1. Introduction
Breast cancer is the second leading cause of cancer death in United States (US), and it is the most frequently diagnosed cancer among US women [
1]. Tumor metastasis is the major cause of more than 90% of breast cancer-related deaths [
2]. The five-year survival rate for metastatic breast cancer (MBC) is only 28.21%. On the other hand, localized breast cancer has a very high (98.9%) five-year survival rate in US. Triple-Negative Breast Cancer (TNBC) is a type of breast cancer characterized by the absence of hormone receptors like estrogen and progesterone and human epidermal growth factor-2 (HER-2) receptor [
2]. TNBCs can have a high rate of recurrences and systematic metastases [
3]. TNBC and MBC can benefit from targeted nanotherapies, reducing tumor angiogenesis and reversing multidrug resistance (MDR) in these cells [
4,
5,
6].
Current therapies of breast cancer include surgery, radiotherapy, chemotherapy, targeted therapy, and immunotherapy [
7]. Small interfering RNAs are increasing explored as a new and evolving class of gene-silencing therapeutics for a variety of cancers including MBC and TNBC [
8,
9,
10,
11]. In spite of the plethora of research on siRNA therapeutics, only one siRNA product is approved in the market. Onpattro
® Patisiran (siRNA), marketed by Alnylam was approved by FDA in 2019 for treatment of hereditary transthyretin amyloidosis. Onpattro
® Patisiran is a lyophilized liposomal formulation. It is the first clinically approved RNAi therapy liposomal therapy administered intravenously [
12,
13]. Administering naked siRNA, cannot be useful as the ribose backbone of RNA is susceptible to hydrolysis by serum endonuclease. In addition, the small size can result in rapid glomerular filtration. These reasons are the main causes for a shot half-life less than 15 min for siRNA [
14,
15]. This requires delivery of siRNA through nanocarriers. Liposomes can be an ideal nanocarrier system for siRNA. They can (i) protects siRNA from degradation, thus increase circulation t1/2 (ii) entrap hydrophilic and hydrophobic cargo, (iii) low immune response, (iv) evasion of reticuloendothelial system, (v) cationic liposomes can protect siRNA from attack by endonucleases (vi) can achieve enhanced permeability and retention (EPR) effect in the tumor, (vii) can deliver siRNA to the cytoplasm, and silence the target protein to hinder tumor growth and progression, (vii) can be used for ligand-targeted delivery to the tumor cells [
14,
16]. An ideal siRNA nanocarrier like liposomes developed for cancer treatment, should selectively target cancer cells, and deliver siRNA to the cytoplasm. In addition to this, the therapy should silence the target protein to obstruct tumor growth and progression [
17].
An important factor aiding the growth, differentiation, and eventual metastasis of tumor cells is angiogenesis. Controlling angiogenesis of tumor cells retards development of new blood vessels and shrinks the existing ones [
18,
19]. Anti-angiogenic agents can reduce oxygen and nutrient supply to the tumors, thus shrinking them [
20,
21]. Tumor cells can metastasize by the activation of epithelial-to-mesenchymal transition (EMT) pathway [
22], EMT is a major contributor to metastasis of breast cancer of epithelial-originated breast cancer, like Luminal A and B type [
23,
24]. EMT pathway is activated during tumor cell migration, metastasis, invasion, and chemotherapy resistance [
25,
26]. Protein factors like matrix metalloproteinases (MMPs), epithelial markers of E-cadherin, and transforming growth factor-β1 (TGF-β1) are some of the important factors that regulate tumor cell migration and invasion in breast cancer and other types of cancer [
27,
28,
29,
30]. Hence, targeting angiogenesis and EMT pathway can be an effective solution in breast cancer metastasis. This can be useful for MBC and TNBC.
Lipocalin-2 (Lcn2) is a 25-kDa protein and a regulator of angiogenesis [
31,
32]. Lcn2 can induce EMT in breast cancer through estrogen receptor α/Slug axis. In addition, Lcn2 can regulate breast cancer angiogenesis. Vascular endothelial growth factor (VEGF), a key angiogenic activator, is significantly increased with higher Lcn2 expression in MCF-7 human breast cancer cells and TNBC cell line MDA-MB-436 and MDA-MB-231 cells [
32]. Lcn2 levels can also be used as a non-invasive biomarker. Urine of breast cancer patients demonstrates elevated Lcn2 levels. This can correlate with progression of breast cancer [
32,
33,
34]. Gao et al. have previously demonstrated that Lcn2 actively promote breast cancer progression via prompting EMT in breast cancer cells and by stimulating neovascularization [
35]. The same group also demonstrated that Lcn2 secreted from TNBC cells increased the levels of vascular endothelial growth factor (VEGF), thus promoting neovascularization. Hence, knockdown of Lcn2 in breast cancer cells can make it an ideal anti-angiogenic target [
32,
36].
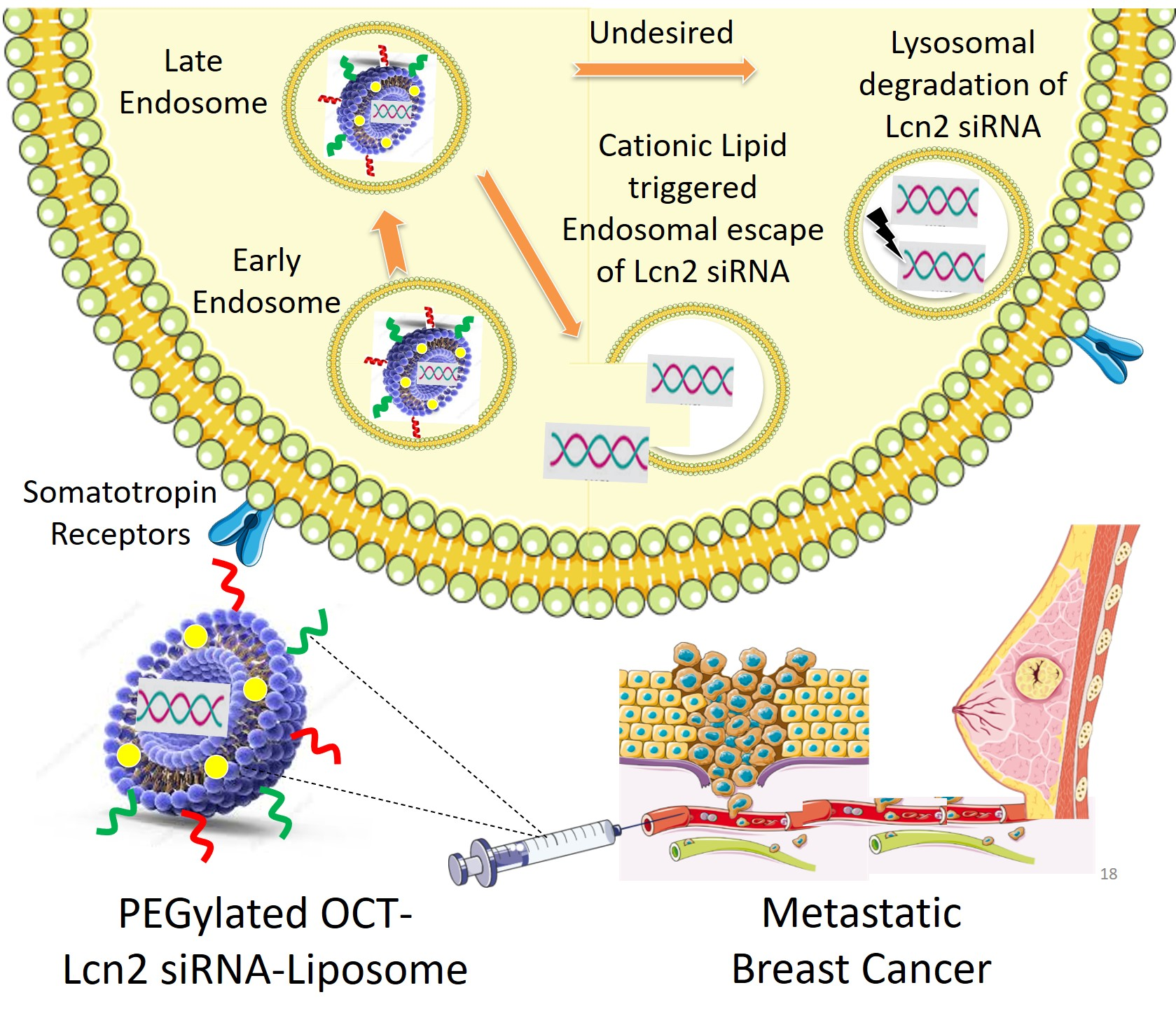
In the present study, we designed a PEGylated cationic liposomal formulation encapsulating Lcn2 siRNA decorated with octreotide (OCT) peptide on its surface for active targeting to breast cancer cells. OCT is an octapeptide analog of somatostatin (SST) growth hormone, having higher affinity for somatostatin receptors (SSTRs). These receptors are widely expressed on cancer cell membranes in including breast cancer [
37]. SSTRs are expressed on MBC cells like MCF-7 and TNBC cells like MDA-MB-231 [
38,
39,
40]. Here we demonstrate formulation development, optimization, and evaluation of bioactivity of OCT-targeted Lcn2 siRNA encapsulated PEGylated cationic liposomes (OCT-Lcn2-Lipo) for drug delivery to breast cancer cells like MCF-7 and MDA-MB-231.
