed]
- Ishihara, M.; Yokote, Y.; Sakagami, H. Quantitative structure-cytotoxicity relationship analysis of coumarin and its derivatives by semiempirical molecular orbital method. Anticancer Res. 2006, 26, 2883–2886. [Google Scholar] [PubMed]
- Sasaki, M.; Okamura, M.; Ideo, A.; Shimada, J.; Suzuki, F.; Ishihara, M.; Kikuchi, H.; Kanda, Y.; Kunii, S.; Sakagami, H. Re-evaluation of tumor-specific cytotoxicity of mitomycin c, bleomycin and peplomycin. Anticancer Res. 2006, 26, 3373–3380. [Google Scholar]
- Ishihara, M.; Kawase, M.; Sakagami, H. Quantitative structure-activity relationship analysis of 4-trifluoromethylimidazole derivatives with the concept of absolute hardness. Anticancer Res. 2007, 27, 4047–4052. [Google Scholar]
- Ishihara, M.; Kawase, M.; Westman, G.; Samuelsson, K.; Motohashi, N.; Sakagami, H. Quantitative structure-cytotoxicity relationship analysis of phenoxazine derivatives by semiempirical molecular-orbital method. Anticancer Res. 2007, 27, 4053–4058. [Google Scholar] [PubMed]
- Ishihara, M.; Sakagami, H. QSAR of molecular structure and cytotoxic activity of vitamin K2 derivatives with concept of absolute hardness. Anticancer Res. 2007, 27, 4059–4064. [Google Scholar]
- Takekawa, F.; Nagumo, T.; Shintani, S.; Hashimoto, K.; Kikuchi, H.; Katayama, T.; Ishihara, M.; Amano, O.; Kawase, M.; Sakagami, H. Tumor-specific cytotoxic activity and type of cell death induced by 4-trifluoromethylimidazoles in human oral squamous cell carcinoma cell lines. Anticancer Res. 2007, 27, 4065–4070. [Google Scholar]
- Suzuki, F.; Hashimoto, K.; Ishihara, M.; Westman, G.; Samuelsson, K.; Kawase, M.; Motohashi, N.; Sakagami, H. Tumor-specificity and type of cell death induced by phenoxazines. Anticancer Res. 2007, 27, 4233–4238. [Google Scholar] [PubMed]
- Sakagami, H.; Hashimoto, K.; Suzuki, F.; Ishihara, M.; Kikuchi, H.; Katayama, T.; Satoh, K. Tumor-specificity and type of cell death induced by vitamin K2 derivatives and prenylalcohols. Anticancer Res. 2008, 28, 151–158. [Google Scholar] [PubMed]
- Ishihara, M.; Sakagami, H. Quantitative structure-cytotoxicity relationship analysis of 3-formylchromone derivatives by a semiempirical molecularorbital method with the concept of absolute hardness. Anticancer Res. 2008, 28, 277–282. [Google Scholar]
- Ishihara, M.; Kawase, M.; Sakagami, H. Quantitative structure-cytotoxicity relationship analysis of 5-trifluoromethyloxazole derivatives by a semiempirical molecular-orbital method with the concept of absolute hardness. Anticancer Res. 2008, 28, 997–1004. [Google Scholar]
- Ishihara, M.; Hatano, H.; Kawase, M.; Sakagami, H. Estimation of relationship between the structure of 1, 2, 3, 4-tetrahydroisoquinoline derivatives determined by a semiempirical molecular-orbital method and their cytotoxicity. Anticancer Res. 2009, 29, 2265–2272. [Google Scholar]
- Hatano, H.; Takekawa, F.; Hashimoto, K.; Ishihara, M.; Kawase, M.; Qing, C.; Qin-Tao, W.; Sakagami, H. Tumor-specific cytotoxic activity of 1, 2, 3, 4-tetrahydroisoquinoline derivatives against human oral squamous cell carcinoma cell lines. Anticancer Res. 2009, 29, 3079–3086. [Google Scholar]
