Hippo Signaling Mediates TGFβ-Dependent Transcriptional Inputs in Cardiac Cushion Mesenchymal Cells to Regulate Extracellular Matrix Remodeling


 ,
, 
Abstract
:
1. Introduction
2. Material and Methods
2.1. Cushion Mesenchymal Cell Culture and Drug Treatment
2.2. Immunofluorescence Staining and Immunoblotting
2.3. Collagen Gel Contraction Assays
2.4. RNA Isolation, cDNA Synthesis, and Quantitative PCR
2.5. Western Blot Analysis
2.6. Statistical Analysis
3. Results
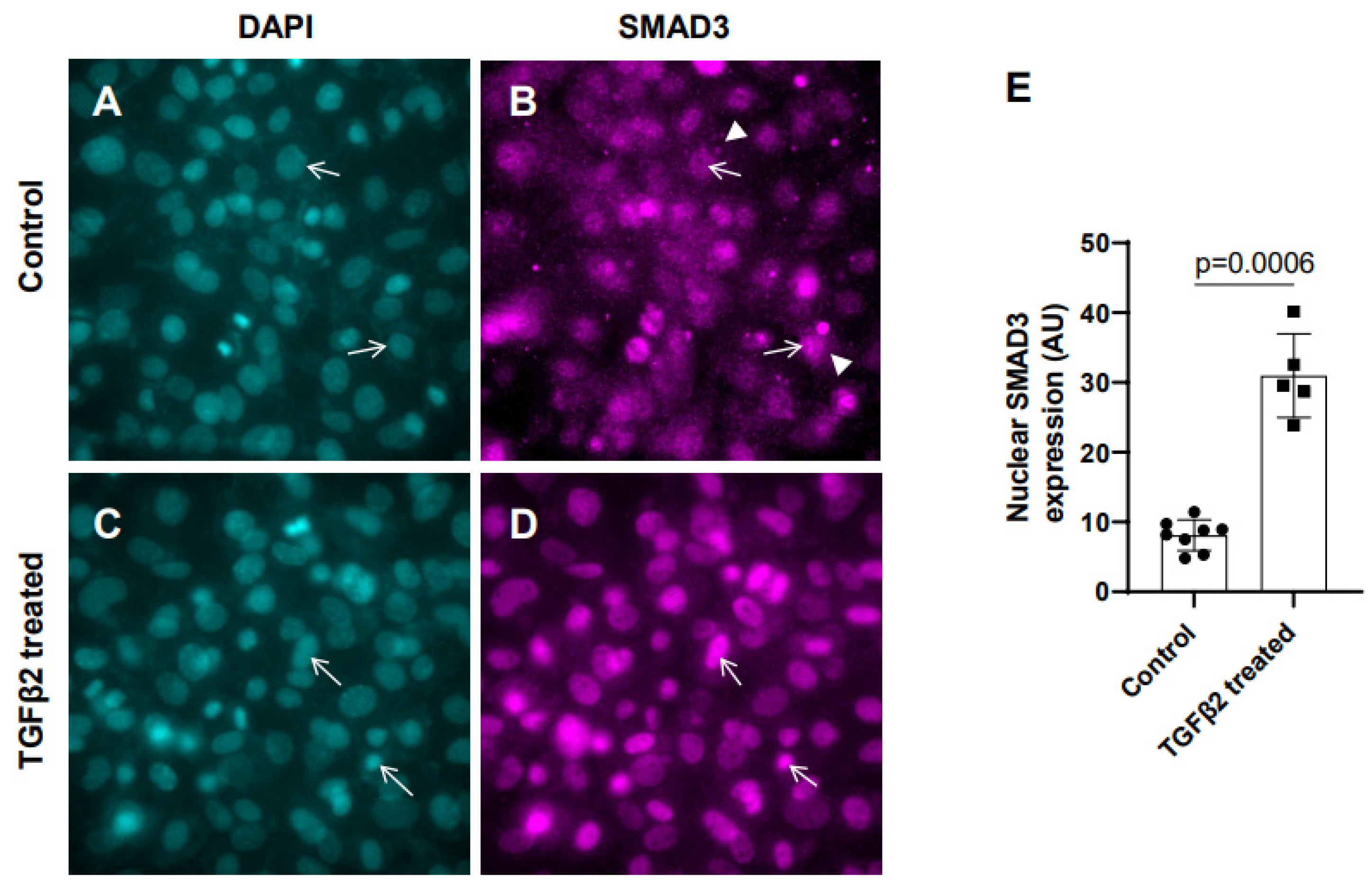
3.1. Exogenous TGFβ2 Induced SMAD3 Activation in Cushion Mesenchymal Cells
3.2. TGFβ2 Treatment Leads to Nuclear Translocation of YAP1 in Cushion Mesenchymal Cells
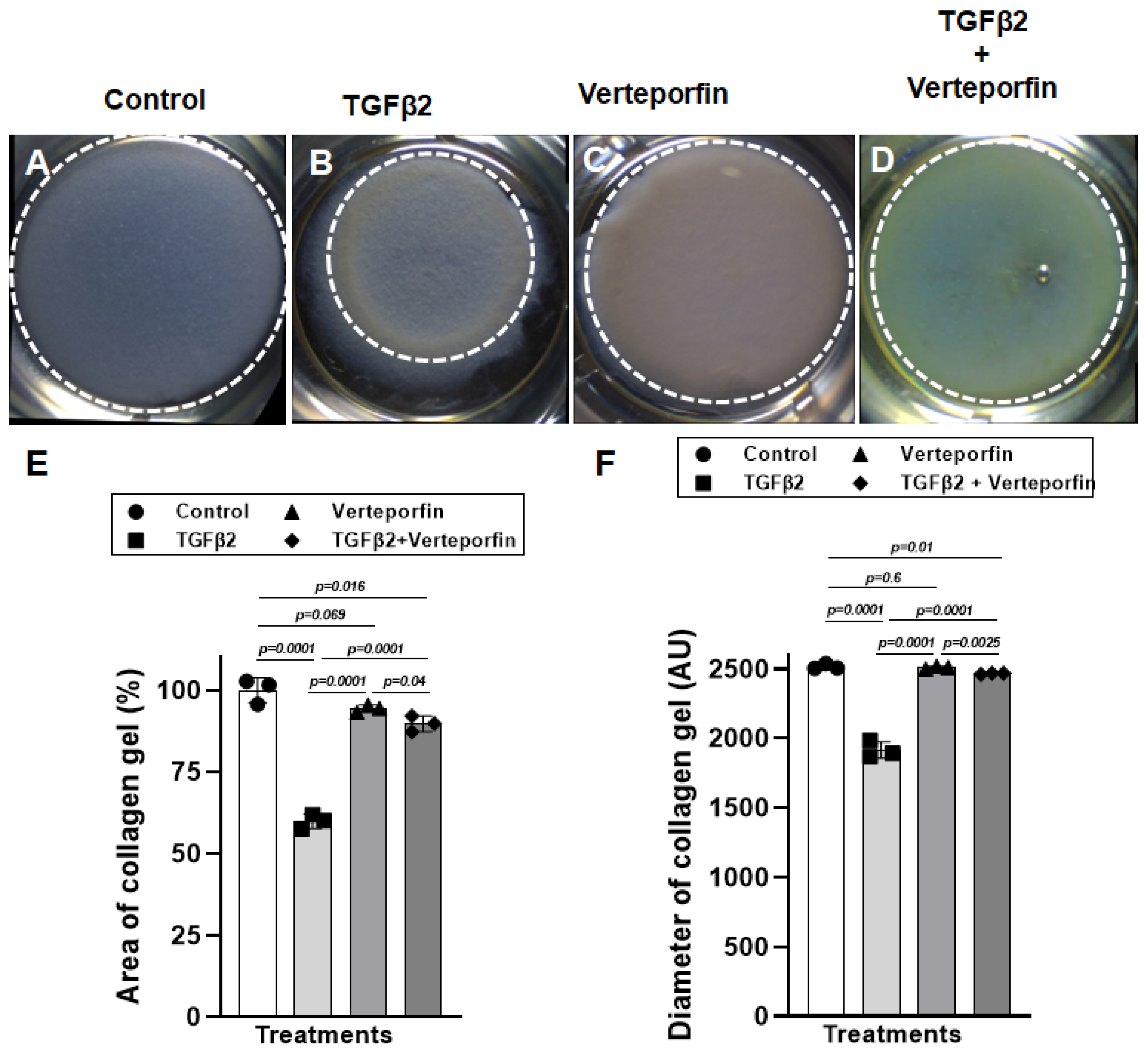
3.3. Suppression of YAP1 Inhibits TGFβ2-Induced Cell-Matrix Remodeling in Cushion Mesenchymal Cells
3.4. YAP1 Inhibitor Attenuated TGFβ2-Induced Expression of Genes Involved in Cushion Mesenchymal Cell-Matrix Remodeling
3.5. YAP1 Inhibition Decreases Activation of SMAD2 and SMAD3
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gittenberger-de Groot, A.C.; Bartelings, M.M.; DeRuiter, M.C.; Poelmann, R.E. Basics of cardiac development for the understanding of congenital heart malformations. Pediatr. Res 2005, 57, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Grewal, N.; DeRuiter, M.C.; Jongbloed, M.R.; Goumans, M.J.; Klautz, R.J.; Poelmann, R.E.; Gittenberger-de Groot, A.C. Normal and abnormal development of the aortic wall and valve: Correlation with clinical entities. Neth. Heart J. 2014, 22, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Bosse, K.; Hans, C.P.; Zhao, N.; Koenig, S.N.; Huang, N.; Guggilam, A.; LaHaye, S.; Tao, G.; Lucchesi, P.A.; Lincoln, J.; et al. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J. Mol. Cell Cardiol. 2013, 60, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Poelmann, R.E.; Gittenberger-de Groot, A.C.; Goerdajal, C.; Grewal, N.; De Bakker, M.A.G.; Richardson, M.K. Ventricular Septation and Outflow Tract Development in Crocodilians Result in Two Aortas with Bicuspid Semilunar Valves. J. Cardiovasc. Dev. Dis. 2021, 8, 132. [Google Scholar] [CrossRef] [PubMed]