2. Materials and Methods
2.1. Materials
Lcn2 siGENOME SMARTpool siRNA was purchased from Dharmacon (Lafayette, CO). Octreotide acetate peptide was purchased from BCN Peptides (Barcelona, Spain). Didodecyldimethylammonium bromide (DDAB), cholesterol acetate and Vitamin E TPGS was purchased from Sigma Aldrich. 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxy(polyethylene glycol)-2000] (DSPE-PEG2000-COOH) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxy(polyethylene glycol)-2000, NHS ester] (sodium salt) (DSPE-PEG2000-NHS) was purchased from Avanti Polar Lipids (Alabaster, AL, USA). N-hydroxysuccinimide (NHS) and 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) was purchased from TCI Chemicals (Tokyo Chemical Industry, Tokyo Japan). Slide-A-Lyzer® dialysis cassette (MWCO 2000 kDa) for conjugation reaction and liposome preparation and Lab-Tek® II Chamber Slide for confocal microscopy was obtained from Thermo Fisher Scientific (Pittsburgh, PA, USA). Non-essential amino acids were purchased from Life Technologies®. Fetal bovine serum (FBS) was purchased from Atlanta Biologics (Lawrenceville, GA, USA). Quant-iT ™ RNA Assay Kit, Gibco® Dulbecco’s Modified Eagle Medium (DMEM), Trypsin (TrypLE) were purchased from Invitrogen (Carlsbad, CA, USA). Dichloromethane (CH2Cl2), ethanol (EtOH), acetonitrile (AcN), and HPLC grade water (H2O) were purchased from Thermo Fisher Scientific (Pittsburgh, PA, USA). Dimethylsulphoxide D-6 for 1HNMR was obtained from Millipore Sigma. Distilled deionized (DDI) water was obtained from Barnstead EasyPure UV Deionization systems Thermo Fisher Scientific (Pittsburgh, PA, USA). Other chemicals and reagents used in this study were purchased from Sigma-Aldrich unless specified.
Cell Culture
Breast cancer cell lines MCF-7, triple-negative breast cancer cell line MDA-MB-231 and normal breast epithelial cell line MCF-12A were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). Cell lines were stored in liquid nitrogen, maintained at −198 °C. T-75 Corning flask purchased from Thermo Fisher Scientific (Pittsburgh, PA, USA) were used for cell culture. Cells were cultured in DMEM (Gibco) containing 10% serum (heat-inactivated FBS), antibiotics (100 IU/mL of Streptomycin and 100 IU/mL of penicillin), 1% sodium pyruvate and 1% of non-essential amino acids. The cells were maintained at 37 °C (normal body temperature), 5% CO2, and 90% relative humidity in a Marshall Scientific, Heracell 150 CO2 incubator. The cells were harvested when they reached 80–90% confluence. The cell growth was observed under a ZEISS Telaval 31 Inverted Phase Contrast Microscope.
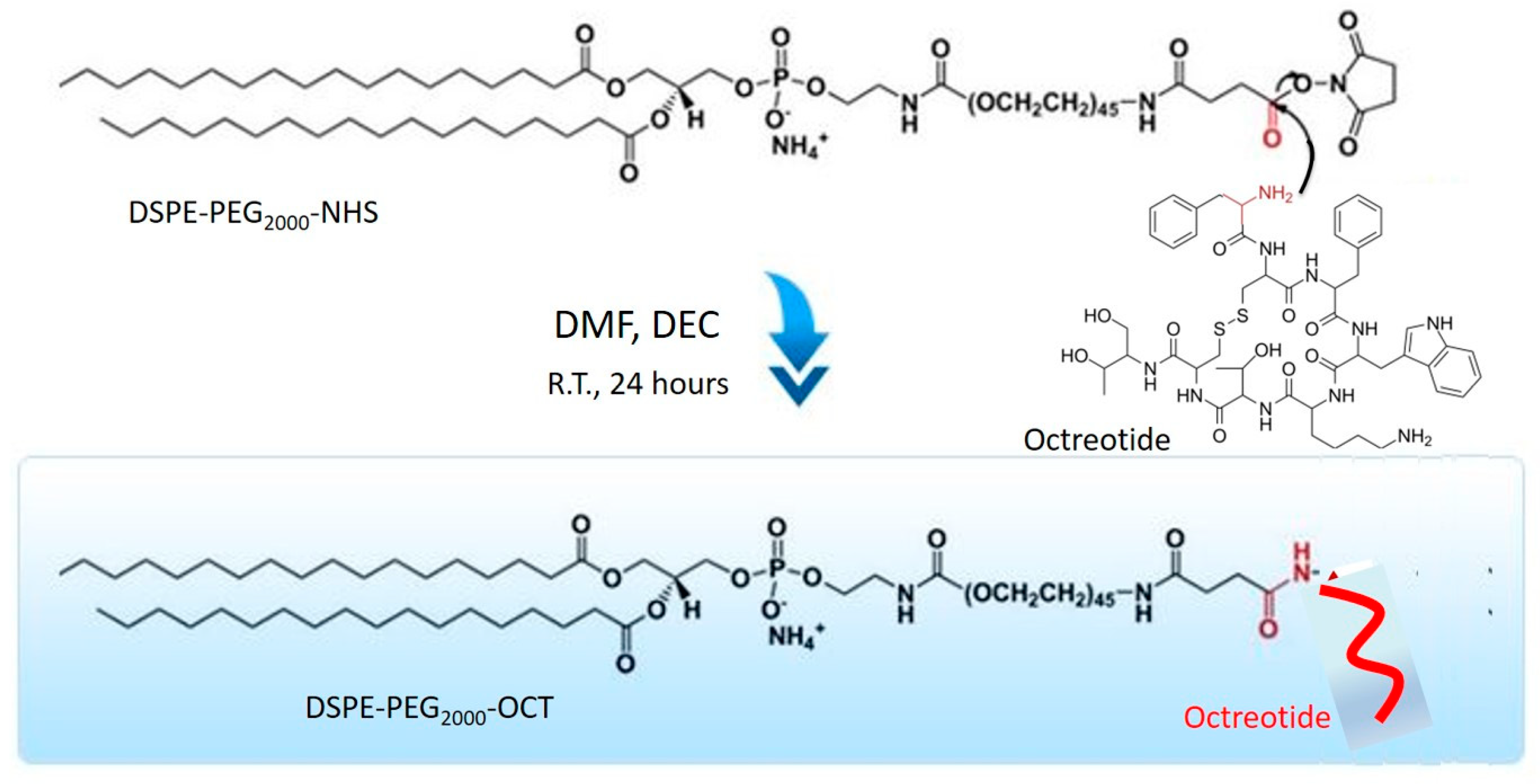
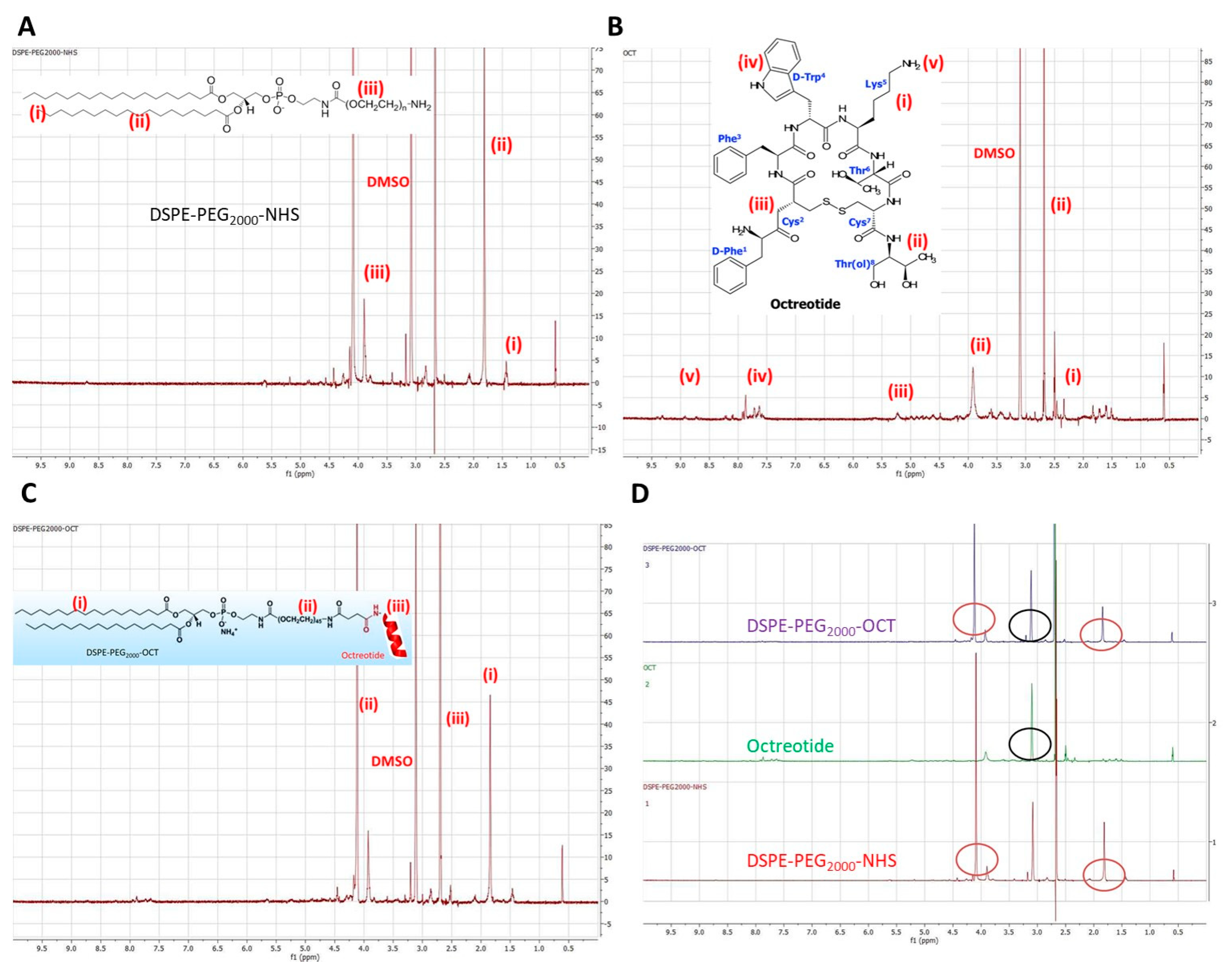
2.2. Synthesis of DSPE-PEG2000-OCT Graft Copolymer
Liposomes containing Lcn2 siRNA and surface targeted with octreotide were prepared by first targeting a particular liposomal cationic lipid with octreotide. DSPE-PEG2000-NHS containing a terminal-NHS group for conjugation was used for octreotide conjugation. Briefly, 3 mg of EDC, 4 mg of NHS and 10 mg of octreotide were mixed in 1 mmol of DSPE-PEG2000-NHS solution in PBS at pH 7.4. The reaction was carried for 12 h at dark at R.T. The conjugated lipid solution was added to Slide-A-Lyzer® dialysis cassette (MWCO 20 kDa) to remove the unreacted EDC, NHS, and octreotide. The resultant polymer solution containing DSPE-PEG2000-NHS conjugated to Octreotide (DSPE-PEG2000-OCT) was lyophilized.