- Takekawa, F.; Sakagami, H.; Ishihara, M. Estimation of relationship between structure of newly synthesized dihydroimidazoles determined by a semiempirical molecular-orbital method and their cytotoxicity. Anticancer Res. 2009, 29, 5019–5022. [Google Scholar] [PubMed]
- Ishihara, M.; Wakabayashi, H.; Motohashi, N.; Sakagami, H. Quantitative structure-cytotoxicity relationship of newly synthesized tropolones determined by a semiempirical molecular-orbital method (PM5). Anticancer Res. 2010, 30, 129–134. [Google Scholar]
- Ishihara, M.; Wakabayashi, H.; Motohashi, N.; Sakagami, H. Quantitative structure–cytotoxicity relationship of newly synthesized trihaloacetylazulenes determined by a semi-empirical molecular-orbital method (PM5). Anticancer Res. 2011, 31, 515–520. [Google Scholar]
- Ishihara, M.; Wakabayashi, H.; Motohashi, N.; Sakagami, H. Estimation of relationship between the structure of trihaloacetylazulene derivatives determined by a semiempirical molecular–orbital method (PM5) and their cytotoxicity. Anticancer Res. 2010, 30, 837–842. [Google Scholar] [PubMed]
- Ohno, H.; Araho, D.; Uesawa, Y.; Kagaya, H.; Ishihara, M.; Sakagami, H.; Yamamoto, M. Evaluation of cytotoxicity and tumor-specificity of licorice flavonoids based on chemical structure. Anticancer Res. 2013, 33, 3061–3068. [Google Scholar]
- Uesawa, Y.; Mohri, K.; Kawase, M.; Ishihara, M.; Sakagami, H. Quantitative structure–activity relationship (QSAR) analysis of tumor-specificity of 1, 2, 3, 4-tetrahydroisoquinoline derivatives. Anticancer Res. 2011, 31, 4231–4238. [Google Scholar] [PubMed]
- Sekine, S.; Shimodaira, C.; Uesawa, Y.; Kagaya, H.; Kanda, Y.; Ishihara, M.; Amano, O.; Sakagami, H.; Wakabayashi, H. Quantitative structure–activity relationship analysis of cytotoxicity and anti-uv activity of 2-aminotropones. Anticancer Res. 2014, 34, 1743–1750. [Google Scholar]
- Shimada, C.; Uesawa, Y.; Ishihara, M.; Kagaya, H.; Kanamoto, T.; Terakubo, S.; Nakashima, H.; Takao, K.; Saito, T.; Sugita, Y.; et al. Quantitative structure–cytotoxicity relationship of phenylpropanoid amides. Anticancer Res. 2014, 34, 3543–3548. [Google Scholar]
- Shimada, C.; Uesawa, Y.; Ishihara, M.; Kagaya, H.; Kanamoto, T.; Terakubo, S.; Nakashima, H.; Takao, K.; Miyashiro, T.; Sugita, Y.; et al. Quantitative structure–cytotoxicity relationship of piperic acid amides. Anticancer Res. 2014, 34, 4877–4884. [Google Scholar]
- Shimada, C.; Uesawa, Y.; Ishii-Nozawa, R.; Ishihara, M.; Kagaya, H.; Kanamoto, T.; Terakubo, S.; Nakashima, H.; Takao, K.; Sugita, Y.; et al. Quantitative structure–cytotoxicity relationship of 3-styrylchromones. Anticancer Res. 2014, 34, 5405–5412. [Google Scholar]
- Uesawa, Y.; Sakagami, H.; Ishihara, M.; Kagaya, H.; Kanamoto, T.; Terakubo, S.; Nakashima, H.; Yahagi, H.; Takao, K.; Sugita, Y. Quantitative structure–cytotoxicity relationship of 3-styryl-2H-chromenes. Anticancer Res. 2015, 35, 5299–5308. [Google Scholar] [PubMed]
