


One Billion hiPSC-Cardiomyocytes: Upscaling Engineered Cardiac Tissues to Create High Cell Density Therapies for Clinical Translation in Heart Regeneration


 ,
,  , , , ,
, , , ,  ,
,
Abstract
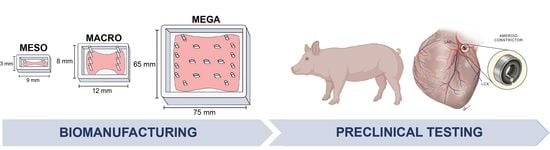
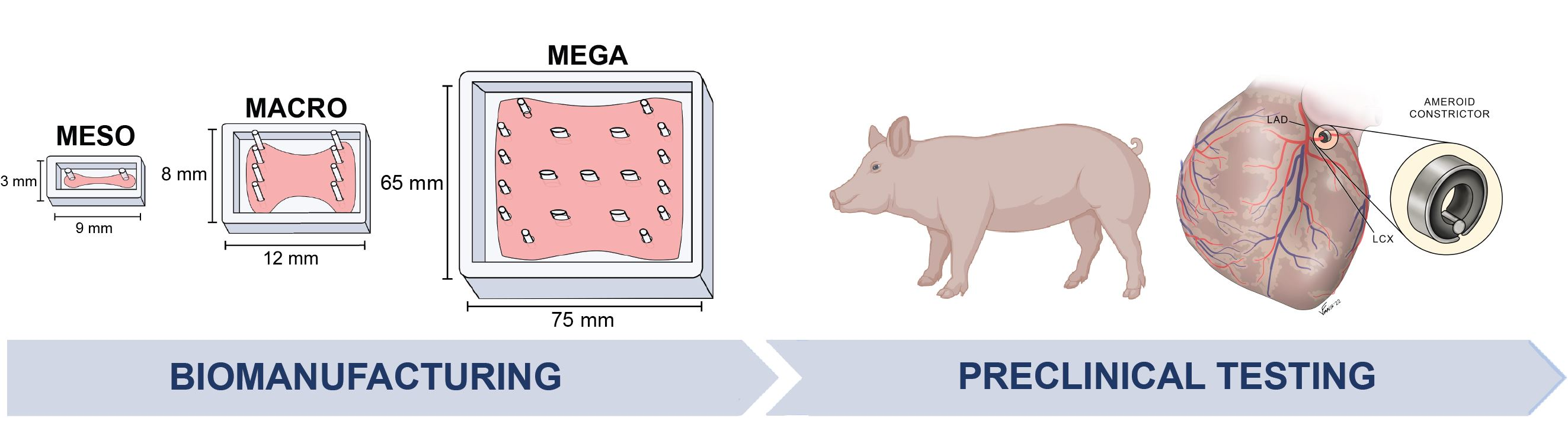
:
1. Introduction
2. Materials and Methods
2.1. HiPSC Maintenance
2.2. HiPSC-CM Differentiation, Freezing/Thawing, Expansion and Selection
2.3. Cardiac Fibroblast Maintenance
2.4. Molding System and Modification for Scale Up
2.5. Fabrication of Engineered Cardiac Tissue (ECTs)
2.6. ECT Survival, Compaction and Pacing Analysis during Culture
2.7. Mechanical Analysis
2.8. Curling Angle and Prestrain Measurement
2.9. Immunohistochemical Staining, Imaging and Analysis of ECTs
2.10. Optical Mapping of Calcium and Voltage Transients
2.11. Animal Methods
2.12. Statistical Analysis
3. Results
3.1. Cell Density Impacts Meso-ECT Syncytium Formation, Structural Integrity and Collagen Remodeling
3.2. Physiological Analysis Reveals Cell-Dose Dependent Effects on Meso-ECT Contractile Stress and Sarcomere Structure
3.3. Increasing the Surface Area of ECTs Reveals Scale Impacts Formation and Cell Density Impacts Excitability without Inducing Arrhythmias
3.4. Clinical Scale Mega-ECT Enables Delivery of 1 Billion hiPSC-CMs to the Chronically Ischemic Swine Heart
4. Discussion
4.1. Challenge #1: Structural Integrity for Surgical Handling of Cell-Dense ECTs
4.2. Challenge 2: Maturation State of PSC-CMs
4.3. Challenge 3: Delivery of PSC-CMs
4.4. Challenge 4: Treatment of Comorbidities
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Laflamme, M.A.; Murry, C.E. Regenerating the heart. Nat. Biotechnol. 2005, 23, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Eschenhagen, T.; Bolli, R.; Braun, T.; Field, L.J.; Fleischmann, B.K.; Frisén, J.; Giacca, M.; Hare, J.M.; Houser, S.; Lee, R.T.; et al. Cardiomyocyte Regeneration. Circulation 2017, 136, 680–686. [Google Scholar] [CrossRef]
- Hsieh, P.C.; Segers, V.F.; Davis, M.E.; MacGillivray, C.; Gannon, J.; Molkentin, J.D.; Robbins, J.; Lee, R.T. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat. Med. 2007, 13, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Rao, G.; Sheth, S.; Grines, C. Percutaneous coronary intervention: 2017 in review. J. Interv. Cardiol. 2018, 31, 117–128. [Google Scholar] [CrossRef]
- Namdar, P.; YekeFallah, L.; Jalalian, F.; Barikani, A.; Razaghpoor, A. Improving Door-to-Balloon Time for Patients With Acute ST-Elevation Myocardial Infarction: A Controlled Clinical Trial. Curr. Probl. Cardiol. 2021, 46, 100674. [Google Scholar] [CrossRef]
- Gu, Y.L.; van der Horst, I.C.C.; Douglas, Y.L.; Svilaas, T.; Mariani, M.A.; Zijlstra, F. Role of coronary artery bypass grafting during the acute and subacute phase of ST-elevation myocardial infarction. Neth. Heart J. 2010, 18, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Jenča, D.; Melenovský, V.; Stehlik, J.; Staněk, V.; Kettner, J.; Kautzner, J.; Adámková, V.; Wohlfahrt, P. Heart failure after myocardial infarction: Incidence and predictors. ESC Heart Fail. 2021, 8, 222–237. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.; Teng, T.H.K.; Finn, J.; Knuiman, M.; Briffa, T.; Stewart, S.; Sanfilippo, F.M.; Ridout, S.; Hobbs, M. Trends From 1996 to 2007 in Incidence and Mortality Outcomes of Heart Failure After Acute Myocardial Infarction: A Population-Based Study of 20 812 Patients With First Acute Myocardial Infarction in W estern A ustralia. J. Am. Heart Assoc. 2013, 2, e000172. [Google Scholar] [CrossRef]
- Sulo, G.; Igland, J.; Vollset, S.E.; Nygård, O.; Ebbing, M.; Sulo, E.; Egeland, G.M.; Tell, G.S. Heart failure complicating acute myocardial infarction; burden and timing of occurrence: A nation-wide analysis including 86,771 patients from the Cardiovascular Disease in Norway (CVDNOR) Project. J. Am. Heart Assoc. 2016, 5, e002667. [Google Scholar] [CrossRef]
- Stone, G.W.; Selker, H.P.; Thiele, H.; Patel, M.R.; Udelson, J.E.; Ohman, E.M.; Maehara, A.; Eitel, I.; Granger, C.B.; Jenkins, P.L.; et al. Relationship Between Infarct Size and Outcomes Following Primary PCI: Patient-Level Analysis From 10 Randomized Trials. J. Am. Coll. Cardiol. 2016, 67, 1674–1683. [Google Scholar] [CrossRef]