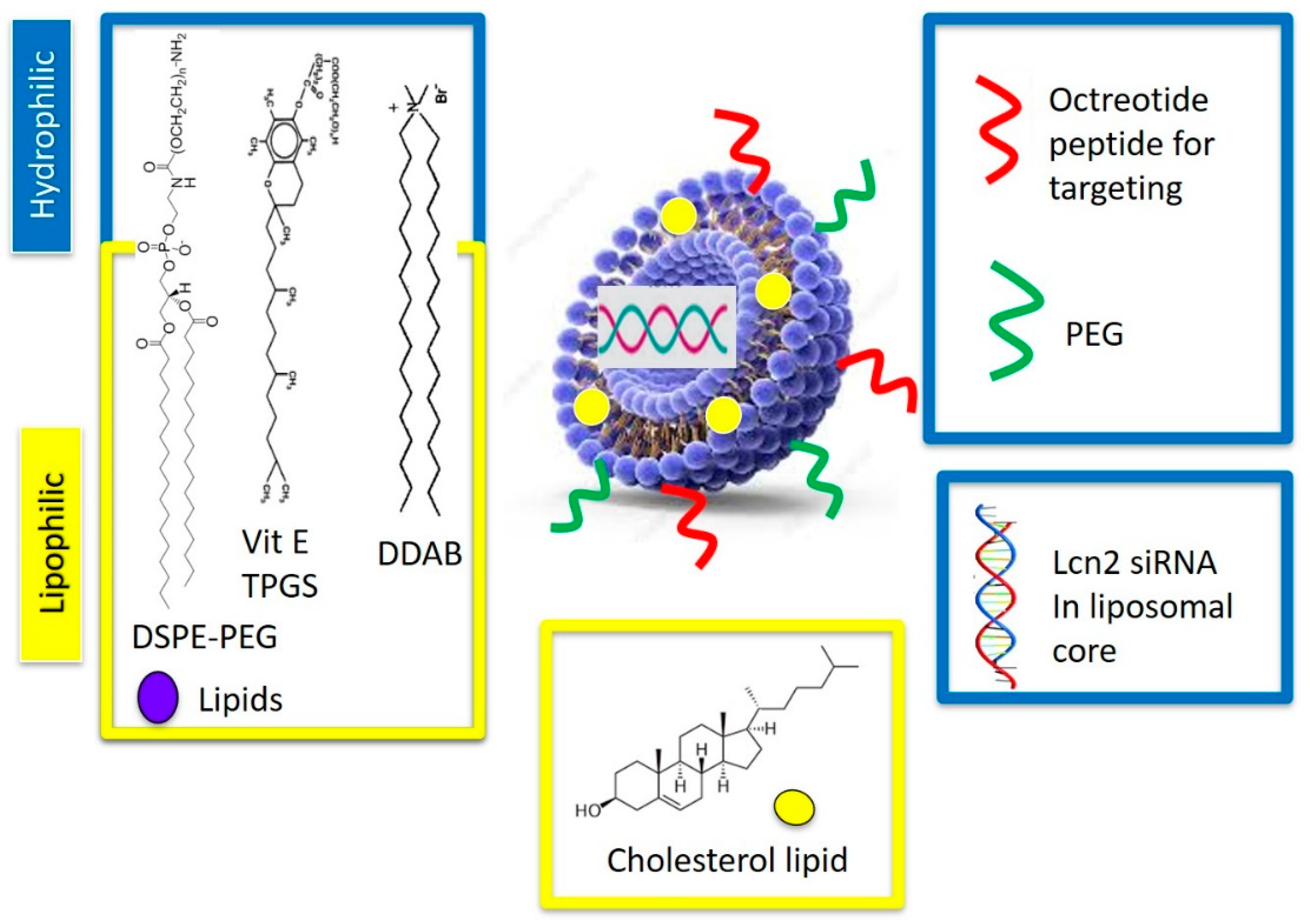
2.3. Liposomal Preparation and Optimization
Octreotide-targeted Lcn2 siRNA encapsulated liposomes (OCT-Lcn2-Lipo) were prepared by thin-film hydration method for liposome preparation. The liposomes were composed of lipids like DDAB, DSPE-PEG
2000-OCT, DSPE-PEG
2000-COOH, Cholesterol and Vitamin E TPGS in different molar ratios as depicted in
Table 1. The liposomal formulations L-1 to L-6 were developed by changing the molar ratios of DDAB and cholesterol lipids in the polymer formulation and keeping other lipids molar ratios constant. An important independent variable of liposomal formulation like the ratio of cationic lipids to cholesterol was changed here to see its effects on the critical quality attributes (CQA’s). Molar ratios of DSPE-PEG
2000-OCT, DSPE-PEG
2000-COOH, and Vitamin E TPGS in the liposomal formulations were constant. This ensured all liposomes had similar OCT targeting and PEG groups on the surface for enhancing tumor targetability and circulation time, respectively. Additionally, molar ratio of Vitamin E TPGS was also kept constant. This ensured that all the formulations could have the same P-gp inhibition potential. Changing fewer parameters of the liposomal formulation enabled preparation of formulations generating more meaningful information.
70 µmolar of the mixture of lipids was solubilized in chloroform and dried under vacuum at high speed under vacuum (Genevac, Ipswich, Suffolk, UK) for approximately 5 h. The resultant lipid film was reconstituted in 1× PBS solution using a bath sonicator. This results in the formation of multilaminar liposomes (MUV) and large laminar liposomes (LUV). A probe sonicator was used for size reduction and generation of single vesicular liposomes (SUV). At this step, 20 μg/mL Lcn2 siRNA or scrambled siRNA (SCR siRNA) was added to the liposomal mixture in PBS solution. Cationic lipids in the liposomes attract the negatively charged siRNA and thus result in encapsulation of the payload. Extrusion of nanomicellar solution was performed by 0.22 µm nylon syringe filter (Tisch Scientific, USA). Liposomal extrusion was followed by dialysis using Slide-A-Lyzer® dialysis cassette (MWCO 20 kDa) for 24 h at R.T. Formulated octreotide-targeted Lcn2 siRNA encapsulated liposomes (OCT-Lcn2-Lipo) were stored at 4 °C until further use. Lcn2-Lipo without octreotide (OCT) targeting were prepared in a similar way, but instead of DSPE-PEG2000-OCT, DSPE-PEG2000-NHS was used. Placebo OCT-Lipo was made with all the above-listed polymers for OCT-Lcn2-Lipo except the addition of Lcn2 siRNA was deleted. In a similar way, Placebo Lipo without OCT targeting, and Lcn2 siRNA targeting was formulated. For imaging studies, FITC-labelled siRNA (Santa Cruz Biotechnology) was used instead of Lcn2 siRNA.
2.3.1. Formulation Characterization: Size, Morphology, Zeta Potential
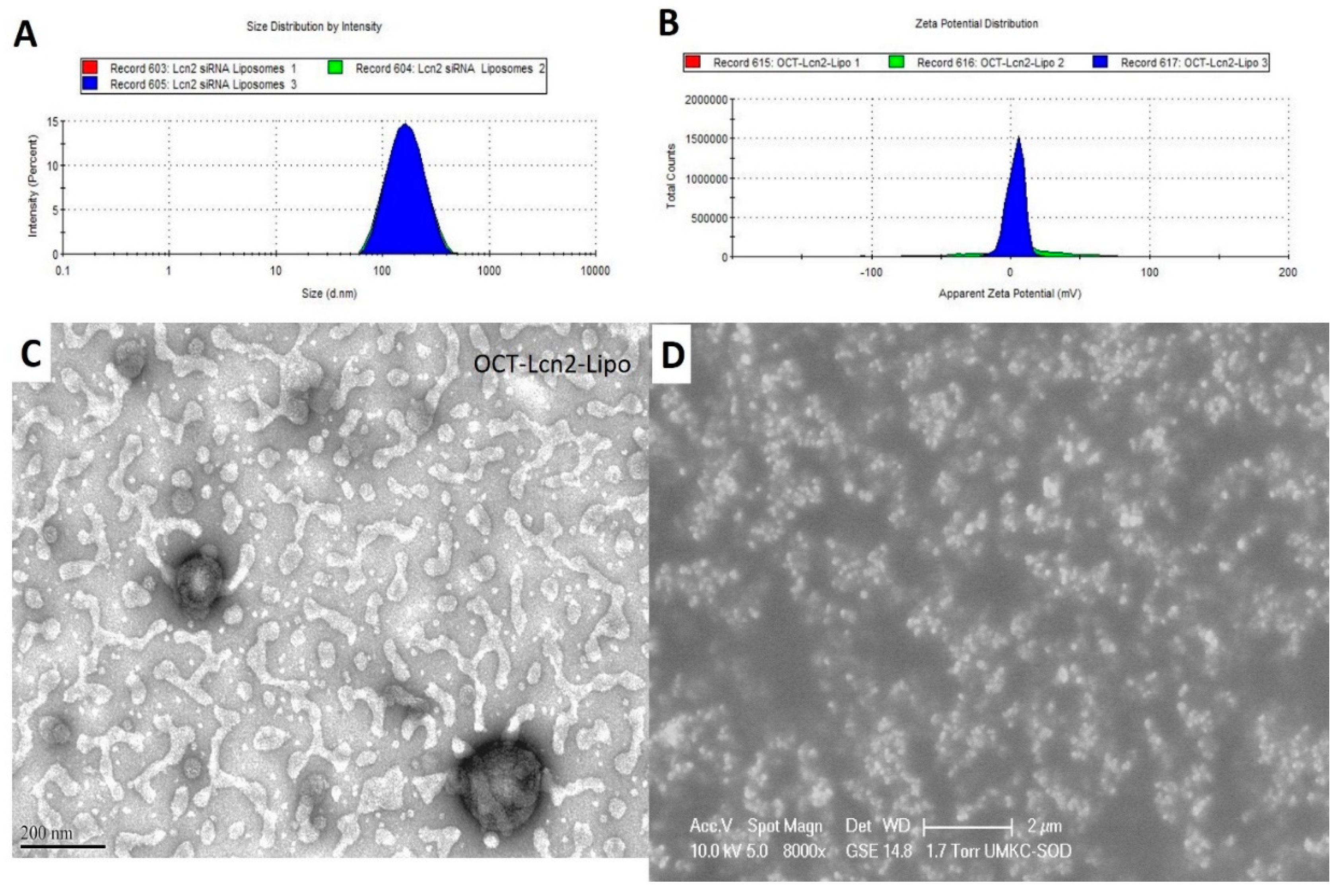
CQAs of OCT-Lcn2-Lipo were (i) hydrodynamic size, (ii) polydispersity index (PDI) and (iii) zeta potential and (iv) entrapment and loading efficiency. Size, PDI and zeta potential of OCT-Lcn2-Lipo was determined by Dynamic Light Scattering (DLS) Zetasizer Nano ZS (Malveran Instrument Ltd., Worcestershire UK). 700 µl of OCT-Lcn2-Lipo was placed in quartz cuvettes. Instrument was calibrated for measuring three values of hydrodynamic size and PDI. Hydrodynamic size was determined in nanometers (nm). For zeta potential, DTS1060 glass cuvettes were used. An average of three values were used for determining the final zeta potential and it was measured in millivolts (mV). The morphological characteristics of OCT-Lcn2-Lipo were visualized using Transmission Electron Microscopy (TEM). A drop of OCT-Lcn2-Lipo was placed on a copper grid sample holder. A layer of carbon and nitrocellulose was applied followed by staining the liposomes by 1% uranyl solution. The photographs were captured by a JEM 1200 EX II TEM at a voltage of 100 kV.