- Burns, T.; Yang, Y.; Hiriart, E.; Wessels, A. The Dorsal Mesenchymal Protrusion and the Pathogenesis of Atrioventricular Septal Defects. J. Cardiovasc. Dev. Dis. 2016, 3, 29. [Google Scholar] [CrossRef] [PubMed]
- Calkoen, E.E.; Hazekamp, M.G.; Blom, N.A.; Elders, B.B.; Gittenberger-de Groot, A.C.; Haak, M.C.; Bartelings, M.M.; Roest, A.A.; Jongbloed, M.R. Atrioventricular septal defect: From embryonic development to long-term follow-up. Int. J. Cardiol. 2016, 202, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Neeb, Z.; Lajiness, J.D.; Bolanis, E.; Conway, S.J. Cardiac outflow tract anomalies. Wiley. Interdiscip. Rev. Dev. Biol. 2013, 2, 499–530. [Google Scholar] [CrossRef]
- Attisano, L.; Wrana, J.L. Signal integration in TGF-beta, WNT, and Hippo pathways. F1000Prime Rep. 2013, 5, 17. [Google Scholar] [CrossRef]
- Tsai, C.R.; Martin, J.F. Hippo signaling in cardiac fibroblasts during development, tissue repair, and fibrosis. Curr. Top. Dev. Biol. 2022, 149, 91–121. [Google Scholar] [CrossRef]
- Piersma, B.; Bank, R.A.; Boersema, M. Signaling in Fibrosis: TGF-beta, WNT, and YAP/TAZ Converge. Front. Med. 2015, 2, 59. [Google Scholar] [CrossRef]
- Nallet-Staub, F.; Yin, X.; Gilbert, C.; Marsaud, V.; Ben Mimoun, S.; Javelaud, D.; Leof, E.B.; Mauviel, A. Cell density sensing alters TGF-beta signaling in a cell-type-specific manner, independent from Hippo pathway activation. Dev. Cell 2015, 32, 640–651. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The role of transforming growth factor (TGF)-beta in the infarcted myocardium. J. Thorac. Dis. 2017, 9, S52–S63. [Google Scholar] [CrossRef] [PubMed]
- Hanna, A.; Humeres, C.; Frangogiannis, N.G. The role of Smad signaling cascades in cardiac fibrosis. Cell Signal. 2021, 77, 109826. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, M.E.; Schepers, D.; Bolar, N.A.; Doyle, J.J.; Gallo, E.; Fert-Bober, J.; Kempers, M.J.; Fishman, E.K.; Chen, Y.; Myers, L.; et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 2012, 44, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Boileau, C.; Guo, D.C.; Hanna, N.; Regalado, E.S.; Detaint, D.; Gong, L.; Varret, M.; Prakash, S.K.; Li, A.H.; d’Indy, H.; et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat. Genet. 2012, 44, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Moustakas, A. Signaling Receptors for TGF-beta Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Zhang, H.; Liu, X. Emerging role of CCN family proteins in fibrosis. J. Cell Physiol. 2021, 236, 4195–4206. [Google Scholar] [CrossRef]
