From Human Pluripotent Stem Cells to 3D Cardiac Microtissues: Progress, Applications and Challenges




Abstract
:1. Introduction
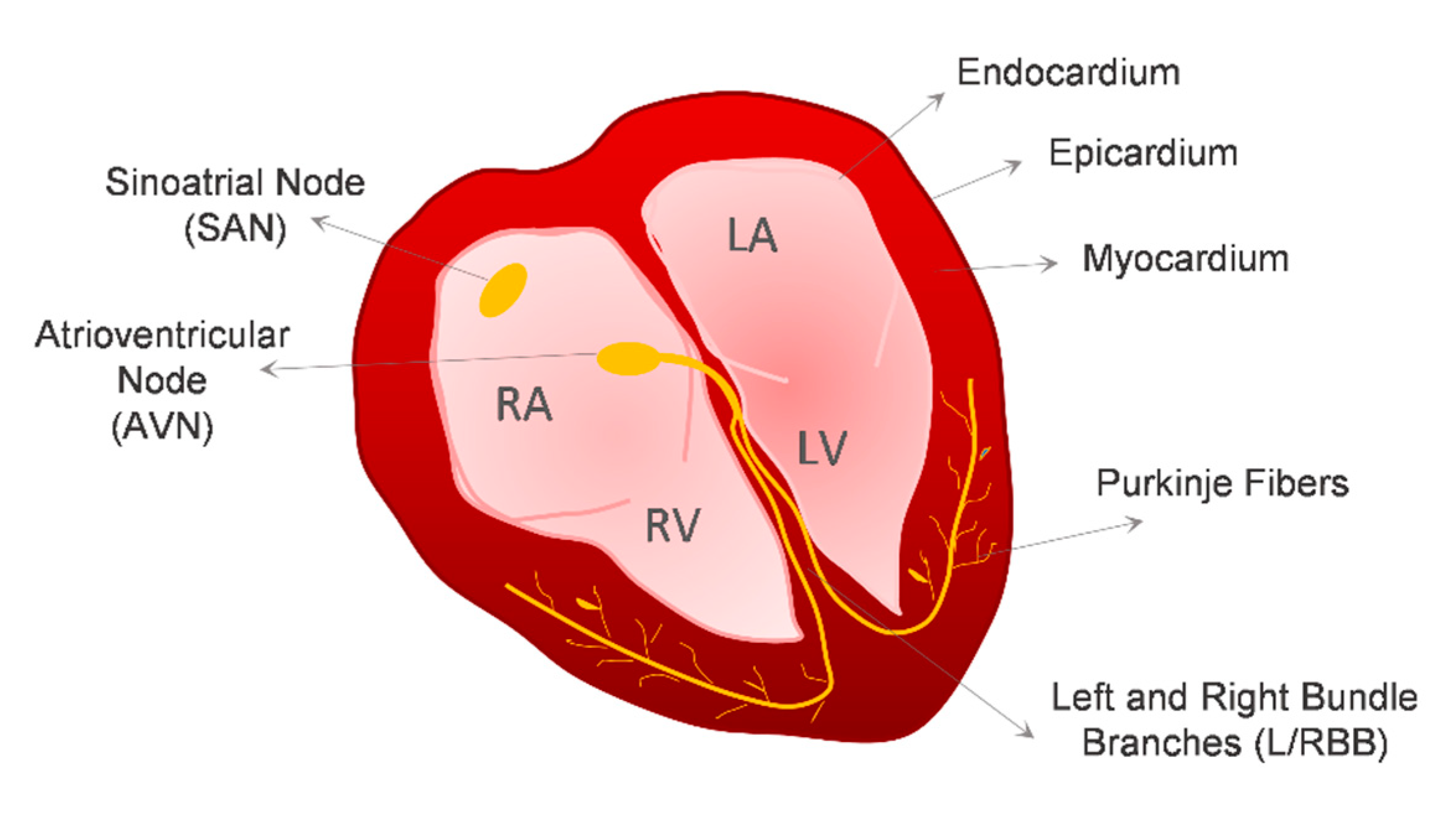
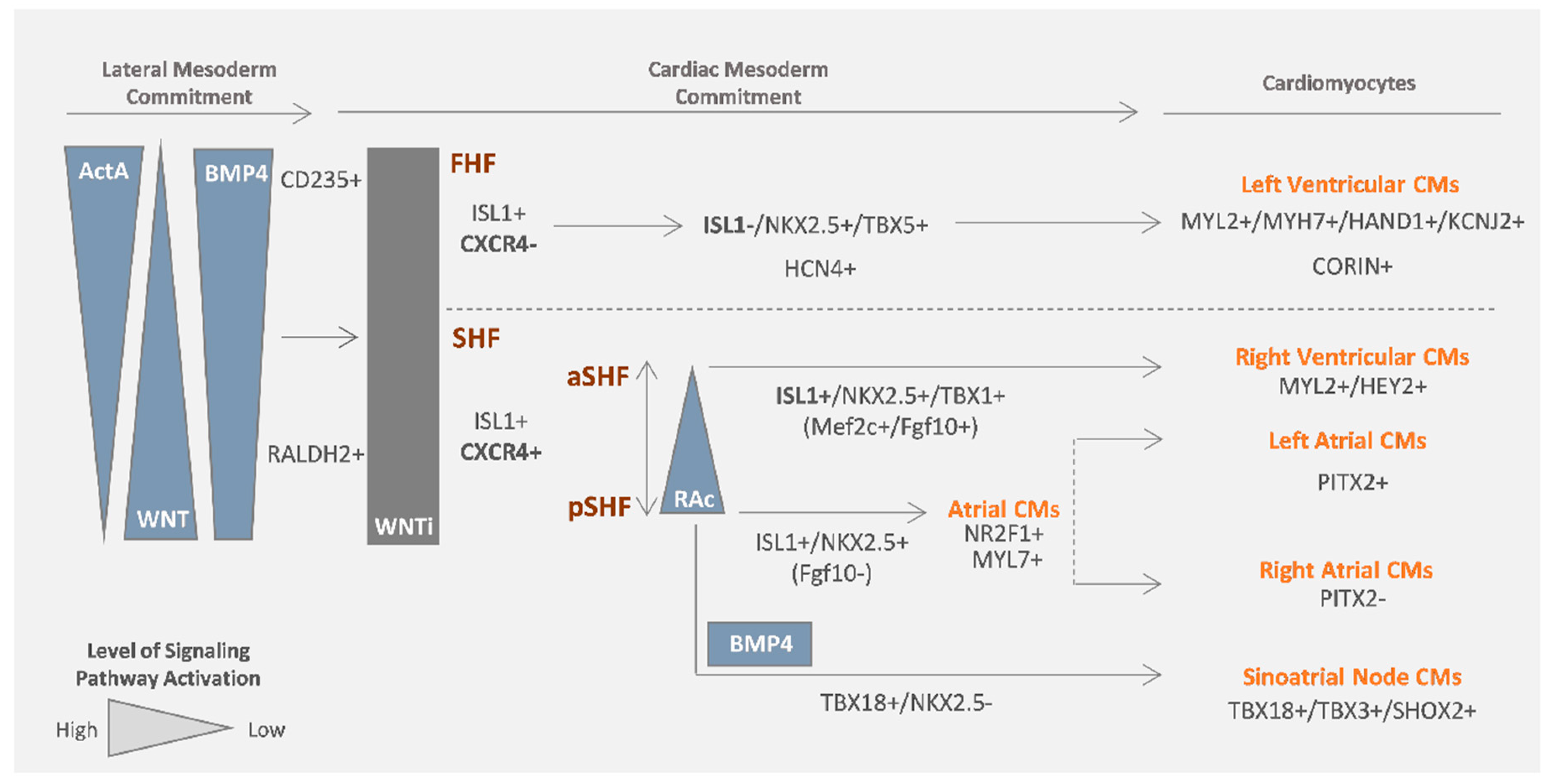
2. Cardiovascular Lineages Specification from hPSCs—Lessons from In Vivo Heart Development
2.1. Cardiomyocytes
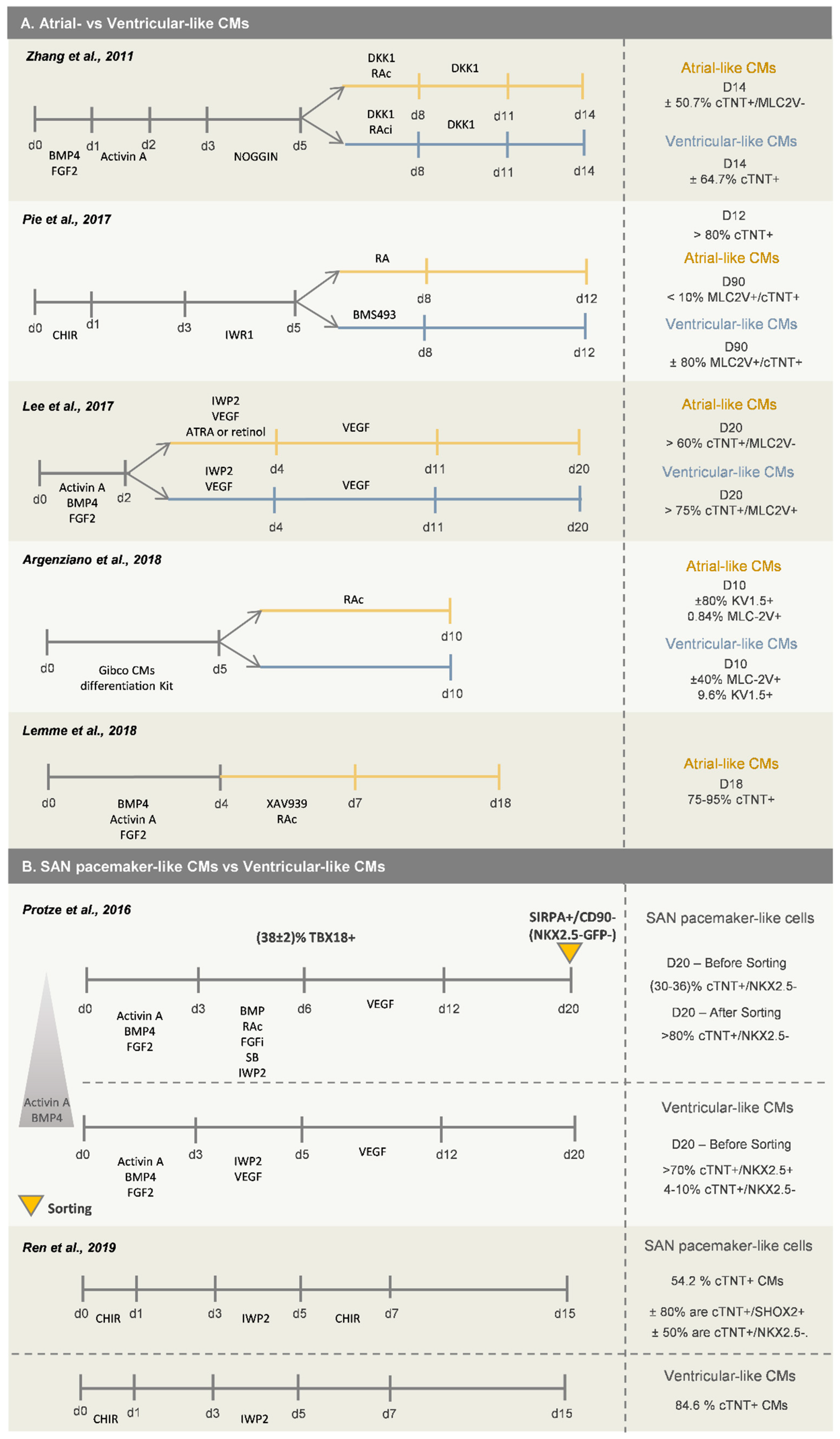
2.1.1. Atrial- Versus Ventricular-like Cardiomyocytes
2.1.2. SAN Cardiomyocytes
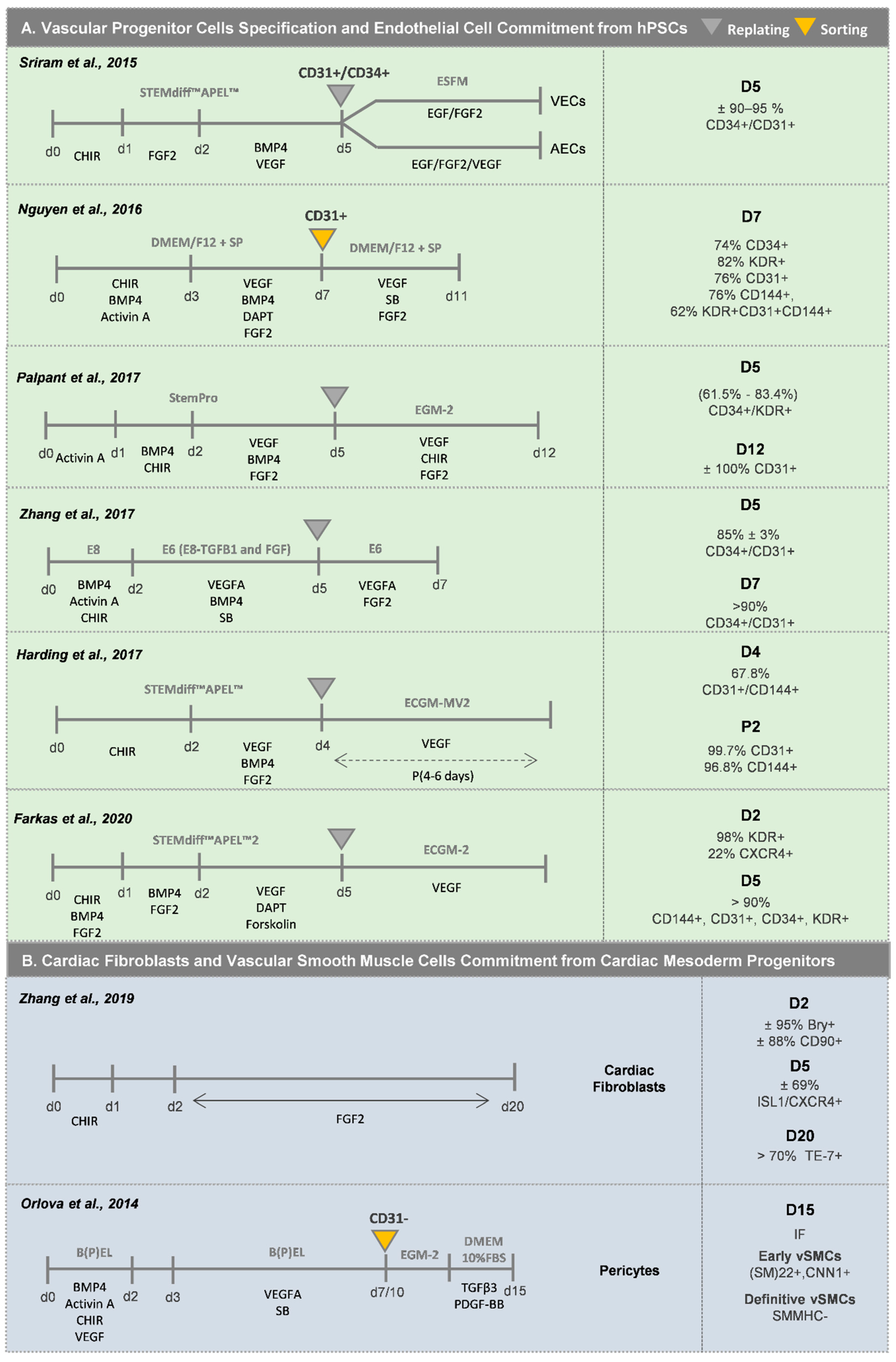
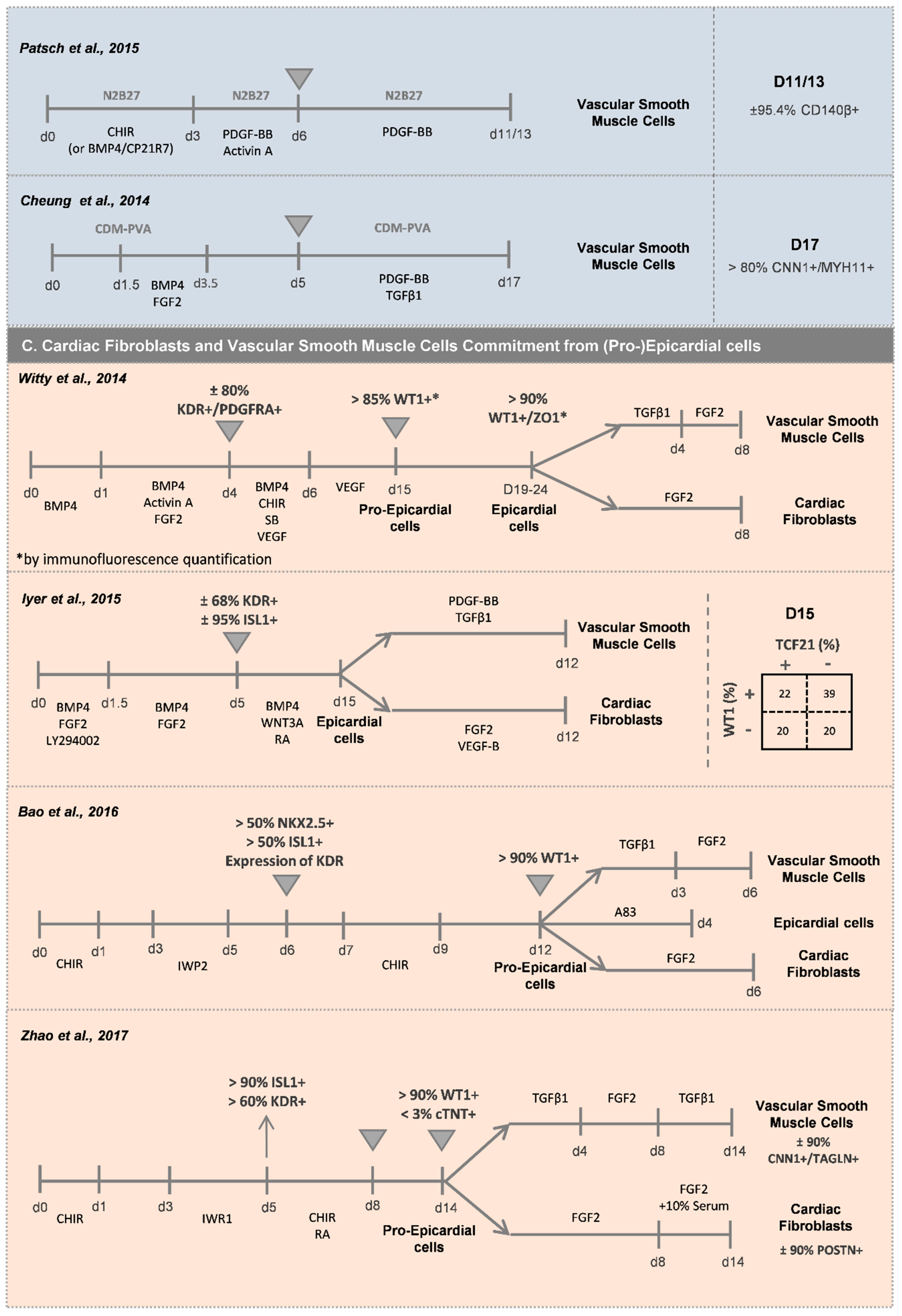
2.2. Vascular Cardiac Cells
2.2.1. Endothelial Cells (ECs)
2.2.2. Vascular Smooth Muscle Cells (vSMCs)
2.3. Cardiac Fibroblasts (CFs)
2.4. Epicardial Cells
3. Impact of 3D Environment on Cardiomyocyte Differentiation of hPSCs
4. Engineering 3D Cardiac Microtissues to Better Mimic the Human Heart Environment
4.1. 3D Hydrogel-Based Engineered Heart Tissues (EHT)
4.2. 3D Scaffold-Free Multicellular Cardiac Microtissues (MTs)
5. In Vitro Applications of hPSC-Derived 3D Cardiac Microtissues (MTs)
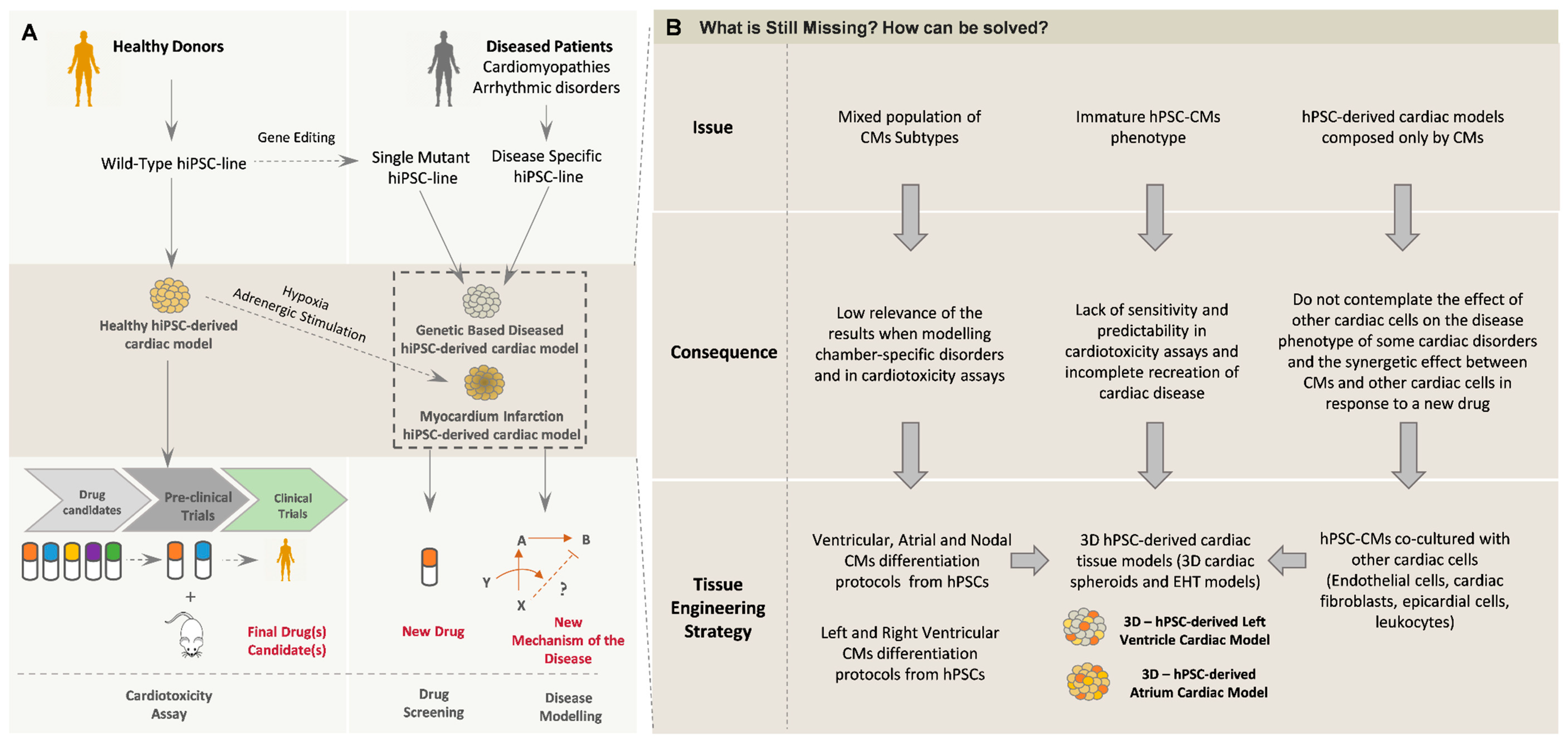
5.1. In Vitro Modeling of Cardiac Disorders—Challenges and Perspectives
5.2. Cardiotoxicity Tests and Drug Screening
6. Conclusions and Future Trends
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Camelliti, P.; Borg, T.K.; Kohl, P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc. Res. 2005, 65, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brutsaert, D.L. Cardiac Endothelial-Myocardial Signaling: Its Role in Cardiac Growth, Contractile Performance, and Rhythmicity. Physiol. Rev. 2003, 83, 59–115. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 22 May 2020).
- Batho, C.A.P.; Mills, R.J.; Hudson, J.E. Metabolic Regulation of Human Pluripotent Stem Cell-Derived Cardiomyocyte Maturation. Curr. Cardiol. Rep. 2020, 22. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.E.; Anzai, T.; Chanthra, N.; Uosaki, H. A Brief Review of Current Maturation Methods for Human Induced Pluripotent Stem Cells-Derived Cardiomyocytes. Front. Cell Dev. Biol. 2020, 8, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, K.M.M.; Chen, A.; Koh, P.W.W.; Deng, T.Z.Z.; Sinha, R.; Tsai, J.M.M.; Barkal, A.A.A.; Shen, K.Y.Y.; Jain, R.; Morganti, R.M.M.; et al. Mapping the Pairwise Choices Leading from Pluripotency to Human Bone, Heart, and Other Mesoderm Cell Types. Cell 2016, 166, 451–467. [Google Scholar] [CrossRef] [Green Version]
- Loh, K.M.; Ang, L.T.; Zhang, J.; Kumar, V.; Ang, J.; Auyeong, J.Q.; Lee, K.L.; Choo, S.H.; Lim, C.Y.Y.; Nichane, M.; et al. Efficient endoderm induction from human pluripotent stem cells by logically directing signals controlling lineage bifurcations. Cell Stem Cell 2014, 14, 237–252. [Google Scholar] [CrossRef] [Green Version]
- Rao, J.; Pfeiffer, M.J.; Frank, S.; Adachi, K.; Piccini, I.; Quaranta, R.; Araúzo-Bravo, M.; Schwarz, J.; Schade, D.; Leidel, S.; et al. Stepwise Clearance of Repressive Roadblocks Drives Cardiac Induction in Human ESCs. Cell Stem Cell 2016, 18, 341–353. [Google Scholar] [CrossRef] [Green Version]
- Kattman, S.J.; Witty, A.D.; Gagliardi, M.; Dubois, N.C.; Niapour, M.; Hotta, A.; Ellis, J.; Keller, G. Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell 2011, 8, 228–240. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Klos, M.; Wilson, G.F.; Herman, A.M.; Lian, X.; Raval, K.K.; Barron, M.R.; Hou, L.; Soerens, A.G.; Yu, J.; et al. Extracellular Matrix Promotes Highly Efficient Cardiac Differentiation of Human Pluripotent Stem Cells. Circ. Res. 2012, 111, 1125–1136. [Google Scholar] [CrossRef]
- Hudson, J.; Titmarsh, D.; Hidalgo, A.; Wolvetang, E.; Cooper-White, J. Primitive Cardiac Cells from Human Embryonic Stem Cells. Stem Cells Dev. 2012, 21, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Uosaki, H.; Fukushima, H.; Takeuchi, A.; Matsuoka, S.; Nakatsuji, N.; Yamanaka, S.; Yamashita, J.K. Efficient and Scalable Purification of Cardiomyocytes from Human Embryonic and Induced Pluripotent Stem Cells by VCAM1 Surface Expression. PLoS ONE 2011, 6, e23657. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Hsiao, C.; Wilson, G.; Zhu, K.; Hazeltine, L.B.; Azarin, S.M.; Raval, K.K.; Zhang, J.; Kamp, T.J.; Palecek, S.P. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1848–E1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Tang, Y.; Zhou, Y.; Zhang, J. Deciphering Role of Wnt Signalling in Cardiac Mesoderm and Cardiomyocyte Differentiation from Human iPSCs: Four-dimensional control of Wnt pathway for hiPSC-CMs differentiation. Sci. Rep. 2019, 9, 19389. [Google Scholar] [CrossRef]
- Halloin, C.; Schwanke, K.; Löbel, W.; Franke, A.; Szepes, M.; Biswanath, S.; Wunderlich, S.; Merkert, S.; Weber, N.; Osten, F.; et al. Continuous WNT Control Enables Advanced hPSC Cardiac Processing and Prognostic Surface Marker Identification in Chemically Defined Suspension Culture. Stem Cell Rep. 2019, 13, 366–379. [Google Scholar] [CrossRef] [Green Version]
- Kempf, H.; Olmer, R.; Haase, A.; Franke, A.; Bolesani, E.; Schwanke, K.; Robles-Diaz, D.; Coffee, M.; Göhring, G.; Dräger, G.; et al. Bulk cell density and Wnt/TGFbeta signalling regulate mesendodermal patterning of human pluripotent stem cells. Nat. Commun. 2016, 7, 13602. [Google Scholar] [CrossRef] [Green Version]
- Burridge, P.W.; Thompson, S.; Millrod, M.A.; Weinberg, S.; Yuan, X.; Peters, A.; Mahairaki, V.; Koliatsos, V.E.; Tung, L.; Zambidis, E.T. A Universal System for Highly Efficient Cardiac Differentiation of Human Induced Pluripotent Stem Cells That Eliminates Interline Variability. PLoS ONE 2011, 6, e18293. [Google Scholar] [CrossRef]
- Laco, F.; Woo, T.L.; Zhong, Q.; Szmyd, R.; Ting, S.; Khan, F.J.; Chai, C.L.L.; Reuveny, S.; Chen, A.; Oh, S. Unraveling the Inconsistencies of Cardiac Differentiation Efficiency Induced by the GSK3β Inhibitor CHIR99021 in Human Pluripotent Stem Cells. Stem Cell Rep. 2018, 10, 1851–1866. [Google Scholar] [CrossRef] [Green Version]
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M.; et al. Chemically defined generation of human cardiomyocytes. Nat. Methods 2014, 11, 855–860. [Google Scholar] [CrossRef] [Green Version]
- Lian, X.; Bao, X.; Zilberter, M.; Westman, M.; Fisahn, A.; Hsiao, C.; Hazeltine, L.B.; Dunn, K.K.; Kamp, T.J.; Palecek, S.P. Chemically defined, albumin-free human cardiomyocyte generation. Nat. Methods 2015, 12, 595–596. [Google Scholar] [CrossRef]