2.3.2. Lcn2 siRNA Entrapment and Loading Efficiency
Lcn2 siRNA entrapment and loading efficiency for OCT-Lcn2-Lipo was determined by Quant-It RiboGreen RNA assay according to the manufactures’ protocol. Human Lcn2 siRNA siGENOME SMARTpool is composed of a mixture of four Lcn2 siRNAs with the following sequence: (i) D-003679-03, GAAGACAAGAGCUACAAUG, (ii) D-003679-01, GAGCUGACUUCGGAACUAA, (iii) D-003679-02, GGAGCUGACUUCGGAACUA and (iv) D-003679-05, UGGGCAACAUUAAGAGUUA. Lcn2 siRNA standard calibration curve was plotted by serially diluted Lcn2 siRNA standard solutions on a on a Spectra-MaxPlus 384 UV-Visible Spectrophotometer at an excitation wavelength of 500 nm and emission wavelength of 525 nm. OCT-Lcn2-Lipo samples were prepared by mixing the liposomes in Triton X-100 solution. This causes lysis of the liposomes and the entrapped siRNA can be measured. Briefly, OCT-Lcn2-Lipo was centrifuged at 12,000 r.p.m. for 10 min, following by discarding the supernatant. Liposomal palette was resuspended in a lysis buffer containing 1.0 mL of 0.5% Triton X-100 and vortexed in a bath sonnicator for 5 min. This solution was incubated at 37 °C water bath for 1 h. 200 µL of the lysed OCT-Lcn2-Lipo solution was mixed with 200 μL diluted Quant-It RiboGreen RNA reagent working solution (diluted 200-fold) for 10 min incubation. 200 μL of this solution was added to a black clear-bottomed 96-well plate for measuring the fluorescence. 200 μL of 1.0 mL of 0.5% Triton X-100 mixed with 200 μL 200-fold diluted RiboGreen RNA reagent working solution was used as a blank control. entrapment and loading efficiencies were calculated by the following formula:
2.4. Dissolution Analysis
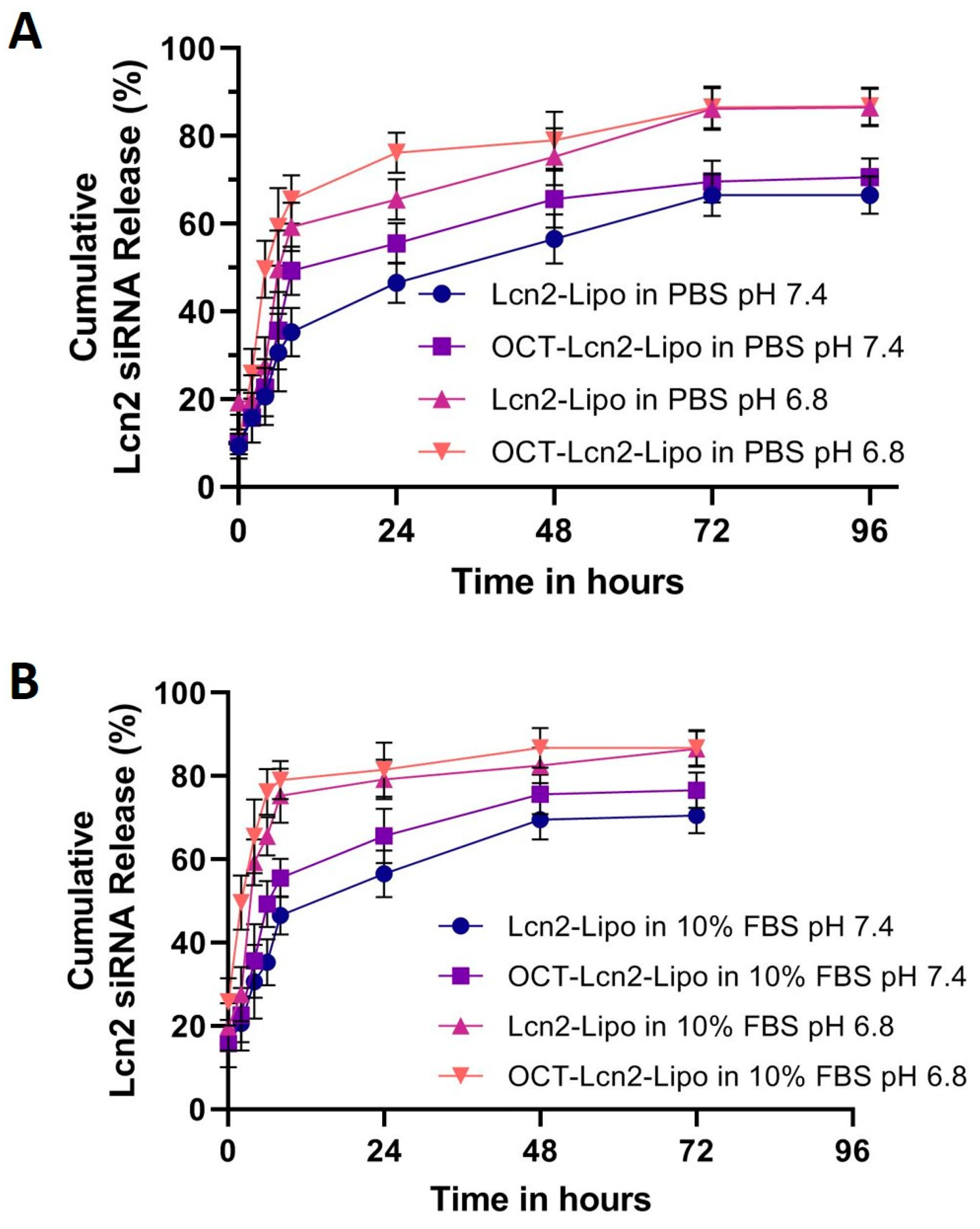
Dissolution study of OCT-Lcn2-Lipo and Lcn2-Lipo formulations was determined by calculating Lcn2 siRNA release from the formulations at predetermined time-points. 1000 µL of the formulations were suspended in 5.0 mL of (i) 1× PBS (pH 7.4 and pH 6.8) at and (ii) 10% FBS in 1× PBS (pH 7.4 and pH 6.8) separately, both maintained at 37 °C in a water-bath. At each time-point, the liposomal-diluted solution was centrifuged at 14,000 RPM for 20 min. 1.0 mL of the supernatant was collected, and fresh PBS was added. This helped in maintaining sink conditions, similar to physiological environment. Amount of Lcn2 siRNA released in the buffer medium was quantified by Quant-It RiboGreen RNA assay, as described for calculating OCT-Lcn2-Lipo encapsulation efficiency.
2.5. Dilution Studies
Dilution of OCT-Lcn2-Lipo formulation was carried out to determine the structural integrity of the liposomal formulation on dilution. OCT-Lcn2-Lipo dilution effects were compared to Lcn2-Lipo formulation. This study was carried out in two dilution media, (i) formulation buffer at R.T. and (ii) 10% FBS in PBS at 37 °C in a water-bath. Briefly, 1.0 mL of the liposomal formulations were diluted separately with both the dilution solutions. Dilutions up to 200-fold were carried out. This was followed by analysis of liposomal size by DLS Zetasizer Nano ZS (Malveran Instrument Ltd., Worcestershire UK).