- Mia, M.M.; Singh, M.K. New Insights into Hippo/YAP Signaling in Fibrotic Diseases. Cells 2022, 11, 2065. [Google Scholar] [CrossRef]
- Mia, M.M.; Singh, M.K. The Hippo Signaling Pathway in Cardiac Development and Diseases. Front. Cell Dev. Biol. 2019, 7, 211. [Google Scholar] [CrossRef]
- Ma, S.; Meng, Z.; Chen, R.; Guan, K.L. The Hippo Pathway: Biology and Pathophysiology. Annu. Rev. Biochem. 2019, 88, 577–604. [Google Scholar] [CrossRef]
- Misra, J.R.; Irvine, K.D. The Hippo Signaling Network and Its Biological Functions. Annu. Rev. Genet. 2018, 52, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; von Gise, A.; Liu, Q.; Hu, T.; Tian, X.; He, L.; Pu, W.; Huang, X.; He, L.; Cai, C.L.; et al. Yap1 is required for endothelial to mesenchymal transition of the atrioventricular cushion. J. Biol. Chem. 2014, 289, 18681–18692. [Google Scholar] [CrossRef] [PubMed]
- Pobbati, A.V.; Hong, W. Emerging roles of TEAD transcription factors and its coactivators in cancers. Cancer Biol. Ther. 2013, 14, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef]
- Lockhart, M.; Wirrig, E.; Phelps, A.; Wessels, A. Extracellular matrix and heart development. Birth Defects Res. A Clin. Mol. Teratol. 2011, 91, 535–550. [Google Scholar] [CrossRef]
- Labibi, B.; Bashkurov, M.; Wrana, J.L.; Attisano, L. Modeling the Control of TGF-beta/Smad Nuclear Accumulation by the Hippo Pathway Effectors, Taz/Yap. iScience 2020, 23, 101416. [Google Scholar] [CrossRef]
- Gori, I.; George, R.; Purkiss, A.G.; Strohbuecker, S.; Randall, R.A.; Ogrodowicz, R.; Carmignac, V.; Faivre, L.; Joshi, D.; Kjaer, S.; et al. Mutations in SKI in Shprintzen-Goldberg syndrome lead to attenuated TGF-beta responses through SKI stabilization. Elife 2021, 10, e63545. [Google Scholar] [CrossRef]
- Peng, Y.; Song, L.; Li, D.; Kesterson, R.; Wang, J.; Wang, L.; Rokosh, G.; Wu, B.; Wang, Q.; Jiao, K. Sema6D acts downstream of bone morphogenetic protein signalling to promote atrioventricular cushion development in mice. Cardiovasc. Res. 2016, 112, 532–542. [Google Scholar] [CrossRef]