- Cai, C.-L.; Liang, X.; Shi, Y.; Chu, P.-H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef] [Green Version]
- Galdos, F.X.; Guo, Y.; Paige, S.L.; Vandusen, N.J.; Wu, S.M.; Pu, W.T. Cardiac Regeneration: Lessons from Development. Circ. Res. 2017, 120, 941–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Später, D.; Abramczuk, M.K.; Buac, K.; Zangi, L.; Stachel, M.W.; Clarke, J.; Sahara, M.; Ludwig, A.; Chien, K.R. A HCN4+ cardiomyogenic progenitor derived from the first heart field and human pluripotent stem cells. Nat. Cell Biol. 2013, 15, 1098–1106. [Google Scholar] [CrossRef]
- Bruneau, B.G.; Logan, M.; Davis, N.; Levi, T.; Tabin, C.J.; Seidman, J.G.; Seidman, C.E. Chamber-specific cardiac expression of Tbx5 and heart defects in Holt- Oram syndrome. Dev. Biol. 1999, 211, 100–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meilhac, S.M.; Esner, M.; Kelly, R.G.; Nicolas, J.F.; Buckingham, M.E. The clonal origin of myocardial cells in different regions of the embryonic mouse heart. Dev. Cell 2004, 6, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Andersen, P.; Tampakakis, E.; Jimenez, D.V.; Kannan, S.; Miyamoto, M.; Shin, H.K.; Saberi, A.; Murphy, S.; Sulistio, E.; Chelko, S.P.; et al. Precardiac organoids form two heart fields via Bmp/Wnt signaling. Nat. Commun. 2018, 9, 3140. [Google Scholar] [CrossRef]
- Huynh, T.; Chen, L.; Terrell, P.; Baldini, A. A fate map of Tbx1 expressing cells reveals heterogeneity in the second cardiac field. Genesis 2007, 45, 470–475. [Google Scholar] [CrossRef]
- Zhang, J.Z.; Termglinchan, V.; Shao, N.Y.; Itzhaki, I.; Liu, C.; Ma, N.; Tian, L.; Wang, V.Y.; Chang, A.C.Y.; Guo, H.; et al. A Human iPSC Double-Reporter System Enables Purification of Cardiac Lineage Subpopulations with Distinct Function and Drug Response Profiles. Cell Stem Cell 2019, 24, 802–811. [Google Scholar] [CrossRef]
- Galli, D.; Domínguez, J.N.; Zaffran, S.; Munk, A.; Brown, N.A.; Buckingham, M.E. Atrial myocardium derives from the posterior region of the second heart field, which acquires left-right identity as Pitx2c is expressed. Development 2008, 135, 1157–1167. [Google Scholar] [CrossRef] [Green Version]
- Verzi, M.P.; McCulley, D.J.; De Val, S.; Dodou, E.; Black, B.L. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev. Biol. 2005, 287, 134–145. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Protze, S.I.; Laksman, Z.; Backx, P.H.; Keller, G.M. Human Pluripotent Stem Cell-Derived Atrial and Ventricular Cardiomyocytes Develop from Distinct Mesoderm Populations. Cell Stem Cell 2017, 21, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Protze, S.I.; Liu, J.; Nussinovitch, U.; Ohana, L.; Backx, P.H.; Gepstein, L.; Keller, G.M. Sinoatrial node cardiomyocytes derived from human pluripotent cells function as a biological pacemaker. Nat. Biotechnol. 2017, 35, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zheng, Y.; Liu, X.; Yan, L.; Fan, X.; Yong, J.; Hu, Y.; Dong, J.; Li, Q.; Wu, X.; et al. Single-Cell Transcriptome Analysis Maps the Developmental Track of the Human Heart. Cell Rep. 2019, 26, 1934–1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryckebusch, L.; Wang, Z.; Bertrand, N.; Lin, S.C.; Chi, X.; Schwartz, R.; Zaffran, S.; Niederreither, K. Retinoic acid deficiency alters second heart field formation. Proc. Natl. Acad. Sci. USA 2008, 105, 2913–2918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, F.; Jiang, J.; Bai, S.; Cao, H.; Tian, L.; Zhao, Y.; Yang, C.; Dong, H.; Ma, Y. Chemical-defined and albumin-free generation of human atrial and ventricular myocytes from human pluripotent stem cells. Stem Cell Res. 2017, 19, 94–103. [Google Scholar] [CrossRef]
- Zhang, Q.; Jiang, J.; Han, P.; Yuan, Q.; Zhang, J.; Zhang, X.; Xu, Y.; Cao, H.; Meng, Q.; Chen, L.; et al. Direct differentiation of atrial and ventricular myocytes from human embryonic stem cells by alternating retinoid signals. Cell Res. 2011, 21, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Lemme, M.; Ulmer, B.M.; Lemoine, M.D.; Zech, A.T.L.; Flenner, F.; Ravens, U.; Reichenspurner, H.; Rol-Garcia, M.; Smith, G.; Hansen, A.; et al. Atrial-like Engineered Heart Tissue: An In Vitro Model of the Human Atrium. Stem Cell Rep. 2018, 11, 1378–1390. [Google Scholar] [CrossRef] [Green Version]
- Devalla, H.D.; Schwach, V.; Ford, J.W.; Milnes, J.T.; El-Haou, S.; Jackson, C.; Gkatzis, K.; Elliott, D.A.; Chuva de Sousa Lopes, S.M.; Mummery, C.L.; et al. Atrial-like cardiomyocytes from human pluripotent stem cells are a robust preclinical model for assessing atrial-selective pharmacology. EMBO Mol. Med. 2015, 7, 394–410. [Google Scholar] [CrossRef]
- Foo, K.S.; Lehtinen, M.L.; Leung, C.Y.; Lian, X.; Xu, J.; Keung, W.; Geng, L.; Kolstad, T.R.S.; Thams, S.; Wong, A.O.; et al. Human ISL1 + Ventricular Progenitors Self-Assemble into an In Vivo Functional Heart Patch and Preserve Cardiac Function Post Infarction. Mol. Ther. 2018, 26, 1644–1659. [Google Scholar] [CrossRef] [Green Version]
- Veevers, J.; Farah, E.N.; Corselli, M.; Witty, A.D.; Palomares, K.; Vidal, J.G.; Emre, N.; Carson, C.T.; Ouyang, K.; Liu, C.; et al. Cell-Surface Marker Signature for Enrichment of Ventricular Cardiomyocytes Derived from Human Embryonic Stem Cells. Stem Cell Rep. 2018, 11, 828–841. [Google Scholar] [CrossRef] [Green Version]
- Ishida, H.; Saba, R.; Kokkinopoulos, I.; Hashimoto, M.; Yamaguchi, O.; Nowotschin, S.; Shiraishi, M.; Ruchaya, P.; Miller, D.; Harmer, S.; et al. GFRA2 Identifies Cardiac Progenitors and Mediates Cardiomyocyte Differentiation in a RET-Independent Signaling Pathway. Cell Rep. 2016, 16, 1026–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, M.; Kanki, Y.; Masumoto, H.; Funakoshi, S.; Hatani, T.; Fukushima, H.; Izumi-Taguchi, A.; Matsui, Y.; Shimamura, T.; Yoshida, Y.; et al. Identification of Cardiomyocyte-Fated Progenitors from Human-Induced Pluripotent Stem Cells Marked with CD82. Cell Rep. 2018, 22, 546–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mommersteeg, M.T.M.; Domínguez, J.N.; Wiese, C.; Norden, J.; De Gier-De Vries, C.; Burch, J.B.E.; Kispert, A.; Brown, N.A.; Moorman, A.F.M.; Christoffels, V.M. The sinus venosus progenitors separate and diversify from the first and second heart fields early in development. Cardiovasc. Res. 2010, 87, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protze, S.I.; Lee, J.H.; Keller, G.M. Human Pluripotent Stem Cell-Derived Cardiovascular Cells: From Developmental Biology to Therapeutic Applications. Cell Stem Cell 2019, 25, 311–327. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Han, P.; Ma, X.; Farah, E.N.; Bloomekatz, J.; Zeng, X.X.I.; Zhang, R.; Swim, M.M.; Witty, A.D.; Knight, H.G.; et al. Canonical Wnt5b Signaling Directs Outlying Nkx2.5+ Mesoderm into Pacemaker Cardiomyocytes. Dev. Cell 2019, 50, 729–743. [Google Scholar] [CrossRef]