2.6. Stability Studies
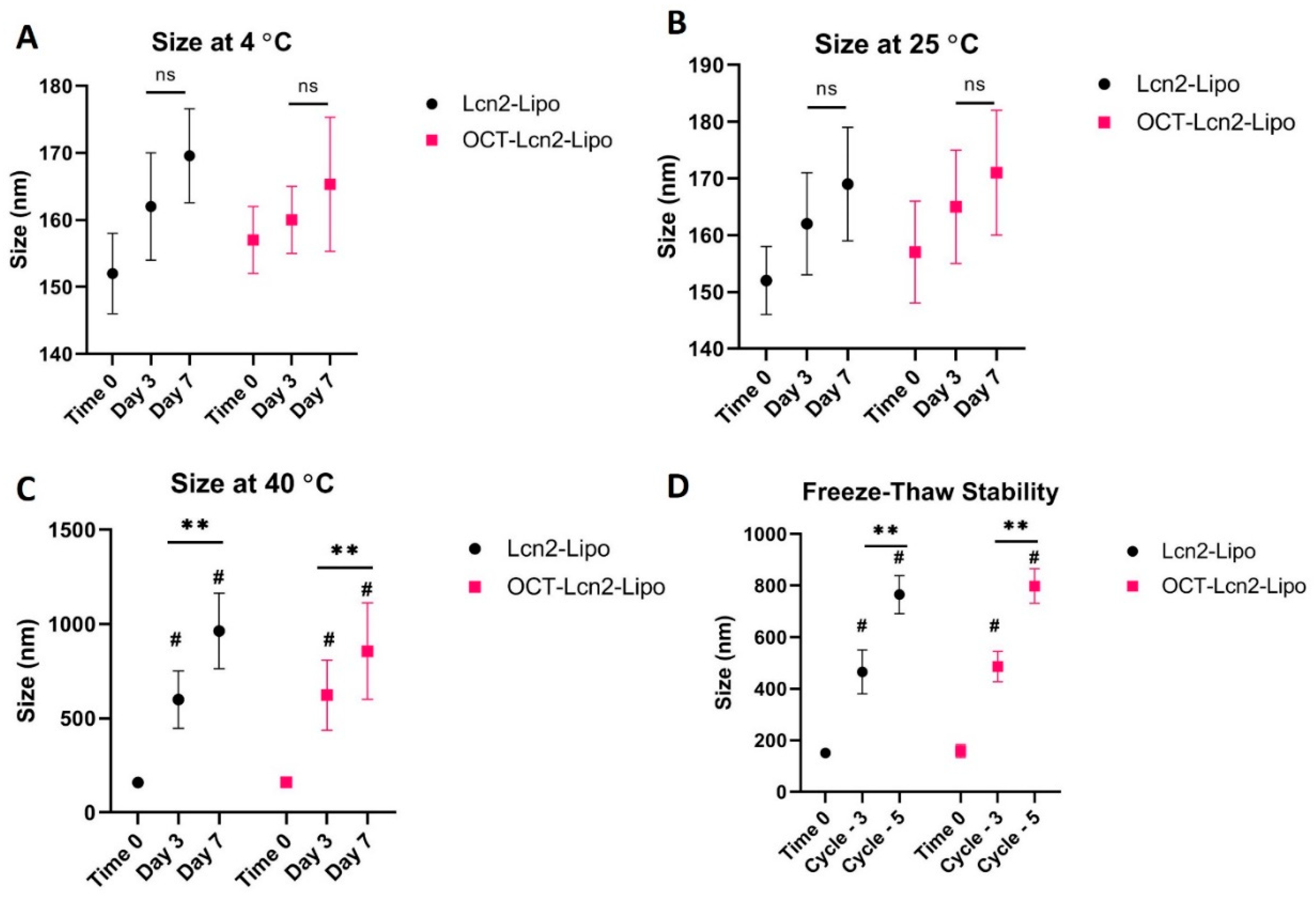
OCT-Lcn2-Lipo was evaluated for its storage stability, stress stability and plasma stability by analyzing its size and comparing it to the non-targeted Lcn2-Lipo formulation. Storage stability involved storage of OCT-Lcn2-Lipo and Lcn2-Lipo at temperatures like 4 °C, 25 °C, and 40 °C. Samples were withdrawn at Time 0, Day 3, and Day 7. Stress stability of the two formulations was evaluated by freeze-thawing the formulations three and five times. The results were compared to time 0. Plasma stability studies involved storage of OCT-Lcn2-Lipo and Lcn2-Lipo formulations in rat plasma at the ratio of; liposomes:10% FBS 1:1 v/v. Formulations were stored at physiological temperature (37 °C) in a water bath with constant shaking. This simulated physiological conditions. Samples were withdrawn at Day 3 and Day 7, which were compared to sample taken at Time 0. These stability studies were assessed by measuring the critical quality attributes like size of the liposomes at various conditions.
2.7. Cellular Uptake and Intracellular Distribution Study
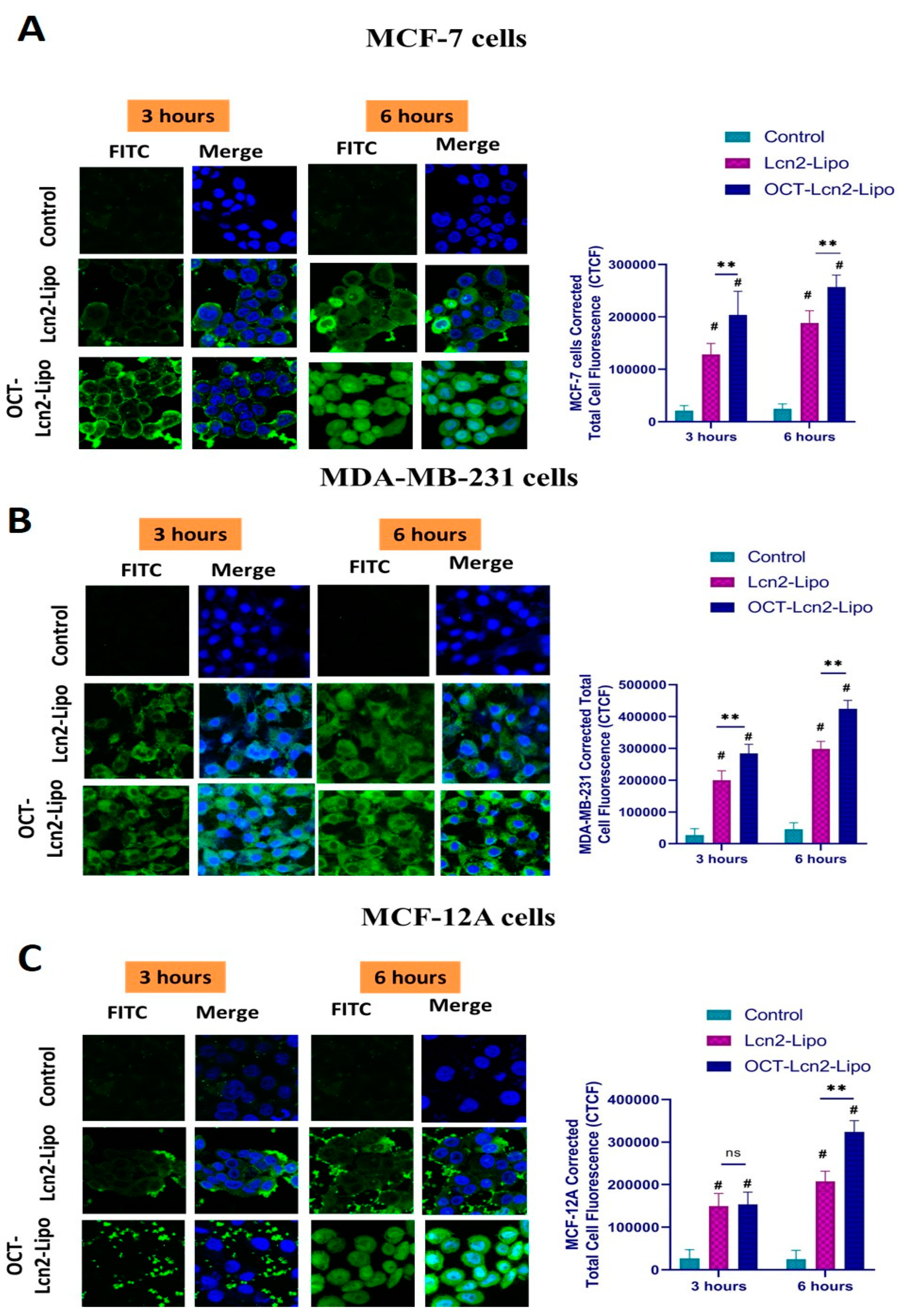
Cellular uptake and intracellular distribution of OCT-Lcn2-Lipo was determined in MCF-7, MDA-MB-231 and MCF-12A cell lines. FITC conjugated siRNA was used along with Lcn2 siRNA in the liposome preparation. Similarly, non-targeted Lcn2-Lipo was prepared having FITC conjugated siRNA entrapped in it along with Lcn2 siRNA. Cellular uptake of the liposomal formulations was determined by analyzing various treated cell lines by fluorescence-assisted cell sorting (FACS). In addition to this, intracellular distribution of the liposomes was determined with confocal laser scanning microscopy (CLSM).
2.7.1. Cellular Uptake by Flow Cytometry
Cellular uptake of OCT-Lcn2-Lipo and Lcn2-Lipo containing FITC Conjugated siRNA was determined by FACS. The uptake studies were carried out in MCF-7, MDA-MB-231 and MCF-12A cell line. Briefly the cells were seeded in a 24-well plate with 5 × 104 cells/well. 20 µL of OCT-Lcn2-Lipo and Lcn2-Lipo was added. All the cell lines used in this study, MCF-7, MBA-MD-231 and MCF-12A received the following three treatments; control, (serum-free media (SFM)), Lcn2-Lipo and OCT-Lcn2-Lipo. Cells were incubated for specific time points like, 1, 3, 6, and 12 h for determining uptake of various formulations. At each time point, the media was removed, and the cells were washed with Dulbecco’s Phosphate-Buffered Saline (DPBS) (Gibco’s). Cells were detached with the help of 200 µL ml of trypsin and collected in a FACS tube followed by centrifugation at 20,000 RPM for 5 min. The supernatant was discarded, and the pellet was washed twice with DPBS. This ensured removal of excess treatment groups. The final sample was made in DPBS to be used for flow cytometry. The mean fluorescence intensity of Lcn2-Lipo and OCT-Lcn2-Lipo were quantified by FACS at an excitation wavelength of 490 nm.
2.7.2. Intracellular Distribution Using CLSM
Intracellular distribution of OCT-Lcn2-Lipo and Lcn2-Lipo was determined in MCF-7, MDA-MD-231 and MCF-12 cells in a time-dependent manner using CLSM. Cells were seeded at 1 × 104 cells/well in an 8-chamber confocal microscopy slide (Nunc Lab-Tek II, Thermo Fisher Scientific). 10.0 µL of OCT-Lcn2-Lipo and Lcn2-Lipo was added. At each time point, the culture media was removed, followed by washing three times with DPBS (3 × 5 min). This ensured removal of excess treatment groups that were not internalized by the cells. This was followed by fixation step. Cold, buffered, 4% paraformaldehyde solution (200 µL) was added to each well. Cells were incubated at 37 °C for 20 min for fixation. After 20 min, the buffered paraformaldehyde solution was removed, and cells were washed with 200 µL DPBS (3 × 5 min). Finally, the cells were stained with mounting media containing DAPI for 15 min (Vectashield Antifade Mounting Medium). A coverslip was placed on top of the cells. The ends of the coverslip were sealed to prevent cellular dehydration and evaporation of the mounting media. Confocal microscopy slides were stored at 4 °C before imaging. Cells were observed under Leica CLSM (Leica TCS SP5, Wetzlar, Germany).