- Sakagami, H.; Uesawa, Y.; Ishihara, M.; Kagaya, H.; Kanamoto, T.; Terakubo, S.; Nakashima, H.; Takao, K.; Sugita, Y. Quantitative structure–cytotoxicity relationship of oleoylamides. Anticancer Res. 2015, 35, 5341–5355. [Google Scholar] [PubMed]
- Uesawa, Y.; Sakagami, H.; Kagaya, H.; Yamashita, M.; Takao, K.; Sugita, Y. Quantitative structure-cytotoxicity relationship of 3-benzylidenechromanones. Anticancer Res. 2016, 36, 5803–5812. [Google Scholar] [CrossRef]
- Fukuchi, K.; Okudaira, N.; Adachi, K.; Odai-Ide, R.; Watanabe, S.; Ohno, H.; Yamamoto, M.; Kanamoto, T.; Terakubo, S.; Nakashima, H.; et al. Antiviral and antitumor activity of licorice root extracts. In Vivo 2016, 30, 777–786. [Google Scholar] [CrossRef]
- Sakagami, H.; Masuda, Y.; Tomomura, M.; Yokose, S.; Uesawa, Y.; Ikezoe, N.; Asahara, D.; Takao, K.; Kanamoto, T.; Terakubo, S.; et al. Quantitative structure–cytotoxicity relationship of chalcones. Anticancer Res. 2017, 37, 1091–1098. [Google Scholar]
- Sakagami, H.; Uesawa, Y.; Masuda, Y.; Tomomura, M.; Yokose, S.; Miyashiro, T.; Murai, J.; Takao, K.; Kanamoto, T.; Terakubo, S.; et al. Quantitative structure–cytotoxicity relationship of newly synthesized piperic acid esters. Anticancer Res. 2017, 37, 6161–6168. [Google Scholar]
- Uesawa, Y.; Sakagami, H.; Ikezoe, N.; Takao, K.; Kagaya, H.; Sugita, Y. Quantitative structure–cytotoxicity relationship of aurones. Anticancer Res. 2017, 37, 6169–6176. [Google Scholar]
- Sakagami, H.; Okudaira, N.; Uesawa, Y.; Takao, K.; Kagaya, H.; Sugita, Y. Quantitative structure–cytotoxicity relationship of 2-azolylchromones. Anticancer Res. 2018, 38, 763–770. [Google Scholar]
- Uesawa, Y.; Sakagami, H.; Okudaira, N.; Toda, K.; Takao, K.; Kagaya, H.; Sugita, Y. Quantitative structure–cytotoxicity relationship of cinnamic acid phenetyl esters. Anticancer Res. 2018, 38, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Maruyama, R.; Irie, Y.; Hashimoto, M.; Wakabayashi, H.; Okudaira, N.; Uesawa, Y.; Kagaya, H.; Sakagami, H. In vitro anti-tumor activity of azulene amide derivatives. In Vivo 2018, 32, 479–486. [Google Scholar] [PubMed]
- Uehara, M.; Minemura, H.; Ohno, T.; Hashimoto, M.; Wakabayashi, H.; Okudaira, N.; Sakagami, H. In vitro antitumor activity of alkylaminoguaiazulenes. In Vivo 2018, 32, 541–547. [Google Scholar] [PubMed]
- Marvin. Available online: https://chemaxon.com/products/marvin (accessed on 15 November 2018).
- MOE. Available online: http://www.chemcomp.com/MOE-Molecular_Operating_Environment.htm (accessed on 15 November 2018).
- Dragon. Available online: https://chm.kode-solutions.net/products_dragon.php (accessed on 15 November 2018).
- Paola, G. Principles of QSAR models validation: Internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar]
- SAS Institute Inc. Chapter 6 Bootstrap Forest. In JMP® 13 Predictive and Specialized Modeling, 2nd ed.; SAS Institute Inc.: Cary, NC, USA, 2017; pp. 107–122. [Google Scholar]
- JMP®. Available online: https://www.jmp.com/en_us/home.html (accessed on 15 November 2018).