- Sahara, M.; Hansson, E.M.; Wernet, O.; Lui, K.O.; Später, D.; Chien, K.R. Manipulation of a VEGF-Notch signaling circuit drives formation of functional vascular endothelial progenitors from human pluripotent stem cells. Cell Res. 2014, 24, 820–841. [Google Scholar] [CrossRef] [Green Version]
- Patsch, C.; Challet-Meylan, L.; Thoma, E.C.; Urich, E.; Heckel, T.; O’Sullivan, J.F.; Grainger, S.J.; Kapp, F.G.; Sun, L.; Christensen, K.; et al. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat. Cell Biol. 2015, 17, 994–1003. [Google Scholar] [CrossRef]
- Zhang, J.; Schwartz, M.P.; Hou, Z.; Bai, Y.; Ardalani, H.; Swanson, S.; Steill, J.; Ruotti, V.; Elwell, A.; Nguyen, B.K.; et al. A Genome-wide Analysis of Human Pluripotent Stem Cell-Derived Endothelial Cells in 2D or 3D Culture. Stem Cell Rep. 2017, 8, 907–918. [Google Scholar] [CrossRef]
- Harding, A.; Cortez-Toledo, E.; Magner, N.L.; Beegle, J.R.; Coleal-Bergum, D.P.; Hao, D.; Wang, A.; Nolta, J.A.; Zhou, P. Highly Efficient Differentiation of Endothelial Cells from Pluripotent Stem Cells Requires the MAPK and the PI3K Pathways. Stem Cells 2017, 35, 909–919. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.T.X.; Okina, E.; Chai, X.; Tan, K.H.; Hovatta, O.; Ghosh, S.; Tryggvason, K. Differentiation of Human Embryonic Stem Cells to Endothelial Progenitor Cells on Laminins in Defined and Xeno-free Systems. Stem Cell Rep. 2016, 7, 802–816. [Google Scholar] [CrossRef] [Green Version]
- Palpant, N.J.; Pabon, L.; Friedman, C.E.; Roberts, M.; Hadland, B.; Zaunbrecher, R.J.; Bernstein, I.; Zheng, Y.; Murry, C.E. Generating high-purity cardiac and endothelial derivatives from patterned mesoderm using human pluripotent stem cells. Nat. Protoc. 2017, 12, 15–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriram, G.; Tan, J.Y.; Islam, I.; Rufaihah, A.J.; Cao, T. Efficient differentiation of human embryonic stem cells to arterial and venous endothelial cells under feeder- and serum-free conditions. Stem Cell Res. Ther. 2015, 6, 261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farkas, S.; Simara, P.; Rehakova, D.; Veverkova, L.; Koutna, I. Endothelial Progenitor Cells Produced From Human Pluripotent Stem Cells by a Synergistic Combination of Cytokines, Small Compounds, and Serum-Free Medium. Front. Cell Dev. Biol. 2020, 8, 309. [Google Scholar] [CrossRef] [PubMed]
- Orlova, V.V.; Van Den Hil, F.E.; Petrus-Reurer, S.; Drabsch, Y.; Ten Dijke, P.; Mummery, C.L. Generation, expansion and functional analysis of endothelial cells and pericytes derived from human pluripotent stem cells. Nat. Protoc. 2014, 9, 1514–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palpant, N.J.; Pabon, L.; Roberts, M.; Hadland, B.; Jones, D.; Jones, C.; Moon, R.T.; Ruzzo, W.L.; Bernstein, I.; Zheng, Y.; et al. Inhibition of β-catenin signaling respecifies anterior-like endothelium into beating human cardiomyocytes. Development 2015, 142, 3198–3209. [Google Scholar] [CrossRef] [Green Version]
- Cheung, C.; Bernardo, A.S.; Pedersen, R.A.; Sinha, S. Directed differentiation of embryonic origin-specific vascular smooth muscle subtypes from human pluripotent stem cells. Nat. Protoc. 2014, 9, 929–938. [Google Scholar] [CrossRef]
- Witty, A.D.; Mihic, A.; Tam, R.Y.; Fisher, S.A.; Mikryukov, A.; Shoichet, M.S.; Li, R.K.; Kattman, S.J.; Keller, G. Generation of the epicardial lineage from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 1026–1035. [Google Scholar] [CrossRef] [Green Version]
- Bao, X.; Lian, X.; Hacker, T.A.; Schmuck, E.G.; Qian, T.; Bhute, V.J.; Han, T.; Shi, M.; Drowley, L.; Plowright, A.T.; et al. Long-term self-renewing human epicardial cells generated from pluripotent stem cells under defined xeno-free conditions. Nat. Biomed. Eng. 2017, 1, 1–12. [Google Scholar] [CrossRef]
- Zhao, J.; Cao, H.; Tian, L.; Huo, W.; Zhai, K.; Wang, P.; Ji, G.; Ma, Y. Efficient Differentiation of TBX18+/WT1+ Epicardial-Like Cells from Human Pluripotent Stem Cells Using Small Molecular Compounds. Stem Cells Dev. 2017, 26, 528–540. [Google Scholar] [CrossRef] [Green Version]
- Iyer, D.; Gambardella, L.; Bernard, W.G.; Serrano, F.; Mascetti, V.L.; Pedersen, R.A.; Talasila, A.; Sinha, S. Robust derivation of epicardium and its differentiated smooth muscle cell progeny from human pluripotent stem cells. Development 2015, 142, 1528–1541. [Google Scholar] [CrossRef] [Green Version]
- Tallquist, M.D.; Molkentin, J.D. Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 2017, 14, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Moore-Morris, T.; Guimarães-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J. Clin. Investig. 2014, 124, 2921–2934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Tao, R.; Campbell, K.F.; Carvalho, J.L.; Ruiz, E.C.; Kim, G.C.; Schmuck, E.G.; Raval, A.N.; da Rocha, A.M.; Herron, T.J.; et al. Functional cardiac fibroblasts derived from human pluripotent stem cells via second heart field progenitors. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, T.C.; Singh, M.K.; Degenhardt, K.; Rivera-Feliciano, J.; Johnson, R.L.; Epstein, J.A.; Tabin, C.J. Distinct Compartments of the Proepicardial Organ Give Rise to Coronary Vascular Endothelial Cells. Dev. Cell 2012, 22, 639–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupu, I.-E.; Redpath, A.N.; Smart, N. Spatiotemporal Analysis Reveals Overlap of Key Proepicardial Markers in the Developing Murine Heart. Stem Cell Rep. 2020, 14, 770–787. [Google Scholar] [CrossRef]
- Cano, E.; Carmona, R.; Ruiz-Villalba, A.; Rojas, A.; Chau, Y.Y.; Wagner, K.D.; Wagner, N.; Hastie, N.D.; Muñoz-Chápuli, R.; Pérez-Pomares, J.M. Extracardiac septum transversum/proepicardial endothelial cells pattern embryonic coronary arterio-venous connections. Proc. Natl. Acad. Sci. USA 2016, 113, 656–661. [Google Scholar] [CrossRef] [Green Version]