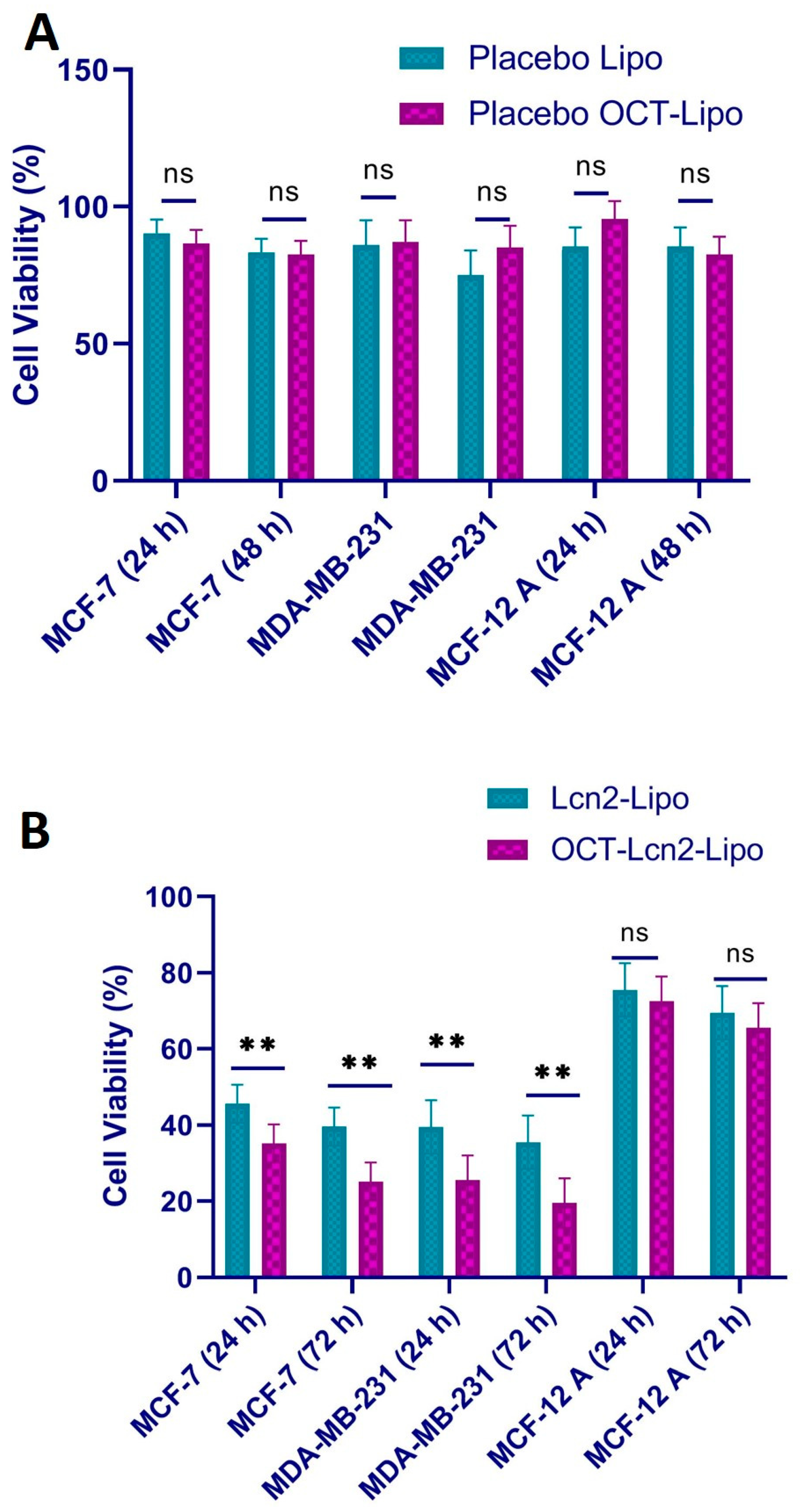
2.8. Cytotoxicity Study
Cellular cytotoxicity of OCT-Lcn2-Lipo, Lcn2-Lipo, placebo OCT-Lipo and placebo Lipo was determined in breast cancer cell line MCF-7, TNBC cell line: MBA-MD-231 and in normal breast epithelium cell line: MCF-12A. In vitro cytotoxicity was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell viability assay. MTT is a yellow tetrazole dye commonly used for determining cell viability. Cell lines were seeded in 96-well plates at a cell density of 1 × 10
4 cells/well suspended in 200 µL of complete media. Cells were then incubated overnight in an incubator maintained at 37 °C in a 5% CO
2 environment and 90% relative humidity. In vitro cytotoxicity of OCT-Lcn2-Lipo, Lcn2-Lipo, placebo OCT-Lipo and placebo Lipo formulations was determined for 24 and 72 h after initial treatment with the liposomes for 6 h. After 6 h, the media was replaced, and fresh media was added to the cells. After 24 and 72 h, cell viability was measured by MTT assay. MTT stock solution was prepared by adding MTT reagent A and reagent B in the ratio 100:1. 20 µl of MTT reagent was added to each well followed by incubation for 3 h. Absorbance of formazan solution was measured using a 96-well microplate reader at an excitation wavelength of 485 nm (BioRad, Hercules, CA, USA). A % Triton-X prepared in serum-free media (SFM) served as the positive control and SFM without any treatment served as the negative control. Cell viability was calculated according to the formula.
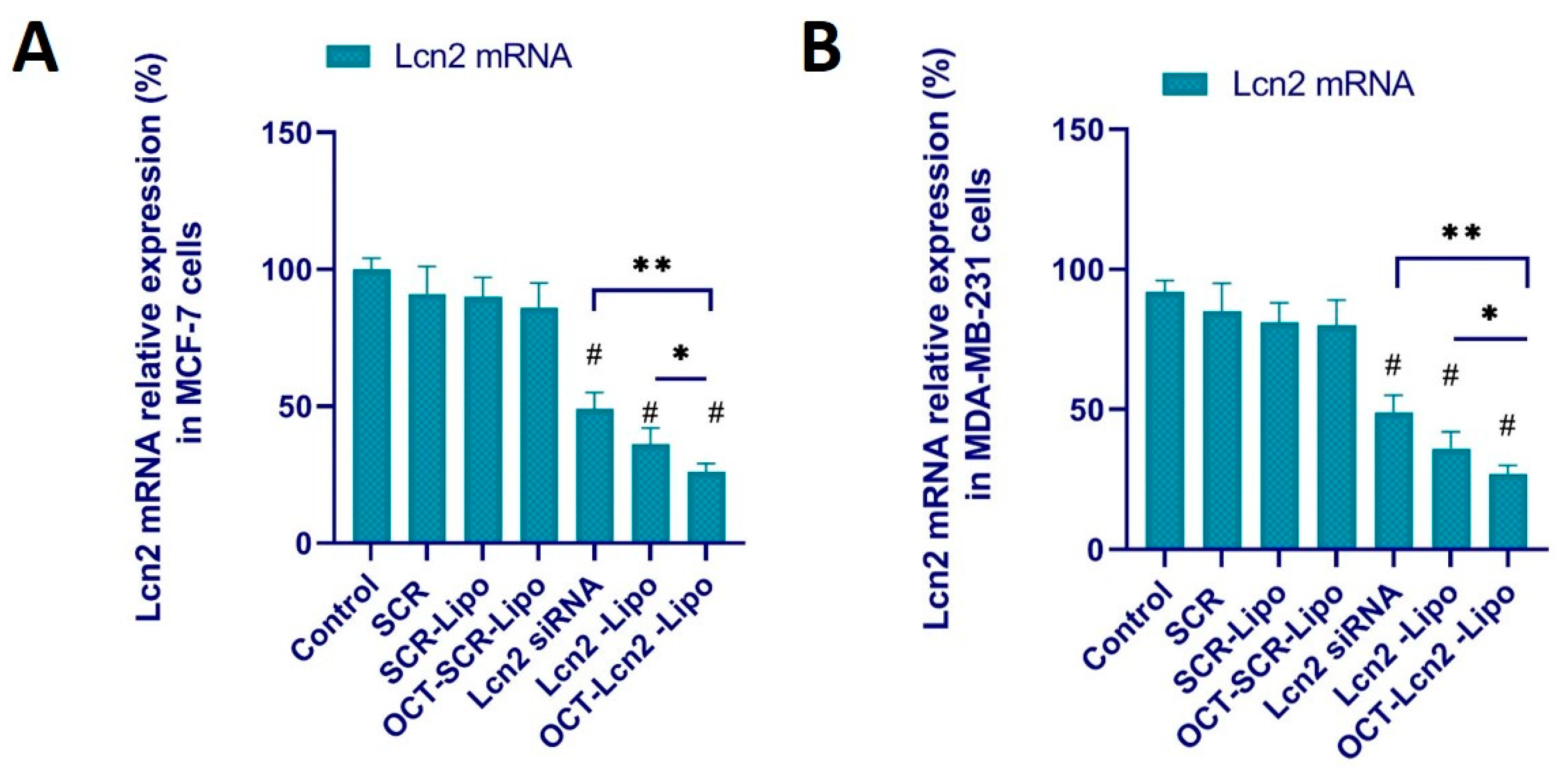
2.9. Lcn2 siRNA Knockdown Efficacy
Real time RT-PCR was used to examine Lcn2 mRNA expression in MCF-7 and MDA-MB-231 cells following OCT-Lcn2-Lipo treatment. Briefly, 5 × 105 MCF-7 and MDA-MB-231 cells were seeded in 6-well plates and incubated for 24 h. Cells were treated (i) PBS (control), (ii) SCR, (iii) SCR-Lipo (iv) OCT-SCR-Lipo, (v) Lcn2, (vi) Lcn2-Lipo and (vii) OCT- Lcn2-Lipo at the final Lcn2 siRNA/SCR concentration of 100 nm. Cells were rinsed thrice with DPBS and further grown for 72 h. RNA was isolated from the breast cancer cells using the RNeasy Mini Kit (QIAGEN) according to the manufacturer’s protocol. cDNA was synthesized in RT-PCR process using the Reverse Transcriptase (BioRad), and levels of Lcn2 mRNA were quantified using a mixture of forward and reverse Lcn2 primers and SYBER green. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the positive control.