- Adler, E.D.; Goldfinger, J.Z.; Kalman, J.; Park, M.E.; Meier, D.E. Palliative Care in the Treatment of Advanced Heart Failure. Circulation 2009, 120, 2597–2606. [Google Scholar] [CrossRef]
- Querdel, E.; Reinsch, M.; Castro, L.; Köse, D.; Bähr, A.; Reich, S.; Geertz, B.; Ulmer, B.; Schulze, M.; Lemoine, M.D.; et al. Human Engineered Heart Tissue Patches Remuscularize the Injured Heart in a Dose-Dependent Manner. Circulation 2021, 143, 1991–2006. [Google Scholar] [CrossRef]
- Eschenhagen, T.; Ridders, K.; Weinberger, F. How to repair a broken heart with pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 2022, 163, 106–117. [Google Scholar] [CrossRef]
- Lian, X.; Zhang, J.; Azarin, S.M.; Zhu, K.; Hazeltine, L.B.; Bao, X.; Hsiao, C.; Kamp, T.J.; Palecek, S.P. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat. Protoc. 2013, 8, 162–175. [Google Scholar] [CrossRef]
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M.; et al. Chemically defined generation of human cardiomyocytes. Nat. Methods 2014, 11, 855–860. [Google Scholar] [CrossRef]
- Rupert, C.E.; Irofuala, C.; Coulombe, K.L.K. Practical adoption of state-of-the-art hiPSC-cardiomyocyte differentiation techniques. PLoS ONE 2020, 15, e0230001. [Google Scholar] [CrossRef]
- Buikema, J.W.; Lee, S.; Goodyer, W.R.; Maas, R.G.; Chirikian, O.; Li, G.; Miao, Y.; Paige, S.L.; Lee, D.; Wu, H.; et al. Wnt Activation and Reduced Cell-Cell Contact Synergistically Induce Massive Expansion of Functional Human iPSC-Derived Cardiomyocytes. Cell Stem Cell 2020, 27, 50–63.e5. [Google Scholar] [CrossRef]
- Tohyama, S.; Hattori, F.; Sano, M.; Hishiki, T.; Nagahata, Y.; Matsuura, T.; Hashimoto, H.; Suzuki, T.; Yamashita, H.; Satoh, Y.; et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 2013, 12, 127–137. [Google Scholar] [CrossRef]
- Rupert, C.E.; Kim, T.Y.; Choi, B.-R.; Coulombe, K.L.K. Human Cardiac Fibroblast Number and Activation State Modulate Electromechanical Function of hiPSC-Cardiomyocytes in Engineered Myocardium. Stem Cells Int. 2020, 2020, 9363809. [Google Scholar] [CrossRef]
- Munarin, F.; Kaiser, N.J.; Kim, T.Y.; Choi, B.R.; Coulombe, K.L.K. Laser-Etched Designs for Molding Hydrogel-Based Engineered Tissues. Tissue Eng. Part C Methods 2017, 23, 311–321. [Google Scholar] [CrossRef]
- Kaiser, N.J.; Bellows, J.A.; Kant, R.J.; Coulombe, K.L.K. Digital Design and Automated Fabrication of Bespoke Collagen Microfiber Scaffolds. Tissue Eng. Part C Methods 2019, 25, 687–700. [Google Scholar] [CrossRef]
- Feyen, D.A.M.; McKeithan, W.L.; Bruyneel, A.A.N.; Spiering, S.; Hörmann, L.; Ulmer, B.; Zhang, H.; Briganti, F.; Schweizer, M.; Hegyi, B.; et al. Metabolic Maturation Media Improve Physiological Function of Human iPSC-Derived Cardiomyocytes. Cell Rep. 2020, 32, 107925. [Google Scholar] [CrossRef]
- Kaiser, N.J.; Kant, R.J.; Minor, A.J.; Coulombe, K.L.K. Optimizing Blended Collagen-Fibrin Hydrogels for Cardiac Tissue Engineering with Human iPSC-derived Cardiomyocytes. ACS Biomater. Sci. Eng. 2019, 5, 887–899. [Google Scholar] [CrossRef]
- Sala, L.; Meer, B.J.v.; Tertoolen, L.G.J.; Bakkers, J.; Bellin, M.; Davis, R.P.; Denning, C.; Dieben, M.A.E.; Eschenhagen, T.; Giacomelli, E.; et al. MUSCLEMOTION. Circ. Res. 2018, 122, e5–e16. [Google Scholar] [CrossRef]
- Shi, X.; Liu, Y.; Copeland, K.M.; McMahan, S.R.; Zhang, S.; Butler, J.R.; Hong, Y.; Cho, M.; Bajona, P.; Gao, H.; et al. Epicardial prestrained confinement and residual stresses: A newly observed heart ventricle confinement interface. J. R. Soc. Interface 2019, 16, 20190028. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.Y.; Kunitomo, Y.; Pfeiffer, Z.; Patel, D.; Hwang, J.; Harrison, K.; Patel, B.; Jeng, P.; Ziv, O.; Lu, Y. Complex excitation dynamics underlie polymorphic ventricular tachycardia in a transgenic rabbit model of long QT syndrome type 1. Heart Rhythm 2015, 12, 220–228. [Google Scholar] [CrossRef]
- Soepriatna, A.H.; Navarrete-Welton, A.; Kim, T.Y.; Daley, M.C.; Bronk, P.; Kofron, C.M.; Mende, U.; Coulombe, K.L.K.; Choi, B.-R. Action potential metrics and automated data analysis pipeline for cardiotoxicity testing using optically mapped hiPSC-derived 3D cardiac microtissues. PLoS ONE 2023, 18, e0280406. [Google Scholar] [CrossRef]