- Niebruegge, S.; Bauwens, C.L.; Peerani, R.; Thavandiran, N.; Masse, S.; Sevaptisidis, E.; Nanthakumar, K.; Woodhouse, K.; Husain, M.; Kumacheva, E.; et al. Generation of human embryonic stem cell-derived mesoderm and cardiac cells using size-specified aggregates in an oxygen-controlled bioreactor. Biotechnol. Bioeng. 2009, 102, 493–507. [Google Scholar] [CrossRef]
- Burridge, P.W.; Anderson, D.; Priddle, H.; Barbadillo Muñoz, M.D.; Chamberlain, S.; Allegrucci, C.; Young, L.E.; Denning, C. Improved Human Embryonic Stem Cell Embryoid Body Homogeneity and Cardiomyocyte Differentiation from a Novel V-96 Plate Aggregation System Highlights Interline Variability. Stem Cells 2007, 25, 929–938. [Google Scholar] [CrossRef]
- Bauwens, C.L.; Peerani, R.; Niebruegge, S.; Woodhouse, K.A.; Kumacheva, E.; Husain, M.; Zandstra, P.W. Control of Human Embryonic Stem Cell Colony and Aggregate Size Heterogeneity Influences Differentiation Trajectories. Stem Cells 2008, 26, 2300–2310. [Google Scholar] [CrossRef]
- Mohr, J.C.; Zhang, J.; Azarin, S.M.; Soerens, A.G.; de Pablo, J.J.; Thomson, J.A.; Lyons, G.E.; Palecek, S.P.; Kamp, T.J. The microwell control of embryoid body size in order to regulate cardiac differentiation of human embryonic stem cells. Biomaterials 2010, 31, 1885–1893. [Google Scholar] [CrossRef] [Green Version]
- Hwang, Y.S.; Bong, G.C.; Ortmann, D.; Hattori, N.; Moeller, H.C.; Khademhosseinia, A. Microwell-mediated control of embryoid body size regulates embryonic stem cell fate via differential expression of WNT5a and WNT11. Proc. Natl. Acad. Sci. USA 2009, 106, 16978–16983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauwens, C.L.; Song, H.; Thavandiran, N.; Ungrin, M.; Massé, S.; Nanthakumar, K.; Seguin, C.; Zandstra, P.W. Geometric control of cardiomyogenic induction in human pluripotent stem cells. Tissue Eng. 2011, 17, 1901–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiao, C.; Tomai, M.; Glynn, J.; Palecek, S.P. Effects of 3-D microwell culture on initial fate specification in human embryonic stem cells. AIChE J. 2014, 60, 1225–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azarin, S.M.; Lian, X.; Larson, E.A.; Popelka, H.M.; de Pablo, J.J.; Palecek, S.P. Modulation of Wnt/β-catenin signaling in human embryonic stem cells using a 3-D microwell array. Biomaterials 2012, 33, 2041–2049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlmann, J.; Kensah, G.; Kempf, H.; Skvorc, D.; Gawol, A.; Elliott, D.A.; Dräger, G.; Zweigerdt, R.; Martin, U.; Gruh, I. The use of agarose microwells for scalable embryoid body formation and cardiac differentiation of human and murine pluripotent stem cells. Biomaterials 2013, 34, 2463–2471. [Google Scholar] [CrossRef]
- Branco, M.A.; Cotovio, J.P.; Rodrigues, C.A.V.; Vaz, S.H.; Fernandes, T.G.; Moreira, L.M.; Cabral, J.M.S.; Diogo, M.M. Transcriptomic analysis of 3D Cardiac Differentiation of Human Induced Pluripotent Stem Cells Reveals Faster Cardiomyocyte Maturation Compared to 2D Culture. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.C.; Ye, J.; Shukla, P.; Hua, G.; Chen, D.; Lin, Z.; Liu, J.; Chai, J.; Gold, J.; Wu, J.; et al. Development of a scalable suspension culture for cardiac differentiation from human pluripotent stem cells. Stem Cell Res. 2015, 15, 365–375. [Google Scholar] [CrossRef]
- Ting, S.; Chen, A.; Reuveny, S.; Oh, S. An intermittent rocking platform for integrated expansion and differentiation of human pluripotent stem cells to cardiomyocytes in suspended microcarrier cultures. Stem Cell Res. 2014, 13, 202–213. [Google Scholar] [CrossRef] [Green Version]
- Fonoudi, H.; Ansari, H.; Abbasalizadeh, S.; Larijani, M.R.; Kiani, S.; Hashemizadeh, S.; Zarchi, A.S.; Bosman, A.; Blue, G.M.; Pahlavan, S.; et al. A Universal and Robust Integrated Platform for the Scalable Production of Human Cardiomyocytes From Pluripotent Stem Cells. Stem Cells Transl. Med. 2015, 4, 1482–1494. [Google Scholar] [CrossRef]
- Kempf, H.; Olmer, R.; Kropp, C.; Rückert, M.; Jara-Avaca, M.; Robles-Diaz, D.; Franke, A.; Elliott, D.A.; Wojciechowski, D.; Fischer, M.; et al. Controlling Expansion and Cardiomyogenic Differentiation of Human Pluripotent Stem Cells in Scalable Suspension Culture. Stem Cell Rep. 2014, 3, 1132–1146. [Google Scholar] [CrossRef] [Green Version]
- Hemmi, N.; Tohyama, S.; Nakajima, K.; Kanazawa, H.; Suzuki, T.; Hattori, F.; Seki, T.; Kishino, Y.; Hirano, A.; Okada, M.; et al. A Massive Suspension Culture System With Metabolic Purification for Human Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cells Transl. Med. 2014, 3, 1473–1483. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Schulte, J.S.; Heinick, A.; Piccini, I.; Rao, J.; Quaranta, R.; Zeuschner, D.; Malan, D.; Kim, K.-P.; Röpke, A.; et al. Universal cardiac induction of human pluripotent stem cells in two and three-dimensional formats: Implications for in vitro maturation. Stem Cells 2015, 33, 1456–1469. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Wang, J.; Loskill, P.; Huebsch, N.; Koo, S.; Svedlund, F.L.; Marks, N.C.; Hua, E.W.; Grigoropoulos, C.P.; Conklin, B.R.; et al. Self-organizing human cardiac microchambers mediated by geometric confinement. Nat. Commun. 2015, 6, 7413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klesen, A.; Jakob, D.; Emig, R.; Kohl, P.; Ravens, U.; Peyronnet, R. Cardiac fibroblasts. Herzschrittmachertherapie + Elektrophysiologie 2018, 29, 62–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warmflash, A.; Sorre, B.; Etoc, F.; Siggia, E.D.; Brivanlou, A.H. A method to recapitulate early embryonic spatial patterning in human embryonic stem cells. Nat. Methods 2014, 11, 847–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thavandiran, N.; Hale, C.; Blit, P.; Sandberg, M.L.; McElvain, M.E.; Gagliardi, M.; Sun, B.; Witty, A.; Graham, G.; Mcintosh, M.; et al. Functional arrays of human pluripotent stem cell-derived cardiac microtissues. bioRxiv 2019, 566059. [Google Scholar] [CrossRef] [Green Version]