2.10. Angiogenic Assay
2.10.1. Generation of Conditioned Media (CM)
Breast cancer cells MCF-7 and TNBC cells MDA-MB-231 were treated with (i) PBS (control), (ii) SCR, (iii) SCR-Lipo (iv) OCT-SCR-Lipo, (v) Lcn2, (vi) Lcn2-Lipo and (vii) OCT- Lcn2-Lipo at the final Lcn2 siRNA/SCR concentration of 100 nm. The cells were seeded at a density of 3 × 10
5 in a 6-well plate following addition of the treatment’s groups to the culture media. After treating the cells for 72 h, DMEM media containing the treatment groups was removed. Cells were washed twice with PBS and 1 mL of fresh serum-free DMEM was added. After 24 h, this conditioned media (CM) was removed and centrifuged at 14,000 RPM for 5 min. This helped in the removal of cellular organelles and cells. This collected supernatant was used for further in vitro anti-angiogenesis studies [
1,
2,
3,
4].
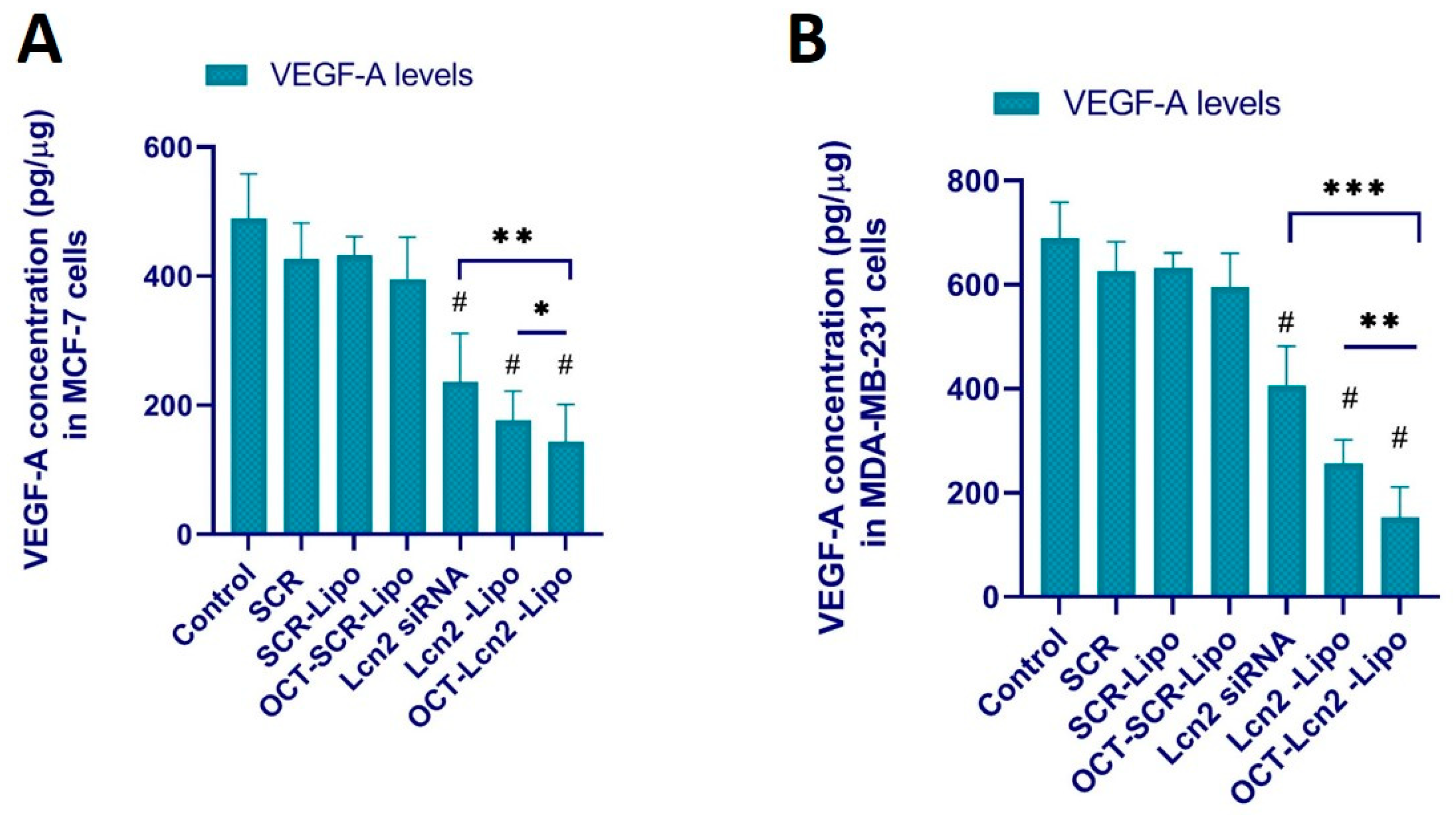
2.10.2. Determination of VEGF-A Levels
CM produced by breast cancer cells MCF-7 and TNBC cells MDA-MB-231 as described above was utilized to measure the human VEGF-A levels by ELISA. VEGF-A levels in the CM was determined by human VEGF-A ELISA kit from R&D Systems (Minneapolis, MN, USA) [
2].
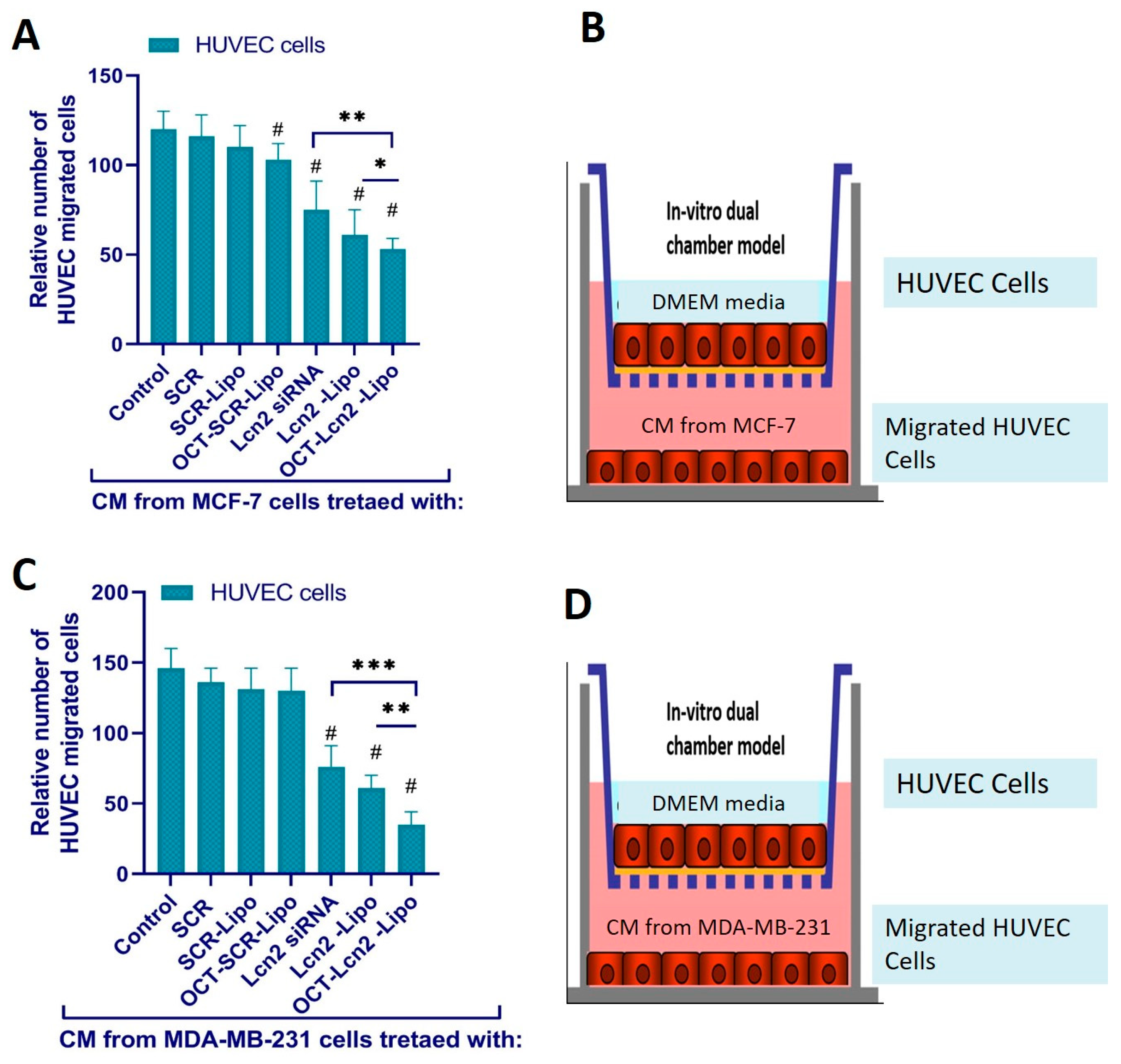
2.10.3. Endothelial Cell Migration Assay
Primary Umbilical Vein Endothelial Cells (HUVEC) (ATCC Mansas VA) were used for this assay. These cells were seeded on the upper chamber of COSTAR transwell filters. The transwell filters had a pore size of 8 µ m and were made from permeable polycarbonate membrane. [
2,
5]. The CM harvested from MCF-7 and TNBC cells MDA-MB-231 cells treated with (i) PBS (control), (ii) SCR, (iii) SCR-Lipo (iv) OCT-SCR-Lipo, (v) Lcn2, (vi) Lcn2-Lipo and (vii) OCT- Lcn2-Lipo at the final Lcn2 siRNA/SCR concentration of 100 nm was used in the is experiment. HUVEC cells seeded on the upper chamber of the trans well inserts were supplemented with CM and serum-free DMEM was added to the bottom chamber. The cells were incubated for 24 h. HUVEC migrated to the opposite/bottom side of the filter insert through the 8 μm pores were counted by Diff-Quik Stain Set. An average of four fields was counted for each sample [
5].