- Carver, W.; Fix, E.; Fix, C.; Fan, D.; Chakrabarti, M.; Azhar, M. Effects of emodin, a plant-derived anthraquinone, on TGF-beta1-induced cardiac fibroblast activation and function. J. Cell Physiol. 2021, 236, 7440–7449. [Google Scholar] [CrossRef]
- Haskett, D.; Doyle, J.J.; Gard, C.; Chen, H.; Ball, C.; Estabrook, M.A.; Encinas, A.C.; Dietz, H.C.; Utzinger, U.; Vande Geest, J.P.; et al. Altered tissue behavior of a non-aneurysmal descending thoracic aorta in the mouse model of Marfan syndrome. Cell Tissue Res. 2012, 347, 267–277. [Google Scholar] [CrossRef]
- Fix, C.; Carver-Molina, A.; Chakrabarti, M.; Azhar, M.; Carver, W. Effects of the isothiocyanate sulforaphane on TGF-beta1-induced rat cardiac fibroblast activation and extracellular matrix interactions. J. Cell Physiol. 2019, 234, 13931–13941. [Google Scholar] [CrossRef] [PubMed]
- Bartram, U.; Molin, D.G.; Wisse, L.J.; Mohamad, A.; Sanford, L.P.; Doetschman, T.; Speer, C.P.; Poelmann, R.E.; Gittenberger-de, G.A. Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in Tgfb2 knockout mice. Circulation 2001, 103, 2745–2752. [Google Scholar] [CrossRef] [PubMed]
- Ishtiaq Ahmed, A.S.; Bose, G.C.; Huang, L.; Azhar, M. Generation of mice carrying a knockout-first and conditional-ready allele of transforming growth factor beta2 gene. Genesis 2014, 52, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Al-Sammarraie, N.; Gebere, M.G.; Johnson, J.; Eberth, J.F.; Azhar, M. Myocardial TGFbeta2 Is Required for Atrioventricular Cushion Remodeling and Myocardial Development. J. Cardiovasc. Dev. Dis. 2021, 8, 26. [Google Scholar] [CrossRef] [PubMed]
- Azhar, M.; Runyan, R.B.; Gard, C.; Sanford, L.P.; Miller, M.L.; Andringa, A.; Pawlowski, S.; Rajan, S.; Doetschman, T. Ligand-specific function of transforming growth factor beta in epithelial-mesenchymal transition in heart development. Dev. Dyn. 2009, 238, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Azhar, M.; Yin, M.; Bommireddy, R.; Duffy, J.J.; Yang, J.; Pawlowski, S.A.; Boivin, G.P.; Engle, S.J.; Sanford, L.P.; Grisham, C.; et al. Generation of mice with a conditional allele for transforming growth factor beta 1 gene. Genesis 2009, 47, 423–431. [Google Scholar] [CrossRef]