- Goldfracht, I.; Efraim, Y.; Shinnawi, R.; Kovalev, E.; Huber, I.; Gepstein, A.; Arbel, G.; Shaheen, N.; Tiburcy, M.; Zimmermann, W.H.; et al. Engineered heart tissue models from hiPSC-derived cardiomyocytes and cardiac ECM for disease modeling and drug testing applications. Acta Biomater. 2019, 92, 145–159. [Google Scholar] [CrossRef]
- Mannhardt, I.; Breckwoldt, K.; Letuffe-Brenière, D.; Schaaf, S.; Schulz, H.; Neuber, C.; Benzin, A.; Werner, T.; Eder, A.; Schulze, T.; et al. Human Engineered Heart Tissue: Analysis of Contractile Force. Stem Cell Rep. 2016, 7, 29–42. [Google Scholar] [CrossRef] [Green Version]
- Lemoine, M.D.; Mannhardt, I.; Breckwoldt, K.; Prondzynski, M.; Flenner, F.; Ulmer, B.; Hirt, M.N.; Neuber, C.; Horváth, A.; Kloth, B.; et al. Human iPSC-derived cardiomyocytes cultured in 3D engineered heart tissue show physiological upstroke velocity and sodium current density. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Lu, H.F.; Leong, M.F.; Lim, T.C.; Chua, Y.P.; Lim, J.K.; Du, C.; Wan, A.C.A. Engineering a functional three-dimensional human cardiac tissue model for drug toxicity screening. Biofabrication 2017, 9, 025011. [Google Scholar] [CrossRef]
- Tiburcy, M.; Hudson, J.E.; Balfanz, P.; Schlick, S.; Meyer, T.; Chang Liao, M.-L.; Levent, E.; Raad, F.; Zeidler, S.; Wingender, E.; et al. Defined Engineered Human Myocardium With Advanced Maturation for Applications in Heart Failure Modeling and Repair. Circulation 2017, 135, 1832–1847. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson-Bouchard, K.; Ma, S.P.; Yeager, K.; Chen, T.; Song, L.J.; Sirabella, D.; Morikawa, K.; Teles, D.; Yazawa, M.; Vunjak-Novakovic, G. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 2018, 556, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Feric, N.T.; Pallotta, I.; Singh, R.; Bogdanowicz, D.R.; Gustilo, M.M.; Chaudhary, K.W.; Willette, R.N.; Chendrimada, T.P.; Xu, X.; Graziano, M.P.; et al. Engineered Cardiac Tissues Generated in the Biowire II: A Platform for Human-Based Drug Discovery. Toxicol. Sci. 2019, 172, 89–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.Y.; Peres Moreno Maia-Joca, R.; Ong, C.S.; Wilson, I.; DiSilvestre, D.; Tomaselli, G.F.; Reich, D.H. Enhancement of human iPSC-derived cardiomyocyte maturation by chemical conditioning in a 3D environment. J. Mol. Cell. Cardiol. 2020, 138, 1–11. [Google Scholar] [CrossRef]
- Voges, H.K.; Mills, R.J.; Elliott, D.A.; Parton, R.G.; Porrello, E.R.; Hudson, J.E. Development of a human cardiac organoid injury model reveals innate regenerative potential. Development 2017, 144, 1118–1127. [Google Scholar] [CrossRef] [Green Version]
- Schaaf, S.; Shibamiya, A.; Mewe, M.; Eder, A.; Stöhr, A.; Hirt, M.N.; Rau, T.; Zimmermann, W.H.; Conradi, L.; Eschenhagen, T.; et al. Human engineered heart tissue as a versatile tool in basic research and preclinical toxicology. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Mills, R.J.; Titmarsh, D.M.; Koenig, X.; Parker, B.L.; Ryall, J.G.; Quaife-Ryan, G.A.; Voges, H.K.; Hodson, M.P.; Ferguson, C.; Drowley, L.; et al. Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proc. Natl. Acad. Sci. USA 2017, 114, E8372–E8381. [Google Scholar] [CrossRef] [Green Version]
- Ruan, J.-L.; Tulloch, N.L.; Razumova, M.V.; Saiget, M.; Muskheli, V.; Pabon, L.; Reinecke, H.; Regnier, M.; Murry, C.E. Mechanical Stress Conditioning and Electrical Stimulation Promote Contractility and Force Maturation of Induced Pluripotent Stem Cell-Derived Human Cardiac Tissue. Circulation 2016, 134, 1557–1567. [Google Scholar] [CrossRef]
- Huebsch, N.; Loskill, P.; Deveshwar, N.; Spencer, C.I.; Judge, L.M.; Mandegar, M.A.; Fox, C.B.; Mohamed, T.M.A.; Ma, Z.; Mathur, A.; et al. Miniaturized iPS-Cell-Derived Cardiac Muscles for Physiologically Relevant Drug Response Analyses. Sci. Rep. 2016, 6, 24726. [Google Scholar] [CrossRef] [Green Version]
- Saleem, U.; van Meer, B.J.; Katili, P.A.; Yusof, N.A.N.M.; Mannhardt, I.; Garcia, A.K.; Tertoolen, L.; de Korte, T.; Vlaming, M.L.H.; McGlynn, K.; et al. Blinded, multi-centre evaluation of drug-induced changes in contractility using human induced pluripotent stem cell-derived cardiomyocytes. Toxicol. Sci. 2020. [Google Scholar] [CrossRef]
- Mills, R.J.; Parker, B.L.; Quaife-Ryan, G.A.; Voges, H.K.; Needham, E.J.; Bornot, A.; Ding, M.; Andersson, H.; Polla, M.; Elliott, D.A.; et al. Drug Screening in Human PSC-Cardiac Organoids Identifies Pro-proliferative Compounds Acting via the Mevalonate Pathway. Cell Stem Cell 2019, 24, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, B.M.; Stoehr, A.; Schulze, M.L.; Patel, S.; Gucek, M.; Mannhardt, I.; Funcke, S.; Murphy, E.; Eschenhagen, T.; Hansen, A. Contractile Work Contributes to Maturation of Energy Metabolism in hiPSC-Derived Cardiomyocytes. Stem Cell Rep. 2018, 10, 834–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacomelli, E.; Meraviglia, V.; Campostrini, G.; Cochrane, A.; Cao, X.; van Helden, R.W.J.; Krotenberg Garcia, A.; Mircea, M.; Kostidis, S.; Davis, R.P.; et al. Human-iPSC-Derived Cardiac Stromal Cells Enhance Maturation in 3D Cardiac Microtissues and Reveal Non-cardiomyocyte Contributions to Heart Disease. Cell Stem Cell 2020, 26, 862–879. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.J.; Coyle, R.C.; Tan, Y.; Jia, J.; Wong, K.; Toomer, K.; Menick, D.R.; Mei, Y. Inspiration from heart development: Biomimetic development of functional human cardiac organoids. Biomaterials 2017, 142, 112–123. [Google Scholar] [CrossRef]
- Pitaktong, I.; Lui, C.; Lowenthal, J.; Mattson, G.; Jung, W.-H.; Bai, Y.; Yeung, E.; Ong, C.S.; Chen, Y.; Gerecht, S.; et al. Early Vascular Cells Improve Microvascularization Within 3D Cardiac Spheroids. Tissue Eng. Part C Methods 2020, 26, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Ravenscroft, S.M.; Pointon, A.; Williams, A.W.; Cross, M.J.; Sidaway, J.E. Cardiac non-myocyte cells show enhanced pharmacological function suggestive of contractile maturity in stem cell derived cardiomyocyte microtissues. Toxicol. Sci. 2016, 152, 99–112. [Google Scholar] [CrossRef]
- Giacomelli, E.; Bellin, M.; Sala, L.; van Meer, B.J.; Tertoolen, L.G.J.; Orlova, V.V.; Mummery, C.L. Three-dimensional cardiac microtissues composed of cardiomyocytes and endothelial cells co-differentiated from human pluripotent stem cells. Development 2017, 144, 1008–1017. [Google Scholar] [CrossRef] [Green Version]