2.11. Statistical Analysis
Significance in each study was determined by using at least three replicated for each experiment. The data is represented as mean ± standard deviation (SD). Statistical significance between different study groups was analyzed by two-way ANOVA or Student’s t-test. P < 0.05 indicated statistical significance in all experiments.
4. Discussion
Targeting mRNA and reducing the protein expression in breast cancer using RNA interference is under intensive investigation [
45,
46,
47]. Many such approaches primarily use liposomal drug delivery systems or lipid nanoparticles to ensure higher entrapment efficiency of siRNA [
45,
46,
47]. The lower rate of survival for metastatic breast cancer is mainly due to its rapid progression into organs like lungs, bone, and blood. To combat this, a targeted therapy specifically targeting breast cancer cells and decreasing their angiogenic and metastatic behavior is the need of the hour. Here, we have recognized Lcn2 as a therapeutic to treat metastatic breast cancer. Lcn2 protein abnormal expression initiates the epithelial-to-mesenchymal transition (EMT) process, cell migration and invasion, and angiogenesis in breast cancer. Particularly, Lcn2 functions as an initiator of metastasis and carcinogenesis by involving multiple signaling pathways, including PI
3K/AKT/NF-κB and HIF-1α/Erk [
48]. Hence, silencing Lcn2 in breast cancer cells can help to reduce metastatic breast cancer and triple-negative breast cancer progression [
4,
44,
49]. Here, effective delivery of Lcn2 siRNA to breast cancer cells MCF-7 and TNBC MDA-MB-231 was achieved by encapsulating Lcn2 siRNA in cationic liposomes and decorating them on the surface with Octreotide (OCT) peptide ligand. Here, we demonstrated that somatostatin receptor overexpression on MCF-7 and MDA-MB-231 cells can be used for selective delivery of cargo to breast cancer cells. Here we engineered, a liposomal drug delivery carrier for Lcn2 siRNA for its effective delivery to breast cancer cells. The liposome was targeted with OCT for selective uptake by breast cancer cells due to abnormally higher expression of somatostatin receptors on their surface.
The liposome consisted of double chain cationic polymers like DDAB, DSPE-PEG
2000-OCT, DSPE-PEG
2000-COOH were utilized in the liposomal preparation. Cationic lipids aid in effective encapsulation and entrapment of negatively charged siRNA as compared to neutral lipids. Such lipids also help in achieving a net positive charge to the liposomes [
14,
15]. This helps in effective binding and internalization of the cationic liposomes in the negatively charged cell membrane. [
16,
17]. Double aliphatic chain of these polymers assists in liposomal formation, which forms a lipid bilayer separating inner aqueous environment from the outer aqueous environment. These polymers are amphiphilic in nature. The hydrophobic aliphatic chains form the inner portion of the lipid bilayer, while the hydrophobic groups form the outer portion [
18]. DDAB is pH-sensitive lipid that is incorporated into the lipid bilayer of OCT-Lcn2-Lipo formulation. The pH-sensitive nature aids in effective transfection of the cells by triggering an early endosomal escape of the liposomes into the cytoplasm [
50,
51]. Terminal 2000 KDa PEG group in the polymer: DSPE-PEG
2000-COOH, has been proven to improve liposomal serum circulation time and biocompatibility [
19,
20]. They also prevent aggregation of the liposomes by providing a hydrophobic shield over the lipid bilayer and stabilize surface charge of the liposomes. Their property of shielding the liposome’s surface charge prevents the liposome uptake by reticuloendothelial system and degradation by serum proteins when administered in vivo [
21,
22]. PEG also provides “stealth” properties to the liposomes, which can significantly prolong the circulation time [
19,
52]. PEGylation strategy utilized in Doxil liposomal nanoformulation to increase circulation time [
53]. Additionally, cholesterol increases the liposomal stability by conformational ordering of lipid chains. Cholesterol molecules are lipophilic and have a rigid planar steroid structure. They arrange themselves within the liposomal lipids and increase the packing density of the liposome [
23]. Vitamin E TPGS added in the liposomal formulation acts as a P-gp inhibitor to overcome MDR in metastatic breast cancer cells [
24,
26]. Additionally, Vitamin E TPGS can also act as a stabilizing agent, solubilizer and bioavailability enhancer [
27,
28,
29]. Lcn2 siRNA was encapsulated within the cationic liposome by solvent evaporation-film rehydration method of liposomal preparation. Lcn2 siRNA has demonstrated reduction in lipocalin 2 protein, which is a proangiogenic factor in TNBC cell line MDA-MB-231 [
2,
30]. Selective targeting of metastatic breast cancer cells was achieved by targeting somatostatin receptors (SSTRs). SSTRs, are highly expressed on various cancer cells like hepatic carcinoma, neuroendocrine cancers, and ovarian and cervical [
31,
32,
33]. Octreotide peptide is an analog of somatostatin (SST) growth hormone, which targets SSTRs. This octreotide (OCT) can function as a targeting agent to selectively target breast cancer cells [
7].
Guo et al. reported that intercellular adhesion molecule-1 (ICAM)-targeted Lcn2 siRNA- encapsulating liposome (ICAM-Lcn2-LP) can effectively knockdown Lcn2 mRNA and cause a significant reduction in VEGF production from MDA-MB-231 cells. In addition, ICAM-Lcn2-LP demonstrated selective uptake in TNBC cells; MDA-MB-231 as compared to normal breast epithelium MCF-10A [
2]. Guo et al. also demonstrate that Lcn2 silencing, along with inhibiting the C-X-C chemokine receptor type 4 (CXCR4) can significantly reduce migration in TNBC. Here, they formulated liposomes modified with anti-CXCR4 antibodies on the surface, encapsulating Lcn2 siRNA for targeting MBC cells and for blocking the migration along CXCR4-CXCL12 axis in breast cancer. Here they achieved 88% Lcn2 silencing in MDA-MB-436 and 92% Lcn2 silencing in for MDA-MB-231 cells. Such results suggest that liposomes engineered to tackle multiple migratory pathways can be effective to slow progression of MBC [
30]. Ju et al. developed and characterized OCT-modified liposomes containing daunorubicin and dihydroartemisinin as a treatment to prevent MBC. The in vitro results in MDA-MB-435S cells demonstrated higher cellular uptake and targeting by OCT-modified liposomes by OCTSSTRs (somatostatin receptors). In addition, the in vivo results exhibited a higher accumulation in tumor site along with a robust overall antitumor efficacy and negligible toxicity in MDA-MB-435S xenograft mice [
7]. Zhang et al. developed octreotide (Oct)-targeted paclitaxel (PTX)-encapsulated PEG-b-PCL polymeric micelles (Oct-M-PTX) for targeting breast cancer and breast cancer stem cells. Oct-M-PTX produced the strongest antitumor efficacy, in vitro. The therapy also was found to be effective in suppressing breast cancer stem cells in vivo [
6].
Here, OCT-Lcn2-Lipo was formulated and optimized to achieve maximum entrapment efficiency and lowest size. Solvent evaporation-film rehydration method was used for the formulation preparation. A total of 100 nm of Lcn2 siRNA was utilized for formulation preparation. Optimized formulation, L-2 had the least PDI and the highest encapsulation and loading efficiencies for Lcn2 siRNA. L-The ratio of various lipids in the liposome were: DDAB: DSPE-PEG2000OCT: DSPE-PEG2000-COOH: Cholesterol: Vitamin E TPGS: 50:10:10:20:10. OCT-lcn2-Lipo, the optimized formulation had a size of 152 had a size of 152.00 nm, PDI 0.13, zeta potential 4.10 mV, entrapment and loading efficiencies of 69.5% and 7.8 %, respectively. OCT-Lcn2-Lipo dilution study indicated that there was no significant difference in liposomal size when the liposomes were diluted up to 100 times in PBS solution. However, swelling and increase in size was observed when OCT-Lcn2-Lipo was incubated in 10% FBS solution diluted up to 200 times and stored at body temperature. Temperature stability indicated that OCT-Lcn2-Lipo was stable up to Day-7 at 4 °C and 25 °C. The OCT-Lcn2-Lipo demonstrated an increase in size as compared to time 0 when stored at 40 °C and when subjected to freeze-thawing.
In vitro uptake of OCT-Lcn2-Lipo in MCF-7 and MDA-MB-231 cells using flow cytometry determined the effective internalization of the liposomes in a time-dependent manner. There was a significant difference in the uptake of OCT-Lcn2-Lipo as compared to Lcn2-Lipo at 6 h in both the cell lines. Uptake plateaued around 12 h. In vitro uptake was also determined in MCF-12 normal breast epithelium cells. The total uptake for the normal cells was lower than the uptake for the breast cancer cells. Similar results were also obtained for intracellular distribution study using confocal microscopy. In vitro cytotoxicity study depicted that the placebo liposomes were safe on all the above three cell lines. While the OCT-Lcn2-Lipo was reduced cell viability to a significantly greater extent in MCF-7 and MDA-MB-231 cells as compared to Lcn2-Lipo. This can imply that OCT targeting can result in higher accumulation of Lcn2 siRNA in the breast cancer cells as compare to the non-targeted liposomes. The cell viability was 80–90% for MCF-12A cells treated with OCT-Lcn2-Lipo and Lcn2-Lipo. This can imply that OCT-Lcn2-Lipo was safe and non-toxic on breast cancer cells.
OCT-Lcn2-lipo could achieve approximately 55–60% silencing of Lcn2 mRNA in MCF-7 and MDAMB-231 cells, respectively. Several research groups have reported the use of lipid drug delivery system for siRNA as an anti-angiogenic therapy [
54,
55,
56,
57]. To assess the anti-angiogenic potential of OCT-Lcn2-Lipo, VEGF-A levels and HUVEC cell migration assay was performed. First, all the cells were treated with OCT-Lcn2-Lipo and other treatment groups, and the media from the cells was separated. This was the CM, which was used for VEGF-A level detection and HUVEC cell migration assay. VEGF-A expression was significantly reduced in OCT-Lcn2-Lipo treatment group as compared to control group where MCF-7 and MDA-MB-231 cells were untreated. Similarly, the total number of HUVEC cells migrating toward the CM from breast cancer cells treated with OCT-Lcn2-Lipo was significantly lower than the control group of breast cancer cells. These results established the in vitro anti-angiogenic potential of OCT-Lcn2-Lipo in MCF-7 and MDA-MB-231 cells.


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}