- Szeto, S.G.; Narimatsu, M.; Lu, M.; He, X.; Sidiqi, A.M.; Tolosa, M.F.; Chan, L.; De Freitas, K.; Bialik, J.F.; Majumder, S.; et al. YAP/TAZ Are Mechanoregulators of TGF-beta-Smad Signaling and Renal Fibrogenesis. J. Am. Soc. Nephrol. 2016, 27, 3117–3128. [Google Scholar] [CrossRef]
- Ferrigno, O.; Lallemand, F.; Verrecchia, F.; L’Hoste, S.; Camonis, J.; Atfi, A.; Mauviel, A. Yes-associated protein (YAP65) interacts with Smad7 and potentiates its inhibitory activity against TGF-beta/Smad signaling. Oncogene 2002, 21, 4879–4884. [Google Scholar] [CrossRef]
- Huang, D.; Li, X.; Sun, L.; Huang, P.; Ying, H.; Wang, H.; Wu, J.; Song, H. Regulation of Hippo signalling by p38 signalling. J. Mol. Cell Biol. 2016, 8, 328–337. [Google Scholar] [CrossRef]
- Chakrabarti, M.; Al-Sammarraie, N.; Gebere, M.G.; Bhattacharya, A.; Chopra, S.; Johnson, J.; Pena, E.A.; Eberth, J.F.; Poelmann, R.E.; Gittenberger-de Groot, A.C.; et al. Transforming Growth Factor Beta3 is Required for Cardiovascular Development. J. Cardiovasc. Dev. Dis. 2020, 7, 19. [Google Scholar] [CrossRef]
- Wang, J.; Liu, S.; Heallen, T.; Martin, J.F. The Hippo pathway in the heart: Pivotal roles in development, disease, and regeneration. Nat. Rev. Cardiol. 2018, 15, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Mo, F.E.; Lau, L.F. The matricellular protein CCN1 is essential for cardiac development. Circ. Res. 2006, 99, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Perrot, A.; Schmitt, K.R.; Roth, E.M.; Stiller, B.; Posch, M.G.; Browne, E.N.; Timmann, C.; Horstmann, R.D.; Berger, F.; Ozcelik, C. CCN1 mutation is associated with atrial septal defect. Pediatr. Cardiol. 2015, 36, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Dayawansa, N.H.; Baratchi, S.; Peter, K. Uncoupling the Vicious Cycle of Mechanical Stress and Inflammation in Calcific Aortic Valve Disease. Front. Cardiovasc. Med. 2022, 9, 783543. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Company | Catalog No | Dilution |
|---|---|---|---|
| pSMAD2 | Cell Signaling, Inc., Danvers MA, USA | 3108S | 1000 |
| SMAD2 | Cell Signaling, Inc., Danvers MA, USA | 5339S | 1000 |
| pSMAD3 | Cell Signaling, Inc., Danvers MA, USA | 9520S | 1000 |