- Wolfgang, S.; Dina, R. Applicability domain and model acceptability critera. In Applied Chemoinformatics; Thomas, E., Johann, G., Eds.; Wiley-VCH: Weinheim, Germany; Berlin, Germany, 2018; pp. 41–43. [Google Scholar]
- SAS Institute Inc. Chapter 5 Partition Models. In JMP® 13 Predictive and Specialized Modeling, 2nd ed.; SAS Institute Inc.: Cary, NC, USA, 2017; pp. 104–106. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Search Terms | Number of Total Reports (A) | Number of Our Reports (B) | % (B/A) × 100 |
|---|---|---|---|
| OSCC | 8951 (100) | 141 | 1.6 |
| OSCC + Anticancer Drug | 335 (3.70) | 60 | 17.9 |
| OSCC + Anticancer Drug + Tumour-Specificity | 50 (0.56) | 40 | 80.0 |
| OSCC + Anticancer Drug + Tumour-Specificity + Newly Synthesized | 2 (0.02) | 2 | 100.0 |
| OSCC + Anticancer Drug + Keratinocyte Toxicity | 5 (0.06) | 4 | 80.0 |
| OSCC + anticancer drug + QSAR | 27 (0.30) | 25 | 92.6 |
| OSCC + Anticancer Drug + QSAR+ Newly Synthesized | 3 (0.03) | 3 | 100.0 |
| No. | Number of Compounds | Basic Skeleton | Ref. |
|---|---|---|---|
| 1 | 9 | Isoflavones and Isoflavanones | [9] |
| 2 | 3 | Three β-Diketones | [10] |
| 3 | 6 | Styrylchromones | [11] |
| 4 | 3 | Nocobactins NA-a, NA-b and Their Ferric Complexes | [12] |
| 5 | 5 | Betulinic Acid and Its Derivatives | [13] |
| 6 | 2 | Berberines | [14] |
| 7 | 20 | Coumarin and Its Derivatives | [15] |
| 8 | 1 | Mitomycin C, Bleomycin and Peplomycin | [16] |
| 9 | 13 | 4-Trifluoromethylimidazole Derivatives | [17] |
| 10 | 15 | Phenoxazine Derivatives | [18] |
| 11 | 7 | Vitamin K2 Derivatives | [19] |
| 12 | 2 | 4-Trifluoromethylimidazoles | [20] |
| 13 | 10 | Phenoxazines | [21] |
| 14 | 18 | Vitamin K2 Derivatives and Prenylalcohols | [22] |
| 15 | 10 | 3-Formylchromone Derivatives | [23] |
| 16 | 12 | 5-Trifluoromethyloxazole Derivatives | [24] |
| 17 | 19 | 1,2,3,4-Tetrahydroisoquinoline Derivatives | [25] |
| 18 | 19 | 1,2,3,4Tetrahydroisoquinoline Derivatives | [26] |
| 19 | 12 | Dihydroimidazoles | [27] |
| 20 | 24 | Tropolones | [28] |
| 21 | 24 | Trihaloacetylazulenes | [29] |
| 22 | 22 | Trihaloacetylazulene Derivatives | [30] |
| 23 | 10 | Licorice Flavonoids | [31] |
| 24 | 4 | 1,2,3,4-Tetrahydroisoquinoline Derivatives | [32] |
| 25 | 19 | 2-Aminotropones | [33] |
| 26 | 12 | Phenylpropanoid Amides | [34] |
| 27 | 12 | Piperic Acid Amides | [35] |
| 28 | 15 | 3-Styrylchromones | [36] |
| 29 | 16 | 3-Styryl-2H-chromenes | [37] |
| 30 | 18 | Oleoylamides | [38] |
| 31 | 17 | 3-Benzylidenechromanones | [39] |
| 32 | 18 | Licorice Root Extracts | [40] |
| 33 | 15 | Chalcones | [41] |
| 34 | 11 | Piperic Acid Esters | [42] |
| 35 | 17 | Aurones | [43] |
| 36 | 24 | 2-Azolylchromones | [44] |
| 37 | 10 | Cinnamic Acid Phenetyl Esters | [45] |
| 38 | 10 | Azulene Amide Derivatives | [46] |
| 39 | 10 | Alkylaminoguaiazulenes | [47] |
| Parameters | Tumour Cells | Normal Cells | SI | ||||||
|---|---|---|---|---|---|---|---|---|---|
| HSC-2 | HSC-3 | HSC-4 | Mean | HGF | HPC | HPLF | Mean | ||
| Number of Tree | 100 | 300 | 100 | 100 | 100 | 100 | 100 | 100 | 300 |
| Number of Term | 952 | 1000 | 952 | 952 | 952 | 952 | 952 | 952 | 1000 |
| Number of Maximum Split at Tree | 100 | 1000 | 2000 | 2000 | 2000 | 2000 | 2000 | 2000 | 2000 |
| Minimum Node Size | 3 | 5 | 5 | 5 | 5 | 5 | 3 | 5 | 5 |
| Seed Value | 29 | 36 | 44 | 77 | 93 | 91 | 730 | 9045 | 124 |
| Number of Tree | 23 | 8 | 21 | 20 | 9 | 4 | 34 | 12 | 8 |
| Number of Term at a Split | 1000 | 1000 | 952 | 952 | 952 | 952 | 952 | 952 | 1000 |
| R2(Training Set) | 0.904 | 0.847 | 0.868 | 0.876 | 0.862 | 0.815 | 0.908 | 0.858 | 0.817 |