- Polonchuk, L.; Chabria, M.; Badi, L.; Hoflack, J.C.; Figtree, G.; Davies, M.J.; Gentile, C. Cardiac spheroids as promising in vitro models to study the human heart microenvironment. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Archer, C.R.; Sargeant, R.; Basak, J.; Pilling, J.; Barnes, J.R.; Pointon, A. Characterization and Validation of a Human 3D Cardiac Microtissue for the Assessment of Changes in Cardiac Pathology. Sci. Rep. 2018, 8, 10160. [Google Scholar] [CrossRef]
- Brandao, K.O.; Tabel, V.A.; Atsma, D.E.; Mummery, C.L.; Davis, R.P. Human pluripotent stem cell models of cardiac disease: From mechanisms to therapies. Dis. Model. Mech. 2017, 10, 1039–1059. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Pan, H.; Tan, C.; Sun, Y.; Song, Y.; Zhang, X.; Yang, W.; Wang, X.; Li, D.; Dai, Y.; et al. Mitochondrial Dysfunctions Contribute to Hypertrophic Cardiomyopathy in Patient iPSC-Derived Cardiomyocytes with MT-RNR2 Mutation. Stem Cell Rep. 2018, 10, 808–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laksman, Z.; Wauchop, M.; Lin, E.; Protze, S.; Lee, J.; Yang, W.; Izaddoustdar, F.; Shafaattalab, S.; Gepstein, L.; Tibbits, G.F.; et al. Modeling Atrial Fibrillation using Human Embryonic Stem Cell-Derived Atrial Tissue. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Miles, C.; Finocchiaro, G.; Papadakis, M.; Gray, B.; Westaby, J.; Ensam, B.; Basu, J.; Parry-Williams, G.; Papatheodorou, E.; Paterson, C.; et al. Sudden Death and Left Ventricular Involvement in Arrhythmogenic Cardiomyopathy. Circulation 2019, 139, 1786–1797. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; van Tintelen, P.J.; McKenna, W.J.; Hauer, R.N.W.; Anastastakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brunckhorst, C.; Bucciarelli-Ducci, C.; et al. Arrhythmogenic right ventricular cardiomyopathy: Evaluation of the current diagnostic criteria and differential diagnosis. Eur. Heart J. 2020, 41, 1414–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cashman, T.J.; Josowitz, R.; Johnson, B.V.; Gelb, B.D.; Costa, K.D. Human engineered cardiac tissues created using induced pluripotent stem cells reveal functional characteristics of BRAF-mediated hypertrophic cardiomyopathy. PLoS ONE 2016, 11, e0146697. [Google Scholar] [CrossRef]
- Zhao, Y.; Rafatian, N.; Feric, N.T.; Cox, B.J.; Aschar-Sobbi, R.; Wang, E.Y.; Aggarwal, P.; Zhang, B.; Conant, G.; Ronaldson-Bouchard, K.; et al. A Platform for Generation of Chamber-Specific Cardiac Tissues and Disease Modeling. Cell 2019, 176, 913–927. [Google Scholar] [CrossRef] [Green Version]
- Prondzynski, M.; Lemoine, M.D.; Zech, A.T.; Horváth, A.; Di Mauro, V.; Koivumäki, J.T.; Kresin, N.; Busch, J.; Krause, T.; Krämer, E.; et al. Disease modeling of a mutation in α-actinin 2 guides clinical therapy in hypertrophic cardiomyopathy. EMBO Mol. Med. 2019, 11, e11115. [Google Scholar] [CrossRef]
- Shah, D.; Prajapati, C.; Penttinen, K.; Cherian, R.M.; Koivumäki, J.T.; Alexanova, A.; Hyttinen, J.; Aalto-Setälä, K. hiPSC-Derived Cardiomyocyte Model of LQT2 Syndrome Derived from Asymptomatic and Symptomatic Mutation Carriers Reproduces Clinical Differences in Aggregates but Not in Single Cells. Cells 2020, 9, 1153. [Google Scholar] [CrossRef]
- Richards, D.J.; Li, Y.; Kerr, C.M.; Yao, J.; Beeson, G.C.; Coyle, R.C.; Chen, X.; Jia, J.; Damon, B.; Wilson, R.; et al. Human cardiac organoids for the modelling of myocardial infarction and drug cardiotoxicity. Nat. Biomed. Eng. 2020, 4, 446–462. [Google Scholar] [CrossRef]
- Onakpoya, I.J.; Heneghan, C.J.; Aronson, J.K. Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: A systematic review of the world literature. BMC Med. 2016, 14, 10. [Google Scholar] [CrossRef] [Green Version]
- Siramshetty, V.B.; Nickel, J.; Omieczynski, C.; Gohlke, B.O.; Drwal, M.N.; Preissner, R. WITHDRAWN—A resource for withdrawn and discontinued drugs. Nucleic Acids Res. 2016, 44, D1080–D1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Korte, T.; Katili, P.A.; Mohd Yusof, N.A.N.; van Meer, B.J.; Saleem, U.; Burton, F.L.; Smith, G.L.; Clements, P.; Mummery, C.L.; Eschenhagen, T.; et al. Unlocking Personalized Biomedicine and Drug Discovery with Human Induced Pluripotent Stem Cell–Derived Cardiomyocytes: Fit for Purpose or Forever Elusive? Annu. Rev. Pharmacol. Toxicol. 2020, 60, 529–551. [Google Scholar] [CrossRef] [PubMed]
- Milani-Nejad, N.; Janssen, P.M.L. Small and large animal models in cardiac contraction research: Advantages and disadvantages. Pharmacol. Ther. 2014, 141, 235–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salama, G.; London, B. Mouse models of long QT syndrome. J. Physiol. 2007, 578, 43–53. [Google Scholar] [CrossRef]
- Nerbonne, J. Studying Cardiac Arrhythmias in the Mouse—A Reasonable Model for Probing Mechanisms? Trends Cardiovasc. Med. 2004, 14, 83–93. [Google Scholar] [CrossRef]
- Turnbull, I.C.; Karakikes, I.; Serrao, G.W.; Backeris, P.; Lee, J.J.; Xie, C.; Senyei, G.; Gordon, R.E.; Li, R.A.; Akar, F.G.; et al. Advancing functional engineered cardiac tissues toward a preclinical model of human myocardium. FASEB J. 2014, 28, 644–654. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.K.; Tran, D.D.; Keung, W.; Chan, P.; Wong, G.; Chan, C.W.; Costa, K.D.; Li, R.A.; Khine, M. Machine Learning of Human Pluripotent Stem Cell-Derived Engineered Cardiac Tissue Contractility for Automated Drug Classification. Stem Cell Rep. 2017, 9, 1560–1572. [Google Scholar] [CrossRef] [Green Version]
- Sala, L.; Bellin, M.; Mummery, C.L. Integrating cardiomyocytes from human pluripotent stem cells in safety pharmacology: Has the time come? Br. J. Pharmacol. 2017, 174, 3749–3765. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Guo, L.; Zeng, H.; White, S.L.; Furniss, M.; Balasubramanian, B.; Lis, E.; Lagrutta, A.; Sannajust, F.; Zhao, L.L.; et al. Multi-parametric assessment of cardiomyocyte excitation-contraction coupling using impedance and field potential recording: A tool for cardiac safety assessment. J. Pharmacol. Toxicol. Methods 2016, 81, 201–216. [Google Scholar] [CrossRef]
- Mckeithan, W.L.; Savchenko, A.; Yu, M.S.; Cerignoli, F.; Bruyneel, A.A.N.; Price, J.H.; Colas, A.R.; Miller, E.W.; Cashman, J.R.; Mercola, M. An Automated Platform for Assessment of Congenital and Drug-Induced Arrhythmia with hiPSC-Derived. Cardiomyocytes 2017, 8, 776. [Google Scholar] [CrossRef] [Green Version]
- Sala, L.; Van Meer, B.J.; Tertoolen, L.G.J.; Bakkers, J.; Bellin, M.; Davis, R.P.; Denning, C.; Dieben, M.A.E.; Eschenhagen, T.; Giacomelli, E.; et al. Musclemotion: A versatile open software tool to quantify cardiomyocyte and cardiac muscle contraction in vitro and in vivo. Circ. Res. 2018, 122, e5–e16. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.R.; Whittaker, R.; Price, J.H.; Vega, R.; Pfeiffer, E.R.; Cerignoli, F.; Towart, R.; Gallacher, D.J. High throughput measurement of Ca++ dynamics in human stem cell-derived cardiomyocytes by kinetic image cytometery: A cardiac risk assessment characterization using a large panel of cardioactive and inactive compounds. Toxicol. Sci. 2015, 148, 503–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devarasetty, M.; Forsythe, S.; Shupe, T.; Soker, S.; Bishop, C.; Atala, A.; Skardal, A. Optical Tracking and Digital Quantification of Beating Behavior in Bioengineered Human Cardiac Organoids. Biosensors 2017, 7, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, P.; Huebsch, N.; Bang, S.H.; Siemons, B.A.; Conklin, B.R.; Healy, K.E.; Ma, Z.; Jacquir, S. Quantitatively characterizing drug-induced arrhythmic contractile motions of human stem cell-derived cardiomyocytes. Biotechnol. Bioeng. 2018, 115, 1958–1970. [Google Scholar] [CrossRef]
- Van Meer, B.J.; Krotenberg, A.; Sala, L.; Davis, R.P.; Eschenhagen, T.; Denning, C.; Tertoolen, L.G.J.; Mummery, C.L. Simultaneous measurement of excitation-contraction coupling parameters identifies mechanisms underlying contractile responses of hiPSC-derived cardiomyocytes. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Pointon, A.; Abi-gerges, N.; Cross, M.J.; Sidaway, J.E. Phenotypic profiling of structural cardiotoxins in vitro reveals dependency on multiple mechanisms of toxicity. Toxicol. Sci. 2013, 132, 317–326. [Google Scholar] [CrossRef] [Green Version]
- Kopljar, I.; Lu, H.R.; Van Ammel, K.; Otava, M.; Tekle, F.; Teisman, A.; Gallacher, D.J. Development of a Human iPSC Cardiomyocyte-Based Scoring System for Cardiac Hazard Identification in Early Drug Safety De-risking. Stem Cell Rep. 2018, 11, 1365–1377. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; McKeithan, W.L.; Serrano, R.; Kitani, T.; Burridge, P.W.; del Álamo, J.C.; Mercola, M.; Wu, J.C. Use of human induced pluripotent stem cell–derived cardiomyocytes to assess drug cardiotoxicity. Nat. Protoc. 2018, 13, 3018–3041. [Google Scholar] [CrossRef]
- Silva, A.C.; Matthys, O.B.; Joy, D.A.; Kauss, M.A.; Natarajan, V.; Lai, M.H.; Turaga, D.; Alexanian, M.; Bruneau, B.G.; McDevitt, T.C. Developmental co-emergence of cardiac and gut tissues modeled by human iPSC-derived organoids. bioRxiv 2020. [Google Scholar] [CrossRef]
- Miranda, C.C.; Fernandes, T.G.; Diogo, M.M.; Cabral, J.M.S. Towards multi-organoid systems for drug screening applications. Bioengineering 2018, 5, 49. [Google Scholar] [CrossRef] [Green Version]
- Skardal, A.; Murphy, S.V.; Devarasetty, M.; Mead, I.; Kang, H.W.; Seol, Y.J.; Zhang, Y.S.; Shin, S.R.; Zhao, L.; Aleman, J.; et al. Multi-tissue interactions in an integrated three-tissue organ-on-a-chip platform. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Kim, H.J.; Fraser, J.P.; Shea, D.E.; Khan, M.; Bahinski, A.; Hamilton, G.A.; Ingber, D.E. Microfabrication of human organs-on-chips. Nat. Protoc. 2013, 8, 2135–2157. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Pre-Differentiation | Differentiation | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Platform | Time | Media | Aggregate Ø at D0 | Platform | Media | Molecules | Duration | Efficiency | |||||
| Halloin et al., 2019 | Stirred Bioreactor | 2 Days | E8 | ±125 µm | Stirred Bioreactor (500 mL) | CDM3 *1 | CHIR | D0–D1 | 5 µM | 10 Days | ±1 × 106 CMs/mL | ||
| IWP2 | D1–D3 | 5 µM | 93 ± 5% CMs | ||||||||||
| Chen et al., 2015 | Spinner Flask | 2 Days | StemPro hESC SFM + FGF2 | 200 ± 20 μm | Spinner Flask (125 mL–1L) | RPMI + B27-INS | D0–D4 | CHIR | D0–D1 | 6/12 µM *2 | 16 Days | ±1 × 106 CMs/mL (1L) ±2 × 106 CMs/mL (500 mL) | |
| RPMI + B27 | D4–D16 | IWP4 | D2–D4 | 5 µM | >90% CMs | ||||||||
| Fonoudi et al., 2015 | Stirred Bioreactor | 5 Days | DMEM/F12+ FGF2 | 175 ± 25 µm | Stirred Bioreactor (100 mL) | RPMI + B27 | D0–D15 | CHIR | D0–D1 | 12 µM | 15 Days | 0.8 × 106 CMs/mL | |
| SB + Pur + IWP2 | D2–D4 | 5 µM each | >80% CMs | ||||||||||
| Branco et al., 2019 | AggrewellTM800 | 3 Days | mTeSR1 | ±300 µm | AggrewellTM800 | D0–D7 | RPMI + B27-INS | D0–D7 | CHIR | D0–D1 | 11 µM | 12 Days | ±20 × 106 CMs/plate |
| ULA 6-well plate | D0–D12 | RPMI + B27 | D7–D12 | IWP4 | D3–D5 | 5 µM | >85% CMs | ||||||
| Burridge et al., 2011 | - | *6 | 96-V ULA plate | D0–D4 | RPMI | D0–D10 | *5 | 10 Days | ±0.4 × 106 CMs/plate | ||||
| 96-U ULA plate | D4–D10 | >80% CMs | |||||||||||
| Dahlmann et al., 2013 | Agarose Microwell plate | 1 Day | FCM *3 | 400–500 µm *4 (±220 µm (D-3)) | ULA 6-well plate | RPMI + B27-INS | D0–D7 | CHIR | D0–D1 | 8 µM | 10 Days | *6 | |
| ULA 6-well plate | 3 Days | RPMI + B27 | D7–D10 | IWR1 | D3–D5 | 4 µM | Up to 65% CMs | ||||||
| (A) Study | Cell Composition | Cell density (Cardiac Cells/EHT) | Hydrogel | Mechanical Load | Electrical Stimulation | Culture Medium | Model | |
| Goldfracht et al., 2019 | hPSC-CMs (≥80% cTNT+) (D14-D20) | 2 × 106 | Porcine cardiac ECM | Passive stretcher device | No | IMDM + 20% FBS | Ring-shape | |
| Ruan et al., 2016 | hPSC-CMs (73 ± 3% cTNT+) (D14-D21) | 2 × 106 | Collagen Type I (1.25 mg/mL) | Static Stress (constructs at a fixed static length) | 2 Hz, 5 ms pulse (1 week) | RPMI+B27 | Ring-shape | |
| Mannhardt et al., 2016 Lemoine et al., 2017 Ulmer et al., 2018 | hPSC-CMs (D14) | 1 × 106 | Bovine fibrinogen (5 mg/mL) | Yes | No | DMEM + 10% HS + 10 mg/mL insulin | strip-like | |
| Lu et al., 2017 | hPSC-CMs (>90% cTNT+) (D12) | 1.3 ×106 | - | No | No | RPMI + B27 | Rectangular tissue holder | |
| Tiburcy et al., 2017 | hESC-CMs (70%) + Human Foreskin Fibroblasts (30%) | 1.5 × 106 | Bovine Collagen (0.8mg/mL) | Yes | No | IMDM + 4% B27-INS + 100 ng/mL IGF1 + 10 ng/mL FGF2 + 5 ng/mL VEGF + 5 ng/mL TGFβ1 | Ring-shape | |
| Ronaldson-Bouchard et al., 2018 | hiPSC-CMs (75%) (D12) Human Dermal Fibroblasts (25%) | - | Human fibrinogen (20 mg/mL) | Yes | Increment regime of electric stimulation (0.33 Hz per day from 2 Hz to 6 Hz) (4 weeks) | RPMI + B27 | strip-like | |
| (B) Study | Medium- to High-throughput EHT models | |||||||
| Zhao et al., 2019 Feric et al., 2019 | hPSC-CMs + hCFs (10:1) | 1.1 × 105 | Rat tail collagen (3 mg/mL) | Yes | Yes | BiowireTM II (TARA Biosystems) | Strip-like Rectangular Chips (5 mm × 1 mm × 0.3 mm) | |
| Mills et al., 2017 Mills et al., 2019 | hPSC-CMs * (D15) | 5 × 104 | Collagen I (2.6 mg/mL) | Yes | No | low glucose, high palmitate, no insulin | Heart-Dyno platform | Elliptic-shape Two elastomeric posts in each well (1 mm from each other) (96-Well plate) |
| Thavandiran et al., 2019 | hPSC-CMs + hCFs (10:1) | 7 × 104 | Collagen I (2 mg/mL) | Yes | No | Cardiac MicroRings (CaMiRi) | Ring-shape Two elastomeric posts in each well (96-Well plate) | |
| Turnbull et al., 2014 Lee et al., 2017 | hPSC-CMs (D14–16) + human Foreskin Fibroblasts (1:1) | 2 × 105 | Bovine Collagen I (2 mg/mL) | Yes | No | NovoheartTM | Strip-like Rectangular casting molds with two flexible PDMS pillars (10 mm from each other) | |
| Huang et al., 2020 | hiPSC -CMs (75%) (D20–22) + HUVECs (10%) + Human adult ventricular CFs (15%) | 2.5 × 105 cells/cm2 | Human fibrinogen (0.75 mg/mL) + rat tail type I collagen (2.25 mg/mL) | Yes | No | RPMI+B27 + T3 + IGF -1 + Dex | µTUG arrays | Elliptic-shape Two elastomeric posts in each microwell (interpillar spacing of 500 µm) (42 microwells of dimensions 400 μm × 800 μm × 200 μm) |
| Study | Cardiac Microtissue Composition | Control | Cell Seeding Density | Culture Platform | |
|---|---|---|---|---|---|
| Richards et al., 2017 | hPSC-CMs (50%)—α-SA | hiPSC-CMs Spheroids | 1.5 × 105 cells/mold (±4300 cells/well) | Custom-made agarose molds containing 35 microwells (800 µm diameter, 800 µm deep) | |
| Human Ventricular CFs (29%)–Vimentin | |||||
| HUVECs (14%)–CD31 | |||||
| Human Adipose-derived Stem Cells (7%) | |||||
| Ravenscroft et al., 2016 | hPSC-CMs/hESC-CMs (57%)–α-actinin, ACTN2 | hPSC-CMs Spheroids | 500 cells/well | U-bottom ultra-low adhesion 96-well plate | |
| Human CFs (29%)-collagen I, COL1A1 | |||||
| Human Cardiac Microvascular ECs (14%)–CD31 | |||||
| Polonchuk et al., 2017 | hPSC-CMs (50%)–cTNT | - | 1 × 104/drop | 96-well Hanging Drop Plates | |
| Human Coronary Artery ECs (25%)–CD31 | |||||
| hiPSC-CFs (25%)–Vimentin | |||||
| Archer et al., 2018 | hPSC-CMs (57%)–α-actinin and cTnI | hPSC-CMs Monolayer | 500 cells/well | Ultra-low attachment 384-well plate | |
| Primary Human Cardiac Microvascular ECs (29%)–CD31 | |||||
| Primary Human CFs (14%)–Vimentin and Collagen I | |||||
| Giacomelli et al., 2017 | hPSC-CMs (85%)–cTNI | hPSC-CMs Spheroids | hPSC-CMs monolayer | 5000 cells/well | V-bottom ultra-low adhesion 96-well plate |
| hPSC-ECs (15%)–CD31 | |||||
| Giacomelli et al., 2020 | hPSC-CMs (70%)–cTNI | hPSC-CMs (85%) + hPSC-ECs (15%) | hPSC-CMs (70%) + hPSC-ECs (15%) + hACFs (15% | 5000 cells/well | V-bottom ultra-low adhesion 96-well plate |
| hPSC-ECs (15%)–CD31 | hPSC-CMs (85%) + hPSC-CFs (15%) | hPSC-CMs (85%) + hPSC-ECs + hPSC- hSFs (15%) | |||
| hPSC-CFs (15%)–COL1A1 | |||||
| Pitaktong et al., 2020 | hPSC-CMs (70%)–cTNT | hiPSC-CMs (70%) + CFs (15%) + HUVECs (15%) | hiPSC-CMs (40%) + CFs (15%) + hPSC-EVCs (45%) | 3.3 × 104 cells/well | U-bottom ultra-low adhesion 96-well plate |
| Human Adult Ventricular CFs (15%)–Vimentin | |||||
| hiPSC-EVCs * (15%)–Pericytes (NG2); ECs (CD31) | |||||
| Study | Culture Format | Dye | Functional/Morphological Analysis | |
|---|---|---|---|---|
| Optical Recording/Software | Analyzed Parameters | |||
| van Meer et al., 2019 | 2D and 3D Spheroids | Simultaneous combination of three fluorescence dyes:
| >300 fps MuscleMotion [132] |
|
| Turnbull et al., 2014 Lee et al., 2017 | EHT | - | Post deflection (100 fps) LabView software | Force of contraction (mN)-peak Contraction parameters (time to peak, time to 90% relaxation, maximum rate of force increase and decrease) |
| Voltage-sensitive dye: di-4-ANEPPS | Video acquisition | Action Potential Duration (ms) | ||
| Mills et al., 2019 | EHT | - | Post deflection (10s time-lapse capture) Vision.PointTracker (Matlab) | Force of Contraction (µN) (peak) Contraction parameters (rate (bpm), 50% activation (s); 50% relaxation (s)) |
| Lu et al., 2017 | EHT | FluoSpheres polystyrene microspheres | Video acquisition (Fluorescence) Imaris software | Contraction speed (µm/s) Contraction rate (bpm) |
| Zhao et al., 2019 Feric et al., 2019 | EHT |
| Post deflection (500 fps) ImageJ Software (Spot Tracker plugin) |
|
| Archer et al., 2018 | 3D Spheroids |
| Quantitative measurements of average fluorescence intensity Columbus™ Image Data Storage and Analysis platform |
|
| Pointon et al., 2013 | 2D |
| Image acquisition (live-cell fluorescent) (Four image fields per well) metaXpress software |
|
| Richards et al., 2017 | 3D Spheroids | Fluo-4 dye | Video acquisition (Fluorescence) (20 fps) ImageJ Software | - Calcium transient profile (Normalized peak of calcium fluorescence, time to peak calcium (sec) and time to 50% calcium decay (sec)) |
| - | Video acquisition (spontaneously beating spheroids) ImageJ Software | - Beating profile (rate of contraction (bpm) and contraction amplitude) | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Branco, M.A.; Cabral, J.M.S.; Diogo, M.M. From Human Pluripotent Stem Cells to 3D Cardiac Microtissues: Progress, Applications and Challenges. Bioengineering 2020, 7, 92. https://doi.org/10.3390/bioengineering7030092
Branco MA, Cabral JMS, Diogo MM. From Human Pluripotent Stem Cells to 3D Cardiac Microtissues: Progress, Applications and Challenges. Bioengineering. 2020; 7(3):92. https://doi.org/10.3390/bioengineering7030092
Chicago/Turabian StyleBranco, Mariana A., Joaquim M.S. Cabral, and Maria Margarida Diogo. 2020. "From Human Pluripotent Stem Cells to 3D Cardiac Microtissues: Progress, Applications and Challenges" Bioengineering 7, no. 3: 92. https://doi.org/10.3390/bioengineering7030092
APA StyleBranco, M. A., Cabral, J. M. S., & Diogo, M. M. (2020). From Human Pluripotent Stem Cells to 3D Cardiac Microtissues: Progress, Applications and Challenges. Bioengineering, 7(3), 92. https://doi.org/10.3390/bioengineering7030092