

Suppressing Chondrocyte Hypertrophy to Build Better Cartilage

 , ,
, , 
Abstract
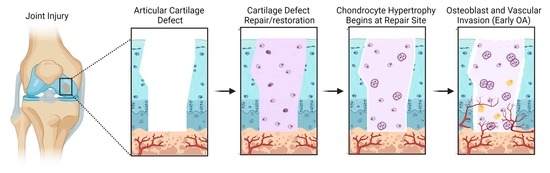
:
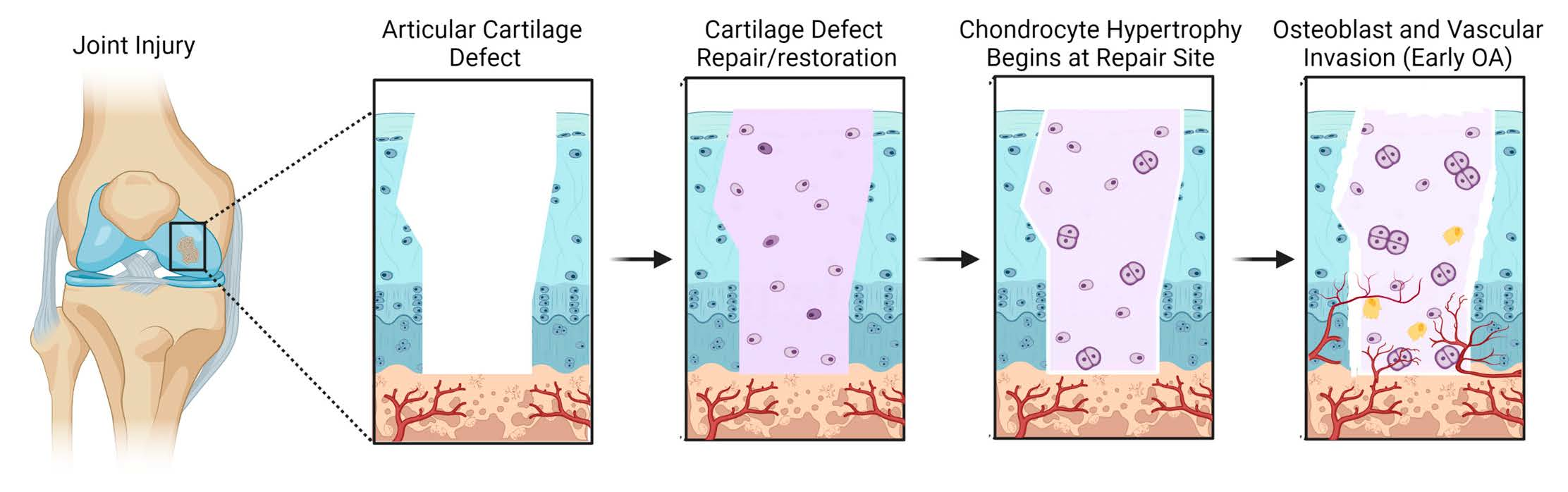{kind=link}
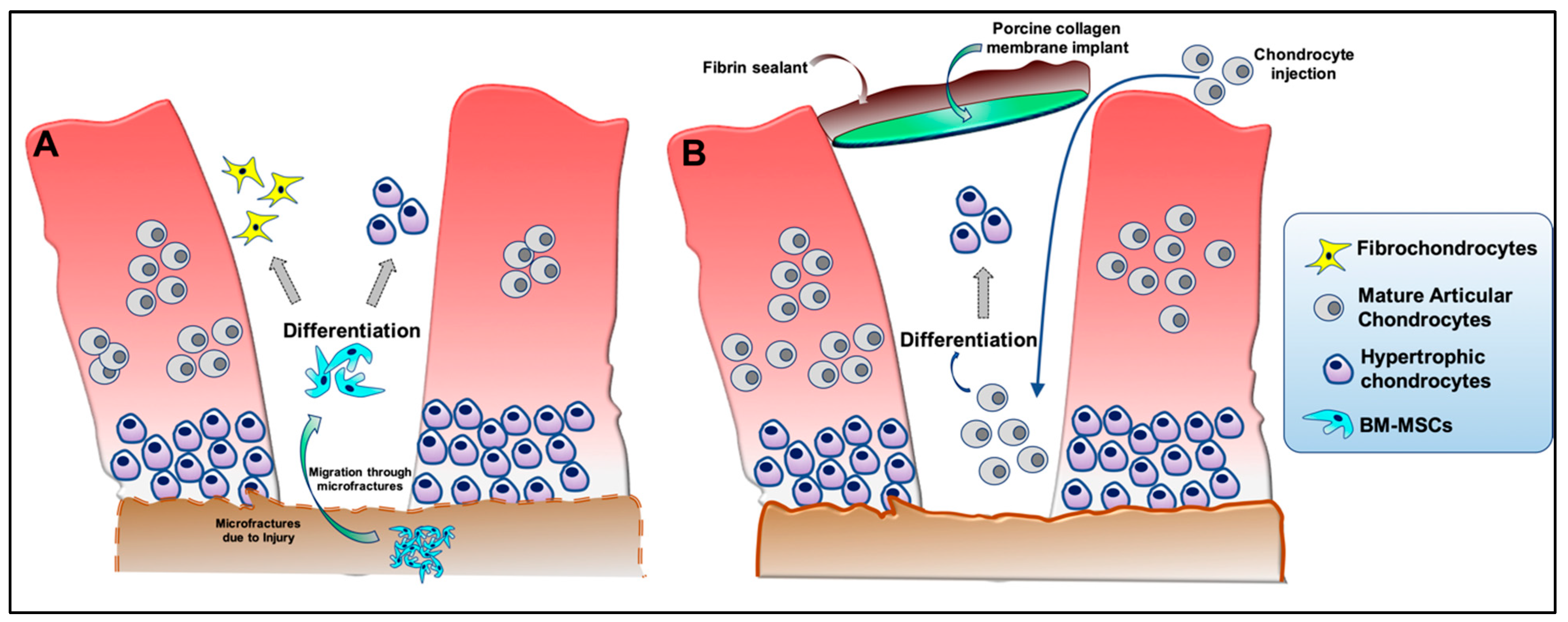{kind=link}
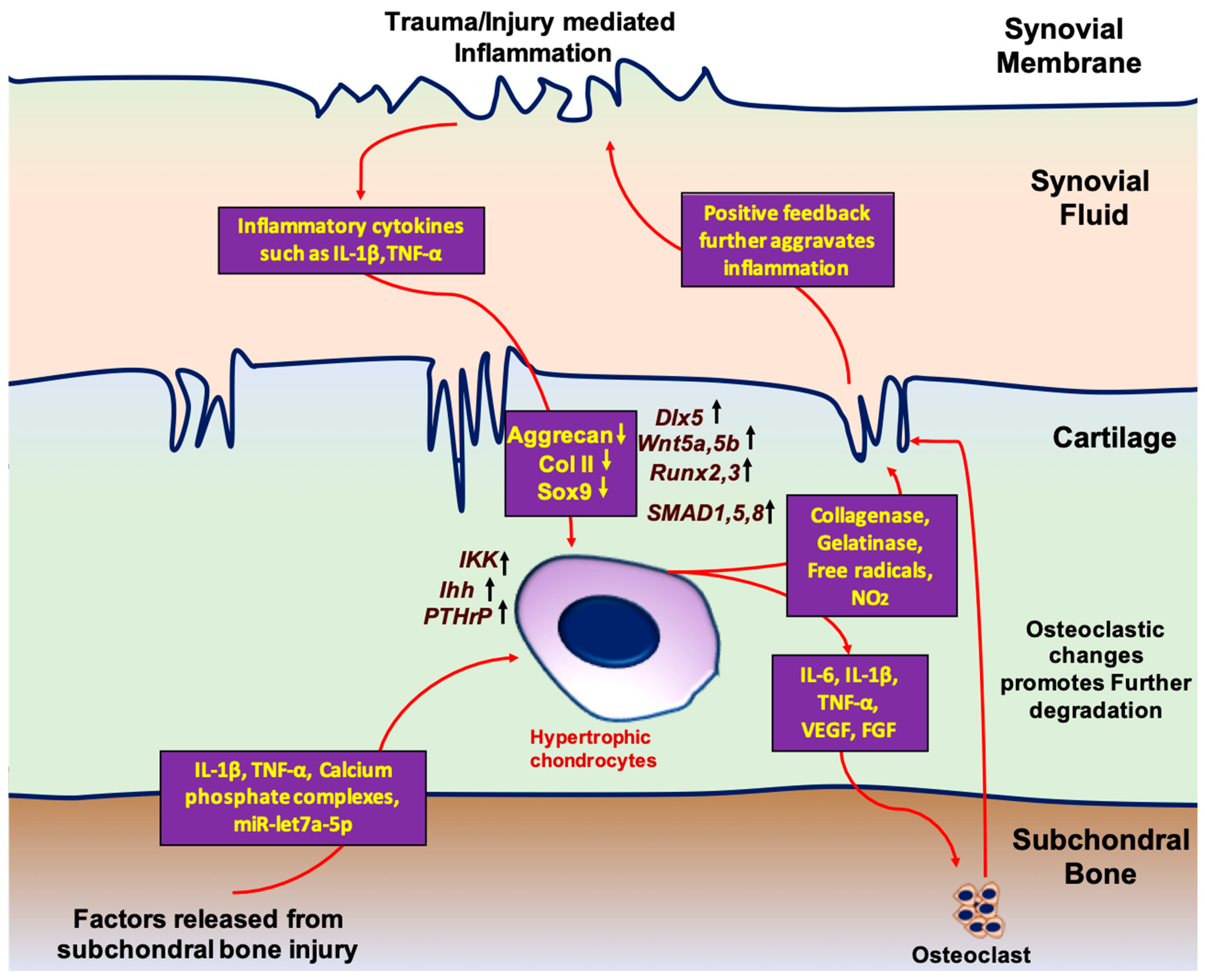{kind=link}
1. Introduction
2. The Healthy State of Articular Cartilage
3. Cartilage Hypertrophy in Injury and Catabolic Joint Disease
4. Pathways That Regulate Chondrocyte Hypertrophy
4.1. Sox9 Signaling
4.2. Runx Family Transcription Factors
4.3. Bone Morphogenetic Protein (BMP) Signaling
4.4. Wnt Signaling
4.5. Indian Hedgehog (Ihh) and Parathyroid Hormone Related Peptide Signaling (PTHrP) Signaling
4.6. IκB Kinase/NF-κB and Inflammatory Cytokines
5. Pre-Clinical Strategies to Inhibit Chondrocyte Hypertrophy in Joint Cartilage and in Applications of Cartilage Tissue Engineering
5.1. Mesenchymal Progenitor Cell Therapy
5.2. Growth Factor Therapy
5.3. Utilization of Anti-Hypertrophic ECM Proteins
5.4. Scaffolding
5.5. Culturing Conditions for Cells Used in Cartilage Tissue Engineering and Repair
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Trivedi, J.; Betensky, D.; Desai, S.; Jayasuriya, C.T. Post-Traumatic Osteoarthritis Assessment in Emerging and Advanced Pre-Clinical Meniscus Repair Strategies: A Review. Front. Bioeng. Biotechnol. 2021, 9, 787330. [Google Scholar] [CrossRef] [PubMed]
- Mason, D.; Englund, M.; Watt, F.E. Prevention of posttraumatic osteoarthritis at the time of injury: Where are we now, and where are we going? J. Orthop. Res. 2021, 39, 1152–1163. [Google Scholar] [CrossRef] [PubMed]
- Camp, C.L.; Stuart, M.J.; Krych, A.J. Current concepts of articular cartilage restoration techniques in the knee. Sports Health 2014, 6, 265–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Bello, A.; Arai, Y.; Ahn, J.; Kim, D.; Cha, K.Y.; Baek, I.; Park, H.; Lee, S.H. Functional Duality of Chondrocyte Hypertrophy and Biomedical Application Trends in Osteoarthritis. Pharmaceutics 2021, 13, 1139. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Fu, P.; Cong, R.; Wu, H.; Pei, M. Strategies to minimize hypertrophy in cartilage engineering and regeneration. Genes Dis. 2015, 2, 76–95. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Wu, X.; Crawford, R.; Xiao, Y.; Prasadam, I. Macro, Micro, and Molecular. Changes of the Osteochondral Interface in Osteoarthritis Development. Front. Cell Dev. Biol. 2021, 9, 659654. [Google Scholar] [CrossRef]
- van der Kraan, P.M.; van den Berg, W.B. Chondrocyte hypertrophy and osteoarthritis: Role in initiation and progression of cartilage degeneration? Osteoarthr. Cartil. 2012, 20, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Vinod, E.; Padmaja, K.; Ramasamy, B.; Sathishkumar, S. Systematic review of articular cartilage derived chondroprogenitors for cartilage repair in animal models. J. Orthop. 2023, 35, 43–53. [Google Scholar] [CrossRef]
- Buckwalter, J.A. Articular cartilage: Injuries and potential for healing. J. Orthop. Sports Phys. Ther. 1998, 28, 192–202. [Google Scholar] [CrossRef] [Green Version]
- Carballo, C.B.; Nakagawa, Y.; Sekiya, I.; Rodeo, S.A. Basic Science of Articular Cartilage. Clin. Sports Med. 2017, 36, 413–425. [Google Scholar] [CrossRef]
- Yildirim, N.; Amanzhanova, A.; Kulzhanova, G.; Mukasheva, F.; Erisken, C. Osteochondral Interface: Regenerative Engineering and Challenges. ACS Biomater. Sci. Eng. 2023, 9, 1205–1223. [Google Scholar] [CrossRef]
- Aghajanian, P.; Mohan, S. The art of building bone: Emerging role of chondrocyte-to-osteoblast transdifferentiation in endochondral ossification. Bone Res. 2018, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Dai, H. Articular cartilage and osteochondral tissue engineering techniques: Recent advances and challenges. Bioact. Mater. 2021, 6, 4830–4855. [Google Scholar] [CrossRef]
- Millward-Sadler, S.J.; Salter, D.M. Integrin-dependent signal cascades in chondrocyte mechanotransduction. Ann. Biomed. Eng. 2004, 32, 435–446. [Google Scholar] [CrossRef]
- Hunziker, E.B. Articular cartilage repair: Are the intrinsic biological constraints undermining this process insuperable? Osteoarthr. Cartil. 1999, 7, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Pattappa, G.; Schewior, R.; Hofmeister, I.; Seja, J.; Zellner, J.; Johnstone, B.; Docheva, D.; Angele, P. Physioxia Has a Beneficial Effect on Cartilage Matrix Production in Interleukin-1 Beta-Inhibited Mesenchymal Stem Cell Chondrogenesis. Cells 2019, 8, 936. [Google Scholar] [CrossRef] [Green Version]
- Seol, D.; McCabe, D.J.; Choe, H.; Zheng, H.; Yu, Y.; Jang, K.; Walter, M.W.; Lehman, A.D.; Ding, L.; Buckwalter, J.A.; et al. Chondrogenic progenitor cells respond to cartilage injury. Arthritis Rheum. 2012, 64, 3626–3637. [Google Scholar] [CrossRef] [Green Version]
- Hu, N.; Gao, Y.; Jayasuriya, C.T.; Liu, W.; Du, H.; Ding, J.; Feng, M.; Chen, Q. Chondrogenic induction of human osteoarthritic cartilage-derived mesenchymal stem cells activates mineralization and hypertrophic and osteogenic gene expression through a mechanomiR. Arthritis Res. 2019, 21, 167. [Google Scholar] [CrossRef] [Green Version]