- Kofron, C.M.; Choi, B.-R.; Coulombe, K.L.K. Arrhythmia Assessment in Heterotypic Human Cardiac Myocyte–Fibroblast Microtissues. In Cardiac Tissue Engineering: Methods and Protocols; Coulombe, K.L.K., Black Iii, L.D., Eds.; Springer: New York, NY, USA, 2022; pp. 147–157. [Google Scholar] [CrossRef]
- Soepriatna, A.H.; Kim, T.Y.; Daley, M.C.; Song, E.; Choi, B.-R.; Coulombe, K.L.K. Human Atrial Cardiac Microtissues for Chamber-Specific Arrhythmic Risk Assessment. Cell. Mol. Bioeng. 2021, 14, 441–457. [Google Scholar] [CrossRef]
- Sabe, S.A.; Xu, C.M.; Sabra, M.; Harris, D.D.; Malhotra, A.; Aboulgheit, A.; Stanley, M.; Abid, M.R.; Sellke, F.W. Canagliflozin Improves Myocardial Perfusion, Fibrosis, and Function in a Swine Model of Chronic Myocardial Ischemia. J. Am. Heart Assoc. 2023, 12, e028623. [Google Scholar] [CrossRef]
- Sabe, S.A.; Xu, C.M.; Potz, B.A.; Malhotra, A.; Sabra, M.; Harris, D.D.; Broadwin, M.; Abid, M.R.; Sellke, F.W. Comparative Analysis of Normoxia- and Hypoxia-Modified Extracellular Vesicle Therapy in Function, Perfusion, and Collateralization in Chronically Ischemic Myocardium. Int. J. Mol. Sci. 2023, 24, 2076. [Google Scholar] [CrossRef]
- Aboulgheit, A.; Karbasiafshar, C.; Sabra, M.; Zhang, Z.; Sodha, N.; Abid, M.R.; Sellke, F.W. Extracellular vesicles improve diastolic function and substructure in normal and high-fat diet models of chronic myocardial ischemia. J. Thorac. Cardiovasc. Surg. 2022, 164, e371–e384. [Google Scholar] [CrossRef] [PubMed]
- Sabe, S.A.; Scrimgeour, L.A.; Xu, C.M.; Sabra, M.; Karbasiafshar, C.; Aboulgheit, A.; Abid, M.R.; Sellke, F.W. Extracellular vesicle therapy attenuates antiangiogenic signaling in ischemic myocardium of swine with metabolic syndrome. J. Thorac. Cardiovasc. Surg. 2022, in press. [CrossRef] [PubMed]
- Potz, B.A.; Sabe, A.A.; Sabe, S.A.; Lawandy, I.J.; Abid, M.R.; Clements, R.T.; Sellke, F.W. Calpain inhibition decreases myocardial fibrosis in chronically ischemic hypercholesterolemic swine. J. Thorac. Cardiovasc. Surg. 2022, 163, e11–e27. [Google Scholar] [CrossRef] [PubMed]
- Aboulgheit, A.; Potz, B.A.; Scrimgeour, L.A.; Karbasiafshar, C.; Shi, G.; Zhang, Z.; Machan, J.T.; Schorl, C.; Brodsky, A.S.; Braga, K.; et al. Effects of High Fat Versus Normal Diet on Extracellular Vesicle–Induced Angiogenesis in a Swine Model of Chronic Myocardial Ischemia. J. Am. Heart Assoc. 2021, 10, e017437. [Google Scholar] [CrossRef] [PubMed]
- Scrimgeour, L.A.; Potz, B.A.; Aboul Gheit, A.; Liu, Y.; Shi, G.; Pfeiffer, M.; Colantuono, B.J.; Sodha, N.R.; Abid, M.R.; Sellke, F.W. Intravenous injection of extracellular vesicles to treat chronic myocardial ischemia. PLoS ONE 2020, 15, e0238879. [Google Scholar] [CrossRef]
- Scrimgeour, L.A.; Potz, B.A.; Aboul Gheit, A.; Shi, G.; Stanley, M.; Zhang, Z.; Sodha, N.R.; Ahsan, N.; Abid, M.R.; Sellke, F.W. Extracellular Vesicles Promote Arteriogenesis in Chronically Ischemic Myocardium in the Setting of Metabolic Syndrome. J. Am. Heart Assoc. 2019, 8, e012617. [Google Scholar] [CrossRef]
- Potz, B.A.; Scrimgeour, L.A.; Pavlov, V.I.; Sodha, N.R.; Abid, M.R.; Sellke, F.W. Extracellular Vesicle Injection Improves Myocardial Function and Increases Angiogenesis in a Swine Model of Chronic Ischemia. J. Am. Heart Assoc. 2018, 7, e008344. [Google Scholar] [CrossRef]
- Potz, B.A.; Sabe, A.A.; Elmadhun, N.Y.; Sabe, S.A.; Braun, B.J.V.; Clements, R.T.; Usheva, A.; Sellke, F.W. Calpain inhibition decreases inflammatory protein expression in vessel walls in a model of chronic myocardial ischemia. Surgery 2017, 161, 1394–1404. [Google Scholar] [CrossRef]
- Sabe, A.A.; Potz, B.A.; Elmadhun, N.Y.; Liu, Y.; Feng, J.; Abid, M.R.; Abbott, J.D.; Senger, D.R.; Sellke, F.W. Calpain inhibition improves collateral-dependent perfusion in a hypercholesterolemic swine model of chronic myocardial ischemia. J. Thorac. Cardiovasc. Surg. 2016, 151, 245–252. [Google Scholar] [CrossRef]
- Potz, B.A.; Sabe, A.A.; Elmadhun, N.Y.; Feng, J.; Liu, Y.; Mitchell, H.; Quesenberry, P.; Abid, M.R.; Sellke, F.W. Calpain inhibition decreases myocardial apoptosis in a swine model of chronic myocardial ischemia. Surgery 2015, 158, 445–452. [Google Scholar] [CrossRef]
- Harada, K.; Grossman, W.; Friedman, M.; Edelman, E.R.; Prasad, P.V.; Keighley, C.S.; Manning, W.J.; Sellke, F.W.; Simons, M. Basic fibroblast growth factor improves myocardial function in chronically ischemic porcine hearts. J. Clin. Investig. 1994, 94, 623–630. [Google Scholar] [CrossRef]
- Ruel, M.; Wu, G.F.; Khan, T.A.; Voisine, P.; Bianchi, C.; Li, J.; Li, J.; Laham, R.J.; Sellke, F.W. Inhibition of the Cardiac Angiogenic Response to Surgical FGF-2 Therapy in a Swine Endothelial Dysfunction Model. Circulation 2003, 108, II-335–II-340. [Google Scholar] [CrossRef] [PubMed]
- Voisine, P.; Bianchi, C.; Ruel, M.; Malik, T.; Rosinberg, A.; Feng, J.; Khan, T.A.; Xu, S.H.; Sandmeyer, J.; Laham, R.J.; et al. Inhibition of the cardiac angiogenic response to exogenous vascular endothelial growth factor. Surgery 2004, 136, 407–415. [Google Scholar] [CrossRef]
- Wulkan, F.; Romagnuolo, R.; Qiang, B.; Laflamme, M.A. Methods for Transepicardial Cell Transplantation in a Swine Myocardial Infarction Model. In Cardiac Tissue Engineering: Methods and Protocols; Humana: New York, NY, USA, 2022; pp. 191–212. [Google Scholar]
- Romagnuolo, R.; Masoudpour, H.; Porta-Sánchez, A.; Qiang, B.; Barry, J.; Laskary, A.; Qi, X.; Massé, S.; Magtibay, K.; Kawajiri, H.; et al. Human Embryonic Stem Cell-Derived Cardiomyocytes Regenerate the Infarcted Pig Heart but Induce Ventricular Tachyarrhythmias. Stem Cell Rep. 2019, 12, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, J.-D.; Khan, A.; Markus, A.; Mohamed, B.A.; Toischer, K.; Alves, F.; Salditt, T. X-ray diffraction and second harmonic imaging reveal new insights into structural alterations caused by pressure-overload in murine hearts. Sci. Rep. 2020, 10, 19317. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef]
- Saraste, A.; Pulkki, K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc. Res. 2000, 45, 528–537. [Google Scholar] [CrossRef]
- Liu, Y.W.; Chen, B.; Yang, X.; Fugate, J.A.; Kalucki, F.A.; Futakuchi-Tsuchida, A.; Couture, L.; Vogel, K.W.; Astley, C.A.; Baldessari, A.; et al. Human embryonic stem cell-derived cardiomyocytes restore function in infarcted hearts of non-human primates. Nat. Biotechnol. 2018, 36, 597–605. [Google Scholar] [CrossRef]
- Shiba, Y.; Gomibuchi, T.; Seto, T.; Wada, Y.; Ichimura, H.; Tanaka, Y.; Ogasawara, T.; Okada, K.; Shiba, N.; Sakamoto, K.; et al. Allogeneic transplantation of iPS cell-derived cardiomyocytes regenerates primate hearts. Nature 2016, 538, 388–391. [Google Scholar] [CrossRef]
- Kant, R.J.; Coulombe, K.L.K. Patterned Arteriole-Scale Vessels Enhance Engraftment, Perfusion, and Maturation of Engineered Human Myocardium for Heart Regeneration. Ph.D. Dissertation, Brown University, Providence, RI, USA, 2022. [Google Scholar]
- Biagi, D.; Fantozzi, E.T.; Campos-Oliveira, J.C.; Naghetini, M.V.; Ribeiro, A.F.; Rodrigues, S.; Ogusuku, I.; Vanderlinde, R.; Christie, M.L.A.; Mello, D.B.; et al. In Situ Maturated Early-Stage Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes Improve Cardiac Function by Enhancing Segmental Contraction in Infarcted Rats. J. Pers. Med. 2021, 11, 374. [Google Scholar] [CrossRef]
- Funakoshi, S.; Fernandes, I.; Mastikhina, O.; Wilkinson, D.; Tran, T.; Dhahri, W.; Mazine, A.; Yang, D.; Burnett, B.; Lee, J.; et al. Generation of mature compact ventricular cardiomyocytes from human pluripotent stem cells. Nat. Commun. 2021, 12, 3155. [Google Scholar] [CrossRef] [PubMed]
- Riegler, J.; Tiburcy, M.; Ebert, A.; Tzatzalos, E.; Raaz, U.; Abilez, O.J.; Shen, Q.; Kooreman, N.G.; Neofytou, E.; Chen, V.C.; et al. Human Engineered Heart Muscles Engraft and Survive Long Term in a Rodent Myocardial Infarction Model. Circ. Res. 2015, 117, 720–730. [Google Scholar] [CrossRef]
- Lorente, M.; Escalona, C.; Zabalza-Cerdeiriña, M.; Álvarez-Moro, J. Left ventricle morphometry in healthy humans. Long axis, contrast enhanced CT study. Sci. Med. Data 2017, 10. [Google Scholar] [CrossRef]
- akshayd21. NIH 3D—Laboratory Rocker/Shaker/Mixer. 2023. Available online: https://3d.nih.gov/entries/3DPX-009288 (accessed on 1 April 2022).
- Gao, L.; Gregorich, Z.R.; Zhu, W.; Mattapally, S.; Oduk, Y.; Lou, X.; Kannappan, R.; Borovjagin, A.V.; Walcott, G.P.; Pollard, A.E.; et al. Large Cardiac Muscle Patches Engineered From Human Induced-Pluripotent Stem Cell–Derived Cardiac Cells Improve Recovery From Myocardial Infarction in Swine. Circulation 2018, 137, 1712–1730. [Google Scholar] [CrossRef]
- Serpooshan, V.; Chen, P.; Wu, H.; Lee, S.; Sharma, A.; Hu, D.A.; Venkatraman, S.; Ganesan, A.V.; Usta, O.B.; Yarmush, M.; et al. Bioacoustic-enabled patterning of human iPSC-derived cardiomyocytes into 3D cardiac tissue. Biomaterials 2017, 131, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Munarin, F.; Kant, R.J.; Rupert, C.E.; Khoo, A.; Coulombe, K.L.K. Engineered human myocardium with local release of angiogenic proteins improves vascularization and cardiac function in injured rat hearts. Biomaterials 2020, 251, 120033. [Google Scholar] [CrossRef] [PubMed]
- Shadrin, I.Y.; Allen, B.W.; Qian, Y.; Jackman, C.P.; Carlson, A.L.; Juhas, M.E.; Bursac, N. Cardiopatch platform enables maturation and scale-up of human pluripotent stem cell-derived engineered heart tissues. Nat. Commun. 2017, 8, 1825. [Google Scholar] [CrossRef]
- Jackman, C.P.; Ganapathi, A.M.; Asfour, H.; Qian, Y.; Allen, B.W.; Li, Y.; Bursac, N. Engineered cardiac tissue patch maintains structural and electrical properties after epicardial implantation. Biomaterials 2018, 159, 48–58. [Google Scholar] [CrossRef]
- von Bibra, C.; Shibamiya, A.; Bähr, A.; Geertz, B.; Köhne, M.; Stuedemann, T.; Starbatty, J.; Hornaschewitz, N.; Li, X.; Wolf, E.; et al. Immature human engineered heart tissues engraft in a guinea pig chronic injury model. bioRxiv 2022. [Google Scholar] [CrossRef]
- Zimmermann, W.-H.; Melnychenko, I.; Wasmeier, G.; Didié, M.; Naito, H.; Nixdorff, U.; Hess, A.; Budinsky, L.; Brune, K.; Michaelis, B.; et al. Engineered heart tissue grafts improve systolic and diastolic function in infarcted rat hearts. Nat. Med. 2006, 12, 452–458. [Google Scholar] [CrossRef]
- Chong, J.J.; Yang, X.; Don, C.W.; Minami, E.; Liu, Y.W.; Weyers, J.J.; Mahoney, W.M.; Van Biber, B.; Cook, S.M.; Palpant, N.J.; et al. Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature 2014, 510, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Nakane, T.; Masumoto, H.; Tinney, J.P.; Yuan, F.; Kowalski, W.J.; Ye, F.; LeBlanc, A.J.; Sakata, R.; Yamashita, J.K.; Keller, B.B. Impact of Cell Composition and Geometry on Human Induced Pluripotent Stem Cells-Derived Engineered Cardiac Tissue. Sci. Rep. 2017, 7, 45641. [Google Scholar] [CrossRef]
- Miyagawa, S.; Kainuma, S.; Kawamura, T.; Suzuki, K.; Ito, Y.; Iseoka, H.; Ito, E.; Takeda, M.; Sasai, M.; Mochizuki-Oda, N.; et al. Case report: Transplantation of human induced pluripotent stem cell-derived cardiomyocyte patches for ischemic cardiomyopathy. Front. Cardiovasc. Med. 2022, 9, 950829. [Google Scholar] [CrossRef]
- Fink, C.; Ergün, S.; Kralisch, D.; Remmers, U.; Weil, J.; Eschenhagen, T. Chronic stretch of engineered heart tissue induces hypertrophy and functional improvement. FASEB J. 2000, 14, 669–679. [Google Scholar] [CrossRef]
- Abilez, O.J.; Tzatzalos, E.; Yang, H.; Zhao, M.T.; Jung, G.; Zöllner, A.M.; Tiburcy, M.; Riegler, J.; Matsa, E.; Shukla, P.; et al. Passive Stretch Induces Structural and Functional Maturation of Engineered Heart Muscle as Predicted by Computational Modeling. Stem Cells 2018, 36, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Legant, W.R.; Pathak, A.; Yang, M.T.; Deshpande, V.S.; McMeeking, R.M.; Chen, C.S. Microfabricated tissue gauges to measure and manipulate forces from 3D microtissues. Proc. Natl. Acad. Sci. USA 2009, 106, 10097–10102. [Google Scholar] [CrossRef]
- Dwyer, K.D.; Coulombe, K.L.K. Cardiac mechanostructure: Using mechanics and anisotropy as inspiration for developing epicardial therapies in treating myocardial infarction. Bioact. Mater. 2021, 6, 2198–2220. [Google Scholar] [CrossRef]
- Power, J.M.; Raman, J.; Dornom, A.; Farish, S.J.; Burrell, L.M.; Tonkin, A.M.; Buxton, B.; Alferness, C.A. Passive ventricular constraint amends the course of heart failure: A study in an ovine model of dilated cardiomyopathy. Cardiovasc. Res. 1999, 44, 549–555. [Google Scholar] [CrossRef]
- Magovern, J.A. Experimental and Clinical Studies with the Paracor Cardiac Restraint Device. Semin. Thorac. Cardiovasc. Surg. 2005, 17, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Moainie, S.L.; Guy, T.S.; Gorman, J.H.; Plappert, T.; Jackson, B.M.; St. John-Sutton, M.G.; Edmunds, L.H.; Gorman, R.C. Infarct restraint attenuates remodeling and reduces chronic ischemic mitral regurgitation after postero-lateral infarction. Ann. Thorac. Surg. 2002, 74, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.-Y.; Siu, C.-W.; Liu, Y.; Zhang, Y.; Chan, W.-S.; Wu, E.X.; Wu, Y.; Nicholls, J.M.; Li, R.A.; Benser, M.E.; et al. Attenuation of Left Ventricular Adverse Remodeling With Epicardial Patching After Myocardial Infarction. J. Card. Fail. 2010, 16, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, H.N. Effects of Cardiac Support Device on reverse remodeling: Molecular, biochemical, and structural mechanisms. J. Card. Fail. 2004, 10, S207–S214. [Google Scholar] [CrossRef] [PubMed]
- Jacot, J.G.; McCulloch, A.D.; Omens, J.H. Substrate stiffness affects the functional maturation of neonatal rat ventricular myocytes. Biophys. J. 2008, 95, 3479–3487. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.J.; Carag-Krieger, C.; Johnson, C.P.; Raab, M.; Tang, H.-Y.; Speicher, D.W.; Sanger, J.W.; Sanger, J.M.; Discher, D.E. Embryonic cardiomyocytes beat best on a matrix with heart-like elasticity: Scar-like rigidity inhibits beating. J. Cell Sci. 2008, 121, 3794–3802. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.T.; Nagarajan, N.; Zorlutuna, P. Effect of Substrate Stiffness on Mechanical Coupling and Force Propagation at the Infarct Boundary. Biophys. J. 2018, 115, 1966–1980. [Google Scholar] [CrossRef]
- McCain, M.L.; Yuan, H.; Pasqualini, F.S.; Campbell, P.H.; Parker, K.K. Matrix elasticity regulates the optimal cardiac myocyte shape for contractility. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1525–H1539. [Google Scholar] [CrossRef] [PubMed]
- Corbin, E.A.; Vite, A.; Peyster, E.G.; Bhoopalam, M.; Brandimarto, J.; Wang, X.; Bennett, A.I.; Clark, A.T.; Cheng, X.; Turner, K.T.; et al. Tunable and Reversible Substrate Stiffness Reveals a Dynamic Mechanosensitivity of Cardiomyocytes. ACS Appl. Mater. Interfaces 2019, 11, 20603–20614. [Google Scholar] [CrossRef]
- Schmitt, P.R.; Dwyer, K.D.; Minor, A.J.; Coulombe, K.L. Wet-Spun Polycaprolactone Scaffolds Provide Customizable Anisotropic Viscoelastic Mechanics for Engineered Cardiac Tissues. Polymers 2022, 14, 4571. [Google Scholar] [CrossRef]
- Estrada, A.C.; Yoshida, K.; Clarke, S.A.; Holmes, J.W. Longitudinal Reinforcement of Acute Myocardial Infarcts Improves Function by Transmurally Redistributing Stretch and Stress. J. Biomech. Eng. 2019, 142, 021009. [Google Scholar] [CrossRef]
- Napiwocki, B.N.; Lang, D.; Stempien, A.; Zhang, J.; Vaidyanathan, R.; Makielski, J.C.; Eckhardt, L.L.; Glukhov, A.V.; Kamp, T.J.; Crone, W.C. Aligned human cardiac syncytium for in vitro analysis of electrical, structural, and mechanical readouts. Biotechnol. Bioeng. 2021, 118, 442–452. [Google Scholar] [CrossRef] [PubMed]
- MacQueen, L.A.; Sheehy, S.P.; Chantre, C.O.; Zimmerman, J.F.; Pasqualini, F.S.; Liu, X.; Goss, J.A.; Campbell, P.H.; Gonzalez, G.M.; Park, S.J.; et al. A tissue-engineered scale model of the heart ventricle. Nat. Biomed. Eng. 2018, 2, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, S.; Shapira, A.; Regev, O.; Nseir, N.; Zussman, E.; Dvir, T. Albumin fiber scaffolds for engineering functional cardiac tissues. Biotechnol. Bioeng. 2014, 111, 1246–1257. [Google Scholar] [CrossRef] [PubMed]
- Adadi, N.; Yadid, M.; Gal, I.; Asulin, M.; Feiner, R.; Edri, R.; Dvir, T. Electrospun Fibrous PVDF-TrFe Scaffolds for Cardiac Tissue Engineering, Differentiation, and Maturation. Adv. Mater. Technol. 2020, 5, 1900820. [Google Scholar] [CrossRef]
- Wanjare, M.; Hou, L.; Nakayama, K.H.; Kim, J.J.; Mezak, N.P.; Abilez, O.J.; Tzatzalos, E.; Wu, J.C.; Huang, N.F. Anisotropic microfibrous scaffolds enhance the organization and function of cardiomyocytes derived from induced pluripotent stem cells. Biomater. Sci. 2017, 5, 1567–1578. [Google Scholar] [CrossRef]
- Stuckey, D.J.; Ishii, H.; Chen, Q.-Z.; Boccaccini, A.R.; Hansen, U.; Carr, C.A.; Roether, J.A.; Jawad, H.; Tyler, D.J.; Ali, N.N.; et al. Magnetic Resonance Imaging Evaluation of Remodeling by Cardiac Elastomeric Tissue Scaffold Biomaterials in a Rat Model of Myocardial Infarction. Tissue Eng. Part A 2010, 16, 3395–3402. [Google Scholar] [CrossRef]
- Vilaeti, A.D.; Dimos, K.; Lampri, E.S.; Mantzouratou, P.; Tsitou, N.; Mourouzis, I.; Oikonomidis, D.L.; Papalois, A.; Pantos, C.; Malamou-Mitsi, V.; et al. Short-term ventricular restraint attenuates post-infarction remodeling in rats. Int. J. Cardiol. 2013, 165, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Liu, Y.; Bai, A.; Cai, H.; Bai, Y.; Jiang, W.; Yang, H.; Wang, X.; Yang, L.; Sun, N.; et al. A viscoelastic adhesive epicardial patch for treating myocardial infarction. Nat. Biomed. Eng. 2019, 3, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Chi, N.-H.; Yang, M.-C.; Chung, T.-W.; Chou, N.-K.; Wang, S.-S. Cardiac repair using chitosan-hyaluronan/silk fibroin patches in a rat heart model with myocardial infarction. Carbohydr. Polym. 2013, 92, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Serpooshan, V.; Zhao, M.; Metzler, S.A.; Wei, K.; Shah, P.B.; Wang, A.; Mahmoudi, M.; Malkovskiy, A.V.; Rajadas, J.; Butte, M.J.; et al. The effect of bioengineered acellular collagen patch on cardiac remodeling and ventricular function post myocardial infarction. Biomaterials 2013, 34, 9048–9055. [Google Scholar] [CrossRef]
- Karbassi, E.; Fenix, A.; Marchiano, S.; Muraoka, N.; Nakamura, K.; Yang, X.; Murry, C.E. Cardiomyocyte maturation: Advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 2020, 17, 341–359. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Garcia, M.J.; Quesnel, E.; Al-Attar, R.; Laskary, A.R.; Laflamme, M.A. Maturation of human pluripotent stem cell derived cardiomyocytes in vitro and in vivo. Semin. Cell Dev. Biol. 2021, 118, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.A.; Al-Siddiqi, H.H.A.A.; Purnama, U.; Iftekhar, S.; Bruyneel, A.A.N.; Kerr, M.; Nazir, R.; da Luz Sousa Fialho, M.; Malandraki-Miller, S.; Alonaizan, R.; et al. Physiological and pharmacological stimulation for in vitro maturation of substrate metabolism in human induced pluripotent stem cell-derived cardiomyocytes. Sci. Rep. 2021, 11, 7802. [Google Scholar] [CrossRef]
- Capulli, A.K.; MacQueen, L.A.; Sheehy, S.P.; Parker, K.K. Fibrous scaffolds for building hearts and heart parts. Adv. Drug. Deliv. Rev. 2016, 96, 83–102. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.C.; Maas, R.G.C.; van Adrichem, I.; Doevendans, P.A.M.; Mercola, M.; Šarić, T.; Buikema, J.W.; van Mil, A.; Chamuleau, S.A.J.; Sluijter, J.P.G.; et al. Metabolic Maturation Increases Susceptibility to Hypoxia-induced Damage in Human iPSC-derived Cardiomyocytes. Stem Cells Transl. Med. 2022, 11, 1040–1051. [Google Scholar] [CrossRef]
- Dhahri, W.; Sadikov Valdman, T.; Wilkinson, D.; Pereira, E.; Ceylan, E.; Andharia, N.; Qiang, B.; Masoudpour, H.; Wulkan, F.; Quesnel, E.; et al. In Vitro Matured Human Pluripotent Stem Cell-Derived Cardiomyocytes Form Grafts With Enhanced Structure and Function in Injured Hearts. Circulation 2022, 145, 1412–1426. [Google Scholar] [CrossRef] [PubMed]
- Restuccia, A.; Seroski, D.T.; Kelley, K.L.; O’Bryan, C.S.; Kurian, J.J.; Knox, K.R.; Farhadi, S.A.; Angelini, T.E.; Hudalla, G.A. Hierarchical self-assembly and emergent function of densely glycosylated peptide nanofibers. Commun. Chem. 2019, 2, 53. [Google Scholar] [CrossRef]
- Conradi, L.; Schmidt, S.; Neofytou, E.; Deuse, T.; Peters, L.; Eder, A.; Hua, X.; Hansen, A.; Robbins, R.C.; Beygui, R.E.; et al. Immunobiology of fibrin-based engineered heart tissue. Stem Cells Transl. Med. 2015, 4, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Terrovitis, J.V.; Smith, R.R.; Marbán, E. Assessment and optimization of cell engraftment after transplantation into the heart. Circ. Res. 2010, 106, 479–494. [Google Scholar] [CrossRef]
- Terrovitis, J.; Lautamäki, R.; Bonios, M.; Fox, J.; Engles, J.M.; Yu, J.; Leppo, M.K.; Pomper, M.G.; Wahl, R.L.; Seidel, J. Noninvasive quantification and optimization of acute cell retention by in vivo positron emission tomography after intramyocardial cardiac-derived stem cell delivery. J. Am. Coll. Cardiol. 2009, 54, 1619–1626. [Google Scholar] [CrossRef]
- Pretorius, D.; Kahn-Krell, A.M.; Lou, X.; Fast, V.G.; Berry, J.L.; Kamp, T.J.; Zhang, J. Layer-by-layer fabrication of large and thick human cardiac muscle patch constructs with superior electrophysiological properties. Front. Cell Dev. Biol. 2021, 9, 670504. [Google Scholar] [CrossRef] [PubMed]
- Deuse, T.; Hu, X.; Gravina, A.; Wang, D.; Tediashvili, G.; De, C.; Thayer, W.O.; Wahl, A.; Garcia, J.V.; Reichenspurner, H.; et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 2019, 37, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Deuse, T.; Tediashvili, G.; Hu, X.; Gravina, A.; Tamenang, A.; Wang, D.; Connolly, A.; Mueller, C.; Mallavia, B.; Looney, M.R.; et al. Hypoimmune induced pluripotent stem cell-derived cell therapeutics treat cardiovascular and pulmonary diseases in immunocompetent allogeneic mice. Proc. Natl. Acad. Sci. USA 2021, 118, e2022091118. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Research Group | Dose(s)/ECT | Dimensions | Experiments |
|---|---|---|---|
| Eschenhagen and Weinberger [12,63] | 450 M (hiPSC-CMs) | 50 × 70 mm | In vivo: healthy swine heart
|
| 4.5 M, 8.5 M, 12 M, 15 M (hiPSC-CMs) | 15 × 25 mm | In vivo: guinea pig cryoinjury model
| |
| Zhang [58] | 4 M/ECT 8 M/2-ECTs (hiPSC-CMs) | 40 × 20 mm | In vivo: swine I/R model of MI
|
| Bursac [61] | 0.5 M/1 M (hiPSC-CMs) | 7 × 7 mm | In vitro/in vivo:
|
| 2 M/10 M (hiPSC-CMs) | 15 × 15 mm 36 × 36 mm | In vitro:
| |
| Coulombe [60] | 7–10 M (hiPSC-CMs) | 18 × 14 mm | In vivo: rat I/R model of MI
|
| Keller [66] | 2.64–5.28 M (hiPSC-CMs) | 15 × 15 mm | In vivo: rat model of MI
|
| 10.56 M (hiPSC-CMs) | 30 × 30 mm | In vitro:
| |
| Zimmerman [64] | 2.5 M/ECT 12.5 M/5-ECT (Neonatal rat CMs) | ~5 × 10 mm | In vivo: rat PL model of MI
|
| Trial Number | Dose | Disease | Description | Phase | Start/End |
|---|---|---|---|---|---|
| NCT03759405 | Not specified | CHF | Autologous iPS-CMs via vein transplantation | 2–3 | December 2022/ December 2024 |
| NCT03763136 | 200 M | CHF | Injection of allogenic hPSC-CM during coronary artery bypass surgery | 1–2 | October 2021/ July 2023 |
| NCT04396899 | Not specified | HFrEF (EF < 35%) | Engineered heart muscle | 1–2 | February 2020/ October 2024 |
| NCT05068674 | 10 M/150 M/300 M | Chronic ischemic LV dysfunction | Dose tolerance study of hESC-CMs | 1 | March 2022/October 2025 |
| NCT04982081 | 100 M/400 M | Congestive HF, CVD, dilated Cardiomyopathy | hiPSC-CMs catheter injection | 1 | September 2021/ July 2023 |
| NCT04696328/ jRCT2053190081 [67] | (3) sheets of 33 M /sheet | Ischemic cardiomyopathy | Allogenic iPSC-CM within a cell sheet | 1 | December 2019/May 2023 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dwyer, K.D.; Kant, R.J.; Soepriatna, A.H.; Roser, S.M.; Daley, M.C.; Sabe, S.A.; Xu, C.M.; Choi, B.-R.; Sellke, F.W.; Coulombe, K.L.K. One Billion hiPSC-Cardiomyocytes: Upscaling Engineered Cardiac Tissues to Create High Cell Density Therapies for Clinical Translation in Heart Regeneration. Bioengineering 2023, 10, 587. https://doi.org/10.3390/bioengineering10050587
Dwyer KD, Kant RJ, Soepriatna AH, Roser SM, Daley MC, Sabe SA, Xu CM, Choi B-R, Sellke FW, Coulombe KLK. One Billion hiPSC-Cardiomyocytes: Upscaling Engineered Cardiac Tissues to Create High Cell Density Therapies for Clinical Translation in Heart Regeneration. Bioengineering. 2023; 10(5):587. https://doi.org/10.3390/bioengineering10050587
Chicago/Turabian StyleDwyer, Kiera D., Rajeev J. Kant, Arvin H. Soepriatna, Stephanie M. Roser, Mark C. Daley, Sharif A. Sabe, Cynthia M. Xu, Bum-Rak Choi, Frank W. Sellke, and Kareen L. K. Coulombe. 2023. "One Billion hiPSC-Cardiomyocytes: Upscaling Engineered Cardiac Tissues to Create High Cell Density Therapies for Clinical Translation in Heart Regeneration" Bioengineering 10, no. 5: 587. https://doi.org/10.3390/bioengineering10050587
APA StyleDwyer, K. D., Kant, R. J., Soepriatna, A. H., Roser, S. M., Daley, M. C., Sabe, S. A., Xu, C. M., Choi, B. -R., Sellke, F. W., & Coulombe, K. L. K. (2023). One Billion hiPSC-Cardiomyocytes: Upscaling Engineered Cardiac Tissues to Create High Cell Density Therapies for Clinical Translation in Heart Regeneration. Bioengineering, 10(5), 587. https://doi.org/10.3390/bioengineering10050587