| R2(External Validation Set) | 0.564 | 0.568 | 0.631 | 0.563 | 0.554 | 0.659 | 0.515 | 0.576 | 0.404 |
| RMSE (External Validation Set) | 0.480 | 0.496 | 0.496 | 0.473 | 0.435 | 0.372 | 0.442 | 0.397 | 0.340 |
| OOB RMSE | 0.808 | 0.778 | 0.742 | 0.760 | 0.593 | 0.587 | 0.618 | 0.573 | 0.579 |
| Maximum Absolute Value of the Residue | 2.052 | 1.875 | 1.424 | 1.408 | 1.347 | 1.758 | 1.331 | 1.582 | 1.188 |
| Mean Absolute Error | 0.236 | 0.255 | 0.232 | 0.216 | 0.216 | 0.199 | 0.240 | 0.198 | 0.191 |
| Cell Type | Descriptor | Meaning | |
|---|---|---|---|
| Tumour Cells | HSC-2 | vsurf_D7 | Lipophilicity |
| vsurf_D2 | Lipophilicity | ||
| GCUT_SMR_0 | Topological shape | ||
| CATS2D_07_LL | Lipophilicity | ||
| SpMin2_Bh(e) | Topological shape and electric state | ||
| HSC-3 | SssNH | Topological shape and electric state | |
| b_max1len | Topological shape | ||
| Mor13s | 3D shape and electric state | ||
| Mor15s | 3D shape and electric state | ||
| F01[C-C] | Topological shape | ||
| HSC-4 | SpMax_L | Topological shape | |
| SpAD_EA(dm) | Topological shape and dipole moment | ||
| ATSC2s | Topological shape and electric state | ||
| vsurf_D7 | Lipophilicity | ||
| ATSC5s | Topological shape and electric state | ||
| Mean | logP(o/w) | Lipophilicity | |
| vsurf_D2 | Lipophilicity | ||
| vsurf_D6 | Lipophilicity | ||
| P_VSA_ppp_L | Topological shape and lipophilicity | ||
| SssNH | Topological shape and electric state | ||
| Normal Cells | HGF | GCUT_SLOGP_0 | Topological shape |
| F10[C-N] | Topological shape | ||
| SssNH | Topological shape and electric state | ||
| SpMin2_Bh(s) | Topological shape | ||
| GCUT_SMR_0 | Topological shape | ||
| HPC | VE1_B(p) | Topological shape and polarizability | |
| b_max1len | Topological shape | ||
| CATS3D_10_PL | 3D shape and electric state | ||
| h_pKb | Topological shape and electric state | ||
| SpMin1_Bh(p) | Topological shape and polarizability | ||
| HPLF | P_VSA_e_3 | Topological shape and electric state | |
| GCUT_SLOGP_0 | Topological shape | ||
| F10[C-N] | Topological shape | ||
| SssNH | Topological shape and electric state | ||
| h_pavgQ | Topological shape and electric state | ||
| Mean | h_pstrain | Topological shape and electric state | |
| h_pavgQ | Topological shape and electric state | ||
| b_max1len | Topological shape | ||
| SssNH | Topological shape and electric state | ||
| F10[C-N] | Topological shape | ||
| SI | TDB08p | 3D shape and polarizability | |
| F06[C-N] | Topological shape | ||
| PEOE_VSA+1 | Topological shape and electric state | ||
| R5u+ | 3D shape and size | ||
| RDF035m | 3D shape and size | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagai, J.; Imamura, M.; Sakagami, H.; Uesawa, Y. QSAR Prediction Model to Search for Compounds with Selective Cytotoxicity Against Oral Cell Cancer. Medicines 2019, 6, 45. https://doi.org/10.3390/medicines6020045
Nagai J, Imamura M, Sakagami H, Uesawa Y. QSAR Prediction Model to Search for Compounds with Selective Cytotoxicity Against Oral Cell Cancer. Medicines. 2019; 6(2):45. https://doi.org/10.3390/medicines6020045
Chicago/Turabian StyleNagai, Junko, Mai Imamura, Hiroshi Sakagami, and Yoshihiro Uesawa. 2019. "QSAR Prediction Model to Search for Compounds with Selective Cytotoxicity Against Oral Cell Cancer" Medicines 6, no. 2: 45. https://doi.org/10.3390/medicines6020045
APA StyleNagai, J., Imamura, M., Sakagami, H., & Uesawa, Y. (2019). QSAR Prediction Model to Search for Compounds with Selective Cytotoxicity Against Oral Cell Cancer. Medicines, 6(2), 45. https://doi.org/10.3390/medicines6020045