- Jayasuriya, C.T.; Hu, N.; Li, J.; Lemme, N.; Terek, R.; Ehrlich, M.G.; Chen, Q. Molecular characterization of mesenchymal stem cells in human osteoarthritis cartilage reveals contribution to the OA phenotype. Sci. Rep. 2018, 8, 7044. [Google Scholar] [CrossRef] [Green Version]
- McCulloch, K.; Litherland, G.J.; Rai, T.S. Cellular senescence in osteoarthritis pathology. Aging Cell 2017, 16, 210–218. [Google Scholar] [CrossRef]
- Mankin, H.J. The reaction of articular cartilage to injury and osteoarthritis (first of two parts). N. Engl. J. Med. 1974, 291, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Mankin, H.J. The reaction of articular cartilage to injury and osteoarthritis (second of two parts). N. Engl. J. Med. 1974, 291, 1335–1340. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.M.; Beier, F. Chondrocyte hypertrophy in skeletal development, growth, and disease. Birth Defects Res. C Embryo Today 2014, 102, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Yoon, B.S.; Pogue, R.; Ovchinnikov, D.A.; Yoshii, I.; Mishina, Y.; Behringer, R.R.; Lyons, K.M. BMPs regulate multiple aspects of growth-plate chondrogenesis through opposing actions on FGF pathways. Development 2006, 133, 4667–4678. [Google Scholar] [CrossRef] [Green Version]
- Serra, R.; Karaplis, A.; Sohn, P. Parathyroid hormone-related peptide (PTHrP)-dependent and -independent effects of transforming growth factor beta (TGF-beta) on endochondral bone formation. J. Cell Biol. 1999, 145, 783–794. [Google Scholar] [CrossRef] [Green Version]
- Mak, K.K.; Kronenberg, H.M.; Chuang, P.T.; Mackem, S.; Yang, Y. Indian hedgehog signals independently of PTHrP to promote chondrocyte hypertrophy. Development 2008, 135, 1947–1956. [Google Scholar] [CrossRef] [Green Version]
- Kamekura, S.; Kawasaki, Y.; Hoshi, K.; Shimoaka, T.; Chikuda, H.; Maruyama, Z.; Komori, T.; Sato, S.; Takeda, S.; Karsenty, G.; et al. Contribution of runt-related transcription factor 2 to the pathogenesis of osteoarthritis in mice after induction of knee joint instability. Arthritis Rheum. 2006, 54, 2462–2470. [Google Scholar] [CrossRef]
- Kamekura, S.; Hoshi, K.; Shimoaka, T.; Chung, U.; Chikuda, H.; Yamada, T.; Uchida, M.; Ogata, N.; Seichi, A.; Nakamura, K.; et al. Osteoarthritis development in novel experimental mouse models induced by knee joint instability. Osteoarthr. Cartil. 2005, 13, 632–641. [Google Scholar] [CrossRef] [Green Version]
- Hoyland, J.A.; Thomas, J.T.; Donn, R.; Marriott, A.; Ayad, S.; Boot-Handford, R.P.; Grant, M.E.; Freemont, A.J. Distribution of type X collagen mRNA in normal and osteoarthritic human cartilage. Bone Min. 1991, 15, 151–163. [Google Scholar] [CrossRef]
- Zheng, Q.; Zhou, G.; Morello, R.; Chen, Y.; Garcia-Rojas, X.; Lee, B. Type X collagen gene regulation by Runx2 contributes directly to its hypertrophic chondrocyte-specific expression in vivo. J. Cell Biol. 2003, 162, 833–842. [Google Scholar] [CrossRef]
- Stephens, M.; Kwan, A.P.; Bayliss, M.T.; Archer, C.W. Human articular surface chondrocytes initiate alkaline phosphatase and type X collagen synthesis in suspension culture. J. Cell Sci. 1992, 103 Pt 4, 1111–1116. [Google Scholar] [CrossRef]
- Troeberg, L.; Nagase, H. Proteases involved in cartilage matrix degradation in osteoarthritis. Biochim. Biophys. Acta 2012, 1824, 133–145. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Ecker, M. Overview of MMP-13 as a Promising Target for the Treatment of Osteoarthritis. Int. J. Mol. Sci. 2021, 22, 1742. [Google Scholar] [CrossRef]
- Mapp, P.I.; Walsh, D.A. Mechanisms and targets of angiogenesis and nerve growth in osteoarthritis. Nat. Rev. Rheumatol. 2012, 8, 390–398. [Google Scholar] [CrossRef]
- Liao, L.; Zhang, S.; Gu, J.; Takarada, T.; Yoneda, Y.; Huang, J.; Zhao, L.; Oh, C.D.; Li, J.; Wang, B.; et al. Deletion of Runx2 in Articular Chondrocytes Decelerates the Progression of DMM-Induced Osteoarthritis in Adult Mice. Sci. Rep. 2017, 7, 2371. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wang, D.; Yuan, Y.; Min, J. New insights on the MMP-13 regulatory network in the pathogenesis of early osteoarthritis. Arthritis Res. 2017, 19, 248. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.; Siegrist, M.; Goodwin, K. Cyclic tensile strain and cyclic hydrostatic pressure differentially regulate expression of hypertrophic markers in primary chondrocytes. Bone 2003, 33, 685–693. [Google Scholar] [CrossRef]
- Beckmann, R.; Houben, A.; Tohidnezhad, M.; Kweider, N.; Fragoulis, A.; Wruck, C.J.; Brandenburg, L.O.; Hermanns-Sachweh, B.; Goldring, M.B.; Pufe, T.; et al. Mechanical forces induce changes in VEGF and VEGFR-1/sFlt-1 expression in human chondrocytes. Int. J. Mol. Sci. 2014, 15, 15456–15474. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.A.; Buckwalter, J.A. Aging, articular cartilage chondrocyte senescence and osteoarthritis. Biogerontology 2002, 3, 257–264. [Google Scholar] [CrossRef]
- Sellam, J.; Berenbaum, F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat. Rev. Rheumatol. 2010, 6, 625–635. [Google Scholar] [CrossRef]
- O’Conor, C.J.; Case, N.; Guilak, F. Mechanical regulation of chondrogenesis. Stem Cell Res. 2013, 4, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelttari, K.; Winter, A.; Steck, E.; Goetzke, K.; Hennig, T.; Ochs, B.G.; Aigner, T.; Richter, W. Premature induction of hypertrophy during in vitro chondrogenesis of human mesenchymal stem cells correlates with calcification and vascular invasion after ectopic transplantation in SCID mice. Arthritis Rheum. 2006, 54, 3254–3266. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, H.M. Developmental regulation of the growth plate. Nature 2003, 423, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Roelofs, A.J.; Kania, K.; Rafipay, A.J.; Sambale, M.; Kuwahara, S.T.; Collins, F.L.; Smeeton, J.; Serowoky, M.A.; Rowley, L.; Wang, H.; et al. Identification of the skeletal progenitor cells forming osteophytes in osteoarthritis. Ann. Rheum. Dis. 2020, 79, 1625–1634. [Google Scholar] [CrossRef] [PubMed]
- Riegger, J.; Brenner, R.E. Pathomechanisms of Posttraumatic Osteoarthritis: Chondrocyte Behavior and Fate in a Precarious Environment. Int. J. Mol. Sci. 2020, 21, 1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumdar, M.K.; Wang, E.; Morris, E.A. BMP-2 and BMP-9 promotes chondrogenic differentiation of human multipotential mesenchymal cells and overcomes the inhibitory effect of IL-1. J. Cell. Physiol. 2001, 189, 275–284. [Google Scholar] [CrossRef]
- Dai, J.; Dong, R.; Han, X.; Li, J.; Gong, X.; Bai, Y.; Kang, F.; Liang, M.; Zeng, F.; Hou, Z.; et al. Osteoclast-derived exosomal let-7a-5p targets Smad2 to promote the hypertrophic differentiation of chondrocytes. Am. J. Physiol. Cell Physiol. 2020, 319, C21–C33. [Google Scholar] [CrossRef]
- Jung, Y.K.; Han, M.S.; Park, H.R.; Lee, E.J.; Jang, J.A.; Kim, G.W.; Lee, S.Y.; Moon, D.; Han, S. Calcium-phosphate complex increased during subchondral bone remodeling affects earlystage osteoarthritis. Sci. Rep. 2018, 8, 487. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.K.; Li, G.W.; Zeng, C.; Lin, C.X.; Huang, L.S.; Huang, G.X.; Zhao, C.; Feng, S.Y.; Fang, H. Mechanical stress contributes to osteoarthritis development through the activation of transforming growth factor beta 1 (TGF-β1). Bone Jt. Res. 2018, 7, 587–594. [Google Scholar] [CrossRef]
- Hu, W.; Chen, Y.; Dou, C.; Dong, S. Microenvironment in subchondral bone: Predominant regulator for the treatment of osteoarthritis. Ann. Rheum. Dis. 2021, 80, 413–422. [Google Scholar] [CrossRef]
- Ayral, X.; Pickering, E.H.; Woodworth, T.G.; Mackillop, N.; Dougados, M. Synovitis: A potential predictive factor of structural progression of medial tibiofemoral knee osteoarthritis—Results of a 1 year longitudinal arthroscopic study in 422 patients. Osteoarthr. Cartil. 2005, 13, 361–367. [Google Scholar] [CrossRef] [Green Version]
- Kozhemyakina, E.; Lassar, A.B.; Zelzer, E. A pathway to bone: Signaling molecules and transcription factors involved in chondrocyte development and maturation. Development 2015, 142, 817–831. [Google Scholar] [CrossRef] [Green Version]
- Long, F.; Zhang, X.M.; Karp, S.; Yang, Y.; McMahon, A.P. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development 2001, 128, 5099–5108. [Google Scholar] [CrossRef]
- Kobayashi, T.; Chung, U.I.; Schipani, E.; Starbuck, M.; Karsenty, G.; Katagiri, T.; Goad, D.L.; Lanske, B.; Kronenberg, H.M. PTHrP and Indian hedgehog control differentiation of growth plate chondrocytes at multiple steps. Development 2002, 129, 2977–2986. [Google Scholar] [CrossRef]
- Chen, H.; Tan, X.N.; Hu, S.; Liu, R.Q.; Peng, L.H.; Li, Y.M.; Wu, P. Molecular Mechanisms of Chondrocyte Proliferation and Differentiation. Front. Cell Dev. Biol. 2021, 9, 664168. [Google Scholar] [CrossRef]
- Cooper, K.L.; Oh, S.; Sung, Y.; Dasari, R.R.; Kirschner, M.W.; Tabin, C.J. Multiple phases of chondrocyte enlargement underlie differences in skeletal proportions. Nature 2013, 495, 375–378. [Google Scholar] [CrossRef] [Green Version]
- Donell, S. Subchondral bone remodelling in osteoarthritis. EFORT Open Rev. 2019, 4, 221–229. [Google Scholar] [CrossRef]
- Plotkin, L.I.; Bruzzaniti, A. Molecular signaling in bone cells: Regulation of cell differentiation and survival. Adv. Protein. Chem. Struct. Biol. 2019, 116, 237–281. [Google Scholar] [CrossRef]
- Lefebvre, V.; Dvir-Ginzberg, M. SOX9 and the many facets of its regulation in the chondrocyte lineage. Connect. Tissue Res. 2017, 58, 2–14. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Liu, G.; Halim, A.; Ju, Y.; Luo, Q.; Song, A.G. Mesenchymal Stem Cell Migration and Tissue Repair. Cells 2019, 8, 784. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, H. Control of chondrogenesis by the transcription factor Sox9. Mod. Rheumatol. 2008, 18, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.; Huang, W.; Whitworth, D.J.; Deng, J.M.; Zhang, Z.; Behringer, R.R.; de Crombrugghe, B. Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc. Natl. Acad. Sci. USA 2001, 98, 6698–6703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, H.; Chaboissier, M.C.; Martin, J.F.; Schedl, A.; de Crombrugghe, B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 2002, 16, 2813–2828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.S.; Yang, H.N.; Woo, D.G.; Jeon, S.Y.; Do, H.J.; Lim, H.Y.; Kim, J.H.; Park, K.H. Chondrogenesis of human mesenchymal stem cells mediated by the combination of SOX trio SOX5, 6, and 9 genes complexed with PEI-modified PLGA nanoparticles. Biomaterials 2011, 32, 3679–3688. [Google Scholar] [CrossRef]
- Zhou, G.; Zheng, Q.; Engin, F.; Munivez, E.; Chen, Y.; Sebald, E.; Krakow, D.; Lee, B. Dominance of SOX9 function over RUNX2 during skeletogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 19004–19009. [Google Scholar] [CrossRef] [Green Version]
- Hattori, T.; Müller, C.; Gebhard, S.; Bauer, E.; Pausch, F.; Schlund, B.; Bösl, M.R.; Hess, A.; Surmann-Schmitt, C.; von der Mark, H.; et al. SOX9 is a major negative regulator of cartilage vascularization, bone marrow formation and endochondral ossification. Development 2010, 137, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.J.; Sun, C.; Liu, Z.; Sun, X.; Zhu, F.; Zhu, Z.Z.; Qiu, Y. Transcription factor Runx2 in the low bone mineral density of girls with adolescent idiopathic scoliosis. Orthop. Surg. 2014, 6, 8–14. [Google Scholar] [CrossRef]
- Komori, T. Runx2, an inducer of osteoblast and chondrocyte differentiation. Histochem. Cell Biol. 2018, 149, 313–323. [Google Scholar] [CrossRef]
- Yoshida, C.A.; Yamamoto, H.; Fujita, T.; Furuichi, T.; Ito, K.; Inoue, K.; Yamana, K.; Zanma, A.; Takada, K.; Ito, Y.; et al. Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 2004, 18, 952–963. [Google Scholar] [CrossRef] [Green Version]
- Javed, A.; Bae, J.S.; Afzal, F.; Gutierrez, S.; Pratap, J.; Zaidi, S.K.; Lou, Y.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; et al. Structural coupling of Smad and Runx2 for execution of the BMP2 osteogenic signal. J. Biol. Chem. 2008, 283, 8412–8422. [Google Scholar] [CrossRef] [Green Version]
- Urist, M.R. Bone morphogenetic protein: The molecularization of skeletal system development. J. Bone Min. Res. 1997, 12, 343–346. [Google Scholar] [CrossRef]
- Duprez, D.; Bell, E.J.; Richardson, M.K.; Archer, C.W.; Wolpert, L.; Brickell, P.M.; Francis-West, P.H. Overexpression of BMP-2 and BMP-4 alters the size and shape of developing skeletal elements in the chick limb. Mech. Dev. 1996, 57, 145–157. [Google Scholar] [CrossRef]
- Guo, P.; Shi, Z.L.; Liu, A.; Lin, T.; Bi, F.; Shi, M.; Yan, S.G. Effects of cartilage oligomeric matrix protein on bone morphogenetic protein-2-induced differentiation of mesenchymal stem cells. Orthop. Surg. 2014, 6, 280–287. [Google Scholar] [CrossRef]
- Kawakami, Y.; Ishikawa, T.; Shimabara, M.; Tanda, N.; Enomoto-Iwamoto, M.; Iwamoto, M.; Kuwana, T.; Ueki, A.; Noji, S.; Nohno, T. BMP signaling during bone pattern determination in the developing limb. Development 1996, 122, 3557–3566. [Google Scholar] [CrossRef]
- Zou, H.; Wieser, R.; Massagué, J.; Niswander, L. Distinct roles of type I bone morphogenetic protein receptors in the formation and differentiation of cartilage. Genes Dev. 1997, 11, 2191–2203. [Google Scholar] [CrossRef] [Green Version]
- Yoon, B.S.; Ovchinnikov, D.A.; Yoshii, I.; Mishina, Y.; Behringer, R.R.; Lyons, K.M. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 5062–5067. [Google Scholar] [CrossRef] [Green Version]
- Nordin, K.; LaBonne, C. Sox5 Is a DNA-binding cofactor for BMP R-Smads that directs target specificity during patterning of the early ectoderm. Dev. Cell 2014, 31, 374–382. [Google Scholar] [CrossRef] [Green Version]
- Papathanasiou, I.; Malizos, K.N.; Tsezou, A. Bone morphogenetic protein-2-induced Wnt/β-catenin signaling pathway activation through enhanced low-density-lipoprotein receptor-related protein 5 catabolic activity contributes to hypertrophy in osteoarthritic chondrocytes. Arthritis Res. 2012, 14, R82. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Mak, K.K.; Taketo, M.M.; Yang, Y. The Wnt/beta-catenin pathway interacts differentially with PTHrP signaling to control chondrocyte hypertrophy and final maturation. PLoS ONE 2009, 4, e6067. [Google Scholar] [CrossRef] [Green Version]
- Bendall, A.J.; Hu, G.; Levi, G.; Abate-Shen, C. Dlx5 regulates chondrocyte differentiation at multiple stages. Int. J. Dev. Biol. 2003, 47, 335–344. [Google Scholar]
- Hsu, S.H.; Noamani, B.; Abernethy, D.E.; Zhu, H.; Levi, G.; Bendall, A.J. Dlx5- and Dlx6-mediated chondrogenesis: Differential domain requirements for a conserved function. Mech. Dev. 2006, 123, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, D.; Kosher, R.A. Dlx5 is a positive regulator of chondrocyte differentiation during endochondral ossification. Dev. Biol. 2002, 252, 257–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robledo, R.F.; Rajan, L.; Li, X.; Lufkin, T. The Dlx5 and Dlx6 homeobox genes are essential for craniofacial, axial, and appendicular skeletal development. Genes Dev. 2002, 16, 1089–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, H.J.; Fisher, M.C.; Li, Y.; Ferrari, D.; Wang, C.K.; Lichtler, A.C.; Dealy, C.N.; Kosher, R.A. Studies on the role of Dlx5 in regulation of chondrocyte differentiation during endochondral ossification in the developing mouse limb. Dev. Growth Differ. 2007, 49, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Bendall, A.J. Dlx5 Is a cell autonomous regulator of chondrocyte hypertrophy in mice and functionally substitutes for Dlx6 during endochondral ossification. PLoS ONE 2009, 4, e8097. [Google Scholar] [CrossRef]
- McCarthy, H.E.; Bara, J.J.; Brakspear, K.; Singhrao, S.K.; Archer, C.W. The comparison of equine articular cartilage progenitor cells and bone marrow-derived stromal cells as potential cell sources for cartilage repair in the horse. Vet. J. 2012, 192, 345–351. [Google Scholar] [CrossRef]
- Twomey-Kozak, J.; Desai, S.; Liu, W.; Li, N.Y.; Lemme, N.; Chen, Q.; Owens, B.D.; Jayasuriya, C.T. Distal-Less Homeobox 5 Is a Therapeutic Target for Attenuating Hypertrophy and Apoptosis of Mesenchymal Progenitor Cells. Int. J. Mol. Sci. 2020, 21, 4823. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [Green Version]
- Ackers, I.; Malgor, R. Interrelationship of canonical and non-canonical Wnt signalling pathways in chronic metabolic diseases. Diabetes Vasc. Dis. Res. 2018, 15, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Hill, T.P.; Später, D.; Taketo, M.M.; Birchmeier, W.; Hartmann, C. Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev. Cell 2005, 8, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Reinhold, M.I.; Kapadia, R.M.; Liao, Z.; Naski, M.C. The Wnt-inducible transcription factor Twist1 inhibits chondrogenesis. J. Biol. Chem. 2006, 281, 1381–1388. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Zhang, E.; Yang, M.; Lu, L. Overexpression of Wnt11 promotes chondrogenic differentiation of bone marrow-derived mesenchymal stem cells in synergism with TGF-β. Mol. Cell. Biochem. 2014, 390, 123–131. [Google Scholar] [CrossRef]
- Yang, Y.; Topol, L.; Lee, H.; Wu, J. Wnt5a and Wnt5b exhibit distinct activities in coordinating chondrocyte proliferation and differentiation. Development 2003, 130, 1003–1015. [Google Scholar] [CrossRef] [Green Version]
- Bradley, E.W.; Drissi, M.H. Wnt5b regulates mesenchymal cell aggregation and chondrocyte differentiation through the planar cell polarity pathway. J. Cell. Physiol. 2011, 226, 1683–1693. [Google Scholar] [CrossRef]
- Randall, R.M.; Shao, Y.Y.; Wang, L.; Ballock, R.T. Activation of Wnt Planar Cell Polarity (PCP) signaling promotes growth plate column formation in vitro. J. Orthop. Res. 2012, 30, 1906–1914. [Google Scholar] [CrossRef]
- Leijten, J.C.; Bos, S.D.; Landman, E.B.; Georgi, N.; Jahr, H.; Meulenbelt, I.; Post, J.N.; van Blitterswijk, C.A.; Karperien, M. GREM1, FRZB and DKK1 mRNA levels correlate with osteoarthritis and are regulated by osteoarthritis-associated factors. Arthritis Res. 2013, 15, R126. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.; Xu, J.; Liu, Z.; Sosic, D.; Shao, J.; Olson, E.N.; Towler, D.A.; Ornitz, D.M. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development 2003, 130, 3063–3074. [Google Scholar] [CrossRef] [Green Version]
- Naski, M.C.; Colvin, J.S.; Coffin, J.D.; Ornitz, D.M. Repression of hedgehog signaling and BMP4 expression in growth plate cartilage by fibroblast growth factor receptor 3. Development 1998, 125, 4977–4988. [Google Scholar] [CrossRef]
- Minina, E.; Kreschel, C.; Naski, M.C.; Ornitz, D.M.; Vortkamp, A. Interaction of FGF, Ihh/Pthlh, and BMP signaling integrates chondrocyte proliferation and hypertrophic differentiation. Dev. Cell 2002, 3, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Hung, I.H.; Yu, K.; Lavine, K.J.; Ornitz, D.M. FGF9 regulates early hypertrophic chondrocyte differentiation and skeletal vascularization in the developing stylopod. Dev. Biol. 2007, 307, 300–313. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Macica, C.M.; Nasiri, A.; Broadus, A.E. Regulation of articular chondrocyte proliferation and differentiation by indian hedgehog and parathyroid hormone-related protein in mice. Arthritis Rheum. 2008, 58, 3788–3797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Wang, C.; Wang, B. Phosphorylation of Gli2 by protein kinase A is required for Gli2 processing and degradation and the Sonic Hedgehog-regulated mouse development. Dev. Biol. 2009, 326, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Lavine, K.J.; Hung, I.H.; Ornitz, D.M. FGF18 is required for early chondrocyte proliferation, hypertrophy and vascular invasion of the growth plate. Dev. Biol. 2007, 302, 80–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivotto, E.; Borzi, R.M.; Vitellozzi, R.; Pagani, S.; Facchini, A.; Battistelli, M.; Penzo, M.; Li, X.; Flamigni, F.; Li, J.; et al. Differential requirements for IKKalpha and IKKbeta in the differentiation of primary human osteoarthritic chondrocytes. Arthritis Rheum. 2008, 58, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Jimi, E.; Fei, H.; Nakatomi, C. NF-κB Signaling Regulates Physiological and Pathological Chondrogenesis. Int. J. Mol. Sci. 2019, 20, 6275. [Google Scholar] [CrossRef] [Green Version]
- Desai, S.; Jayasuriya, C.T. Implementation of Endogenous and Exogenous Mesenchymal Progenitor Cells for Skeletal Tissue Regeneration and Repair. Bioengineering 2020, 7, 86. [Google Scholar] [CrossRef]
- Xiang, X.N.; Zhu, S.Y.; He, H.C.; Yu, X.; Xu, Y.; He, C.Q. Mesenchymal stromal cell-based therapy for cartilage regeneration in knee osteoarthritis. Stem. Cell Res. 2022, 13, 14. [Google Scholar] [CrossRef]
- Ismail, O.M.; Said, U.N.; El-Omar, O.M. Adult Stem Cells for Cartilage Regeneration. Cureus 2022, 14, e32280. [Google Scholar] [CrossRef]
- Nejadnik, H.; Hui, J.H.; Feng Choong, E.P.; Tai, B.C.; Lee, E.H. Autologous bone marrow-derived mesenchymal stem cells versus autologous chondrocyte implantation: An observational cohort study. Am. J. Sports Med. 2010, 38, 1110–1116. [Google Scholar] [CrossRef]
- Brittberg, M.; Lindahl, A.; Nilsson, A.; Ohlsson, C.; Isaksson, O.; Peterson, L. Treatment of deep cartilage defects in the knee with autologous chondrocyte transplantation. N. Engl. J. Med. 1994, 331, 889–895. [Google Scholar] [CrossRef]
- Friedenstein, A.J.; Latzinik, N.W.; Grosheva, A.G.; Gorskaya, U.F. Marrow microenvironment transfer by heterotopic transplantation of freshly isolated and cultured cells in porous sponges. Exp. Hematol. 1982, 10, 217–227. [Google Scholar]
- Martin, J.A.; Klingelhutz, A.J.; Moussavi-Harami, F.; Buckwalter, J.A. Effects of oxidative damage and telomerase activity on human articular cartilage chondrocyte senescence. J. Gerontol. A Biol. Sci. Med. Sci. 2004, 59, 324–337. [Google Scholar] [CrossRef] [Green Version]
- Pizzute, T.; Lynch, K.; Pei, M. Impact of tissue-specific stem cells on lineage-specific differentiation: A focus on the musculoskeletal system. Stem Cell Rev. Rep. 2015, 11, 119–132. [Google Scholar] [CrossRef] [Green Version]
- Jones, B.A.; Pei, M. Synovium-derived stem cells: A tissue-specific stem cell for cartilage engineering and regeneration. Tissue Eng. Part B Rev. 2012, 18, 301–311. [Google Scholar] [CrossRef]
- Cournil-Henrionnet, C.; Huselstein, C.; Wang, Y.; Galois, L.; Mainard, D.; Decot, V.; Netter, P.; Stoltz, J.F.; Muller, S.; Gillet, P.; et al. Phenotypic analysis of cell surface markers and gene expression of human mesenchymal stem cells and chondrocytes during monolayer expansion. Biorheology 2008, 45, 513–526. [Google Scholar] [CrossRef]
- Peterson, L.; Minas, T.; Brittberg, M.; Nilsson, A.; Sjögren-Jansson, E.; Lindahl, A. Two- to 9-year outcome after autologous chondrocyte transplantation of the knee. Clin. Orthop. Relat. Res. 2000, 374, 212–234. [Google Scholar] [CrossRef]
- Roberts, S.; McCall, I.W.; Darby, A.J.; Menage, J.; Evans, H.; Harrison, P.E.; Richardson, J.B. Autologous chondrocyte implantation for cartilage repair: Monitoring its success by magnetic resonance imaging and histology. Arthritis Res. 2003, 5, R60–R73. [Google Scholar] [CrossRef] [Green Version]
- Jayasuriya, C.T.; Twomey-Kozak, J.; Newberry, J.; Desai, S.; Feltman, P.; Franco, J.R.; Li, N.; Terek, R.; Ehrlich, M.G.; Owens, B.D. Human Cartilage-Derived Progenitors Resist Terminal Differentiation and Require CXCR4 Activation to Successfully Bridge Meniscus Tissue Tears. Stem. Cells 2019, 37, 102–114. [Google Scholar] [CrossRef] [Green Version]
- Jukes, J.M.; Both, S.K.; Leusink, A.; Sterk, L.M.; van Blitterswijk, C.A.; de Boer, J. Endochondral bone tissue engineering using embryonic stem cells. Proc. Natl. Acad. Sci. USA 2008, 105, 6840–6845. [Google Scholar] [CrossRef] [Green Version]
- Barry, F.; Boynton, R.E.; Liu, B.; Murphy, J.M. Chondrogenic differentiation of mesenchymal stem cells from bone marrow: Differentiation-dependent gene expression of matrix components. Exp. Cell Res. 2001, 268, 189–200. [Google Scholar] [CrossRef]
- Mueller, M.B.; Fischer, M.; Zellner, J.; Berner, A.; Dienstknecht, T.; Prantl, L.; Kujat, R.; Nerlich, M.; Tuan, R.S.; Angele, P. Hypertrophy in mesenchymal stem cell chondrogenesis: Effect of TGF-beta isoforms and chondrogenic conditioning. Cells Tissues Organs 2010, 192, 158–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, Y.; Feng, W.; Wu, Y.; Lv, H.; Jia, Y.; Jiang, D. Mechano-growth factor accelerates the proliferation and osteogenic differentiation of rabbit mesenchymal stem cells through the PI3K/AKT pathway. BMC Biochem. 2015, 16, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Z.; Jiang, L.; Xu, Y.; Li, H.; Xu, W.; Wu, S.; Wang, Y.; Tang, Z.; Lv, Y.; Yang, L. Mechano growth factor (MGF) and transforming growth factor (TGF)-β3 functionalized silk scaffolds enhance articular hyaline cartilage regeneration in rabbit model. Biomaterials 2015, 52, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Zhang, B.; Wang, K.; Liu, F.; Xiao, H.; Zhao, J.; Liu, P.; Li, Y.; Lin, F.; Wang, Y. Mechano growth factor E peptide promotes osteoblasts proliferation and bone-defect healing in rabbits. Int. Orthop. 2011, 35, 1099–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Xu, K.; Yu, C.; Dong, L.; Chen, P.; Lv, Y.; Chiang, M.Y.M.; Li, L.; Liu, W.; Yang, L. The use of mechano growth factor to prevent cartilage degeneration in knee osteoarthritis. J. Tissue Eng. Regen. Med. 2018, 12, 738–749. [Google Scholar] [CrossRef]
- Caron, M.M.; Emans, P.J.; Cremers, A.; Surtel, D.A.; Coolsen, M.M.; van Rhijn, L.W.; Welting, T.J. Hypertrophic differentiation during chondrogenic differentiation of progenitor cells is stimulated by BMP-2 but suppressed by BMP-7. Osteoarthr. Cartil. 2013, 21, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Krase, A.; Abedian, R.; Steck, E.; Hurschler, C.; Richter, W. BMP activation and Wnt-signalling affect biochemistry and functional biomechanical properties of cartilage tissue engineering constructs. Osteoarthr. Cartil. 2014, 22, 284–292. [Google Scholar] [CrossRef] [Green Version]
- Crecente-Campo, J.; Borrajo, E.; Vidal, A.; Garcia-Fuentes, M. New scaffolds encapsulating TGF-β3/BMP-7 combinations driving strong chondrogenic differentiation. Eur. J. Pharm. Biopharm. 2017, 114, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Caron, M.M.J.; Ripmeester, E.G.J.; van den Akker, G.; Wijnands, N.; Steijns, J.; Surtel, D.A.M.; Cremers, A.; Emans, P.J.; van Rhijn, L.W.; Welting, T.J.M. Discovery of bone morphogenetic protein 7-derived peptide sequences that attenuate the human osteoarthritic chondrocyte phenotype. Mol. Methods Clin. Dev. 2021, 21, 247–261. [Google Scholar] [CrossRef]
- Kronenberg, H.M. PTHrP and skeletal development. Ann. N. Y. Acad. Sci. 2006, 1068, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Kim, H.J.; Im, G.I. PTHrP promotes chondrogenesis and suppresses hypertrophy from both bone marrow-derived and adipose tissue-derived MSCs. Biochem. Biophys. Res. Commun. 2008, 373, 104–108. [Google Scholar] [CrossRef]
- Mueller, M.B.; Fischer, M.; Zellner, J.; Berner, A.; Dienstknecht, T.; Kujat, R.; Prantl, L.; Nerlich, M.; Tuan, R.S.; Angele, P. Effect of parathyroid hormone-related protein in an in vitro hypertrophy model for mesenchymal stem cell chondrogenesis. Int. Orthop. 2013, 37, 945–951. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Im, G.I. PTHrP isoforms have differing effect on chondrogenic differentiation and hypertrophy of mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2012, 421, 819–824. [Google Scholar] [CrossRef]
- Kafienah, W.; Mistry, S.; Dickinson, S.C.; Sims, T.J.; Learmonth, I.; Hollander, A.P. Three-dimensional cartilage tissue engineering using adult stem cells from osteoarthritis patients. Arthritis Rheum. 2007, 56, 177–187. [Google Scholar] [CrossRef]
- Varghese, S.; Hwang, N.S.; Canver, A.C.; Theprungsirikul, P.; Lin, D.W.; Elisseeff, J. Chondroitin sulfate based niches for chondrogenic differentiation of mesenchymal stem cells. Matrix Biol. 2008, 27, 12–21. [Google Scholar] [CrossRef]
- Wu, Y.N.; Yang, Z.; Hui, J.H.; Ouyang, H.W.; Lee, E.H. Cartilaginous ECM component-modification of the micro-bead culture system for chondrogenic differentiation of mesenchymal stem cells. Biomaterials 2007, 28, 4056–4067. [Google Scholar] [CrossRef]
- Pei, M.; Luo, J.; Chen, Q. Enhancing and maintaining chondrogenesis of synovial fibroblasts by cartilage extracellular matrix protein matrilins. Osteoarthr. Cartil. 2008, 16, 1110–1117. [Google Scholar] [CrossRef] [Green Version]
- Jayasuriya, C.T.; Goldring, M.B.; Terek, R.; Chen, Q. Matrilin-3 induction of IL-1 receptor antagonist is required for up-regulating collagen II and aggrecan and down-regulating ADAMTS-5 gene expression. Arthritis Res. 2012, 14, R197. [Google Scholar] [CrossRef] [Green Version]
- Jayasuriya, C.T.; Zhou, F.H.; Pei, M.; Wang, Z.; Lemme, N.J.; Haines, P.; Chen, Q. Matrilin-3 chondrodysplasia mutations cause attenuated chondrogenesis, premature hypertrophy and aberrant response to TGF-β in chondroprogenitor cells. Int. J. Mol. Sci. 2014, 15, 14555–14573. [Google Scholar] [CrossRef] [Green Version]
- Hirvensalo, E.; Böstman, O.; Partio, E.; Törmälä, P.; Rokkanen, P. Fracture of the humeral capitellum fixed with absorbable polyglycolide pins. 1-year follow-up of 8 adults. Acta Orthop. Scand. 1993, 64, 85–86. [Google Scholar] [CrossRef] [Green Version]
- Böstman, O.; Mäkelä, E.A.; Södergård, J.; Hirvensalo, E.; Törmälä, P.; Rokkanen, P. Absorbable polyglycolide pins in internal fixation of fractures in children. J. Pediatr. Orthop. 1993, 13, 242–245. [Google Scholar] [PubMed]
- Kreuz, P.C.; Müller, S.; Ossendorf, C.; Kaps, C.; Erggelet, C. Treatment of focal degenerative cartilage defects with polymer-based autologous chondrocyte grafts: Four-year clinical results. Arthritis Res. 2009, 11, R33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreuz, P.C.; Müller, S.; Freymann, U.; Erggelet, C.; Niemeyer, P.; Kaps, C.; Hirschmüller, A. Repair of focal cartilage defects with scaffold-assisted autologous chondrocyte grafts: Clinical and biomechanical results 48 months after transplantation. Am. J. Sports Med. 2011, 39, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Ramos, T.A.; Damanik, F.; Quang Le, B.; Wieringa, P.; Bennink, M.; van Blitterswijk, C.; de Boer, J.; Moroni, L. A combinatorial approach towards the design of nanofibrous scaffolds for chondrogenesis. Sci. Rep. 2015, 5, 14804. [Google Scholar] [CrossRef] [Green Version]
- Ahtiainen, K.; Sippola, L.; Nurminen, M.; Mannerström, B.; Haimi, S.; Suuronen, R.; Hyttinen, J.; Ylikomi, T.; Kellomäki, M.; Miettinen, S. Effects of chitosan and bioactive glass modifications of knitted and rolled polylactide-based 96/4 L/D scaffolds on chondrogenic differentiation of adipose stem cells. J. Tissue Eng. Regen. Med. 2015, 9, 55–65. [Google Scholar] [CrossRef]
- Skaalure, S.C.; Chu, S.; Bryant, S.J. An enzyme-sensitive PEG hydrogel based on aggrecan catabolism for cartilage tissue engineering. Adv. Healthc. Mater. 2015, 4, 420–431. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhang, F.; Tsang, W.P.; Wan, C.; Wu, C. Fabrication of injectable high strength hydrogel based on 4-arm star PEG for cartilage tissue engineering. Biomaterials 2017, 120, 11–21. [Google Scholar] [CrossRef]
- Mohan, N.; Mohanan, P.V.; Sabareeswaran, A.; Nair, P. Chitosan-hyaluronic acid hydrogel for cartilage repair. Int. J. Biol. Macromol. 2017, 104, 1936–1945. [Google Scholar] [CrossRef]
- Zhao, M.; Chen, Z.; Liu, K.; Wan, Y.Q.; Li, X.D.; Luo, X.W.; Bai, Y.G.; Yang, Z.L.; Feng, G. Repair of articular cartilage defects in rabbits through tissue-engineered cartilage constructed with chitosan hydrogel and chondrocytes. J. Zhejiang Univ. Sci. B 2015, 16, 914–923. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Seitz, D.; Chevalier, Y.; Müller, P.E.; Jansson, V.; Klar, R.M. Synergistic interaction of hTGF-β(3) with hBMP-6 promotes articular cartilage formation in chitosan scaffolds with hADSCs: Implications for regenerative medicine. BMC Biotechnol. 2020, 20, 48. [Google Scholar] [CrossRef]
- Huang, S.; Song, X.; Li, T.; Xiao, J.; Chen, Y.; Gong, X.; Zeng, W.; Yang, L.; Chen, C. Pellet coculture of osteoarthritic chondrocytes and infrapatellar fat pad-derived mesenchymal stem cells with chitosan/hyaluronic acid nanoparticles promotes chondrogenic differentiation. Stem. Cell Res. 2017, 8, 264. [Google Scholar] [CrossRef]
- Manferdini, C.; Gabusi, E.; Sartore, L.; Dey, K.; Agnelli, S.; Almici, C.; Bianchetti, A.; Zini, N.; Russo, D.; Re, F.; et al. Chitosan-based scaffold counteracts hypertrophic and fibrotic markers in chondrogenic differentiated mesenchymal stromal cells. J. Tissue Eng. Regen. Med. 2019, 13, 1896–1911. [Google Scholar] [CrossRef]
- Li, J.; Dong, S. The Signaling Pathways Involved in Chondrocyte Differentiation and Hypertrophic Differentiation. Stem. Cells Int. 2016, 2016, 2470351. [Google Scholar] [CrossRef] [Green Version]
- Leijten, J.; Georgi, N.; Moreira Teixeira, L.; van Blitterswijk, C.A.; Post, J.N.; Karperien, M. Metabolic programming of mesenchymal stromal cells by oxygen tension directs chondrogenic cell fate. Proc. Natl. Acad. Sci. USA 2014, 111, 13954–13959. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Fukai, A.; Mabuchi, A.; Ikeda, T.; Yano, F.; Ohba, S.; Nishida, N.; Akune, T.; Yoshimura, N.; Nakagawa, T.; et al. Transcriptional regulation of endochondral ossification by HIF-2alpha during skeletal growth and osteoarthritis development. Nat. Med. 2010, 16, 678–686. [Google Scholar] [CrossRef]
- Acharya, C.; Adesida, A.; Zajac, P.; Mumme, M.; Riesle, J.; Martin, I.; Barbero, A. Enhanced chondrocyte proliferation and mesenchymal stromal cells chondrogenesis in coculture pellets mediate improved cartilage formation. J. Cell. Physiol. 2012, 227, 88–97. [Google Scholar] [CrossRef]
- McCorry, M.C.; Puetzer, J.L.; Bonassar, L.J. Characterization of mesenchymal stem cells and fibrochondrocytes in three-dimensional co-culture: Analysis of cell shape, matrix production, and mechanical performance. Stem. Cell Res. 2016, 7, 39. [Google Scholar] [CrossRef] [Green Version]
- Studer, D.; Millan, C.; Öztürk, E.; Maniura-Weber, K.; Zenobi-Wong, M. Molecular and biophysical mechanisms regulating hypertrophic differentiation in chondrocytes and mesenchymal stem cells. Eur. Cell Mater. 2012, 24, 118–135; discussion 135. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shigley, C.; Trivedi, J.; Meghani, O.; Owens, B.D.; Jayasuriya, C.T. Suppressing Chondrocyte Hypertrophy to Build Better Cartilage. Bioengineering 2023, 10, 741. https://doi.org/10.3390/bioengineering10060741
Shigley C, Trivedi J, Meghani O, Owens BD, Jayasuriya CT. Suppressing Chondrocyte Hypertrophy to Build Better Cartilage. Bioengineering. 2023; 10(6):741. https://doi.org/10.3390/bioengineering10060741
Chicago/Turabian StyleShigley, Christian, Jay Trivedi, Ozair Meghani, Brett D. Owens, and Chathuraka T. Jayasuriya. 2023. "Suppressing Chondrocyte Hypertrophy to Build Better Cartilage" Bioengineering 10, no. 6: 741. https://doi.org/10.3390/bioengineering10060741
APA StyleShigley, C., Trivedi, J., Meghani, O., Owens, B. D., & Jayasuriya, C. T. (2023). Suppressing Chondrocyte Hypertrophy to Build Better Cartilage. Bioengineering, 10(6), 741. https://doi.org/10.3390/bioengineering10060741