| SMAD3 | Cell Signaling, Inc., Danvers MA, USA | 9523S | 1000 |
| pSMAD1/5 | Cell Signaling, Inc., Danvers MA, USA | 9516S | 1000 |
| YAP1 | abcam | ab205270 | 2000 |
| HRP-anti-mice IgG | Cell Signaling, Inc., Danvers MA, USA | 7076S | 5000 |
| HRP-anti-Rabbit IgG | Cell Signaling, Inc., Danvers MA, USA | 7074S | 5000 |
| Goat anti-Rabbit, Cyanine3 | Invitrogen, Waltham, MA, USA | A10520 | 1 µg/mL |
| β-actin | Sigma-Aldrich, St. Louis, MO, USA | A5441 | 10,000 |
| Sl. No | Target Gene | Biorad UniqueAssayID: |
|---|---|---|
| 1 | Acta2, Mouse | qMmuCID0006375 |
| 2 | Col1a1, Mouse | qMmuCID0021007 |
| 3 | Ccn2 | qMmuCED0003632 |
| 4 | Ccn1 | qMmuCED0026152 |
| 5 | B2m, Mouse | qMmuCID0040553 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakrabarti, M.; Chattha, A.; Nair, A.; Jiao, K.; Potts, J.D.; Wang, L.; Branch, S.; Harrelson, S.; Khan, S.; Azhar, M. Hippo Signaling Mediates TGFβ-Dependent Transcriptional Inputs in Cardiac Cushion Mesenchymal Cells to Regulate Extracellular Matrix Remodeling. J. Cardiovasc. Dev. Dis. 2023, 10, 483. https://doi.org/10.3390/jcdd10120483
Chakrabarti M, Chattha A, Nair A, Jiao K, Potts JD, Wang L, Branch S, Harrelson S, Khan S, Azhar M. Hippo Signaling Mediates TGFβ-Dependent Transcriptional Inputs in Cardiac Cushion Mesenchymal Cells to Regulate Extracellular Matrix Remodeling. Journal of Cardiovascular Development and Disease. 2023; 10(12):483. https://doi.org/10.3390/jcdd10120483
Chicago/Turabian StyleChakrabarti, Mrinmay, Ahad Chattha, Abhijith Nair, Kai Jiao, Jay D. Potts, Lianming Wang, Scotty Branch, Shea Harrelson, Saeed Khan, and Mohamad Azhar. 2023. "Hippo Signaling Mediates TGFβ-Dependent Transcriptional Inputs in Cardiac Cushion Mesenchymal Cells to Regulate Extracellular Matrix Remodeling" Journal of Cardiovascular Development and Disease 10, no. 12: 483. https://doi.org/10.3390/jcdd10120483
APA StyleChakrabarti, M., Chattha, A., Nair, A., Jiao, K., Potts, J. D., Wang, L., Branch, S., Harrelson, S., Khan, S., & Azhar, M. (2023). Hippo Signaling Mediates TGFβ-Dependent Transcriptional Inputs in Cardiac Cushion Mesenchymal Cells to Regulate Extracellular Matrix Remodeling. Journal of Cardiovascular Development and Disease, 10(12), 483. https://doi.org/10.3390/jcdd10120483