
Massively Parallel Reporter Assays for High-Throughput In Vivo Analysis of Cis-Regulatory Elements

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
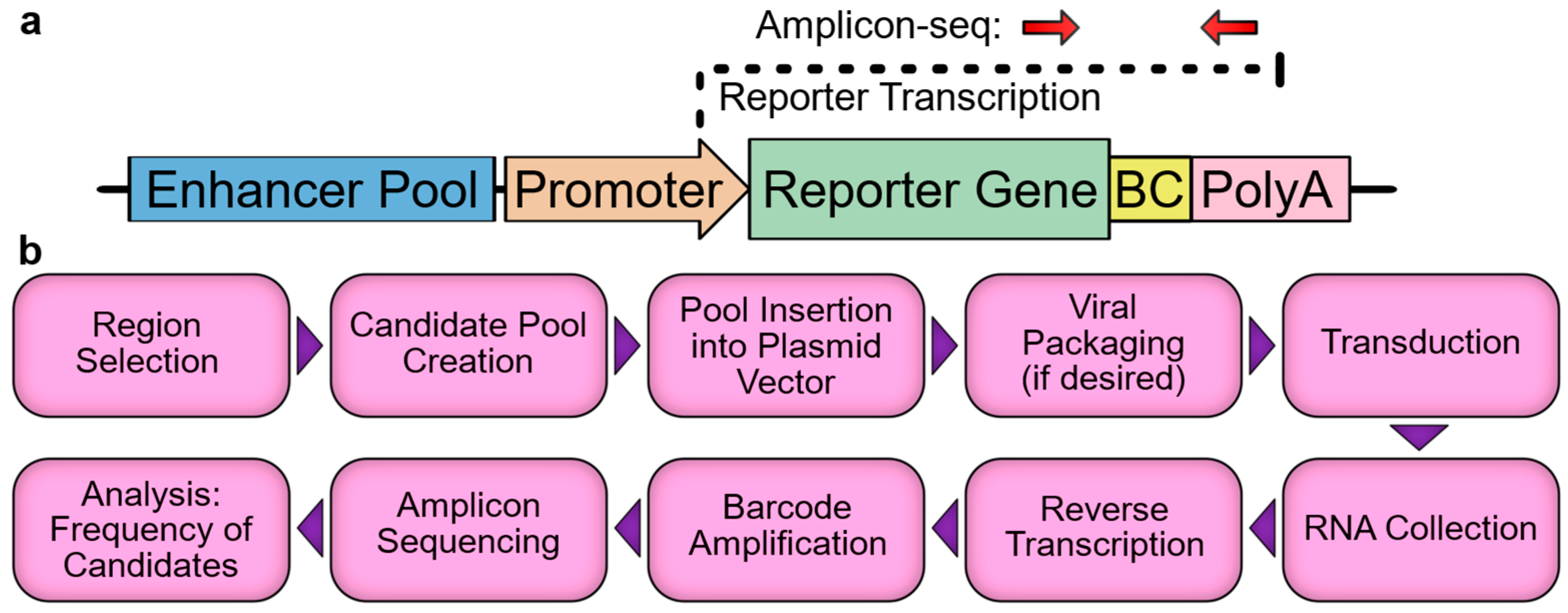
2. Massively Parallel Reporter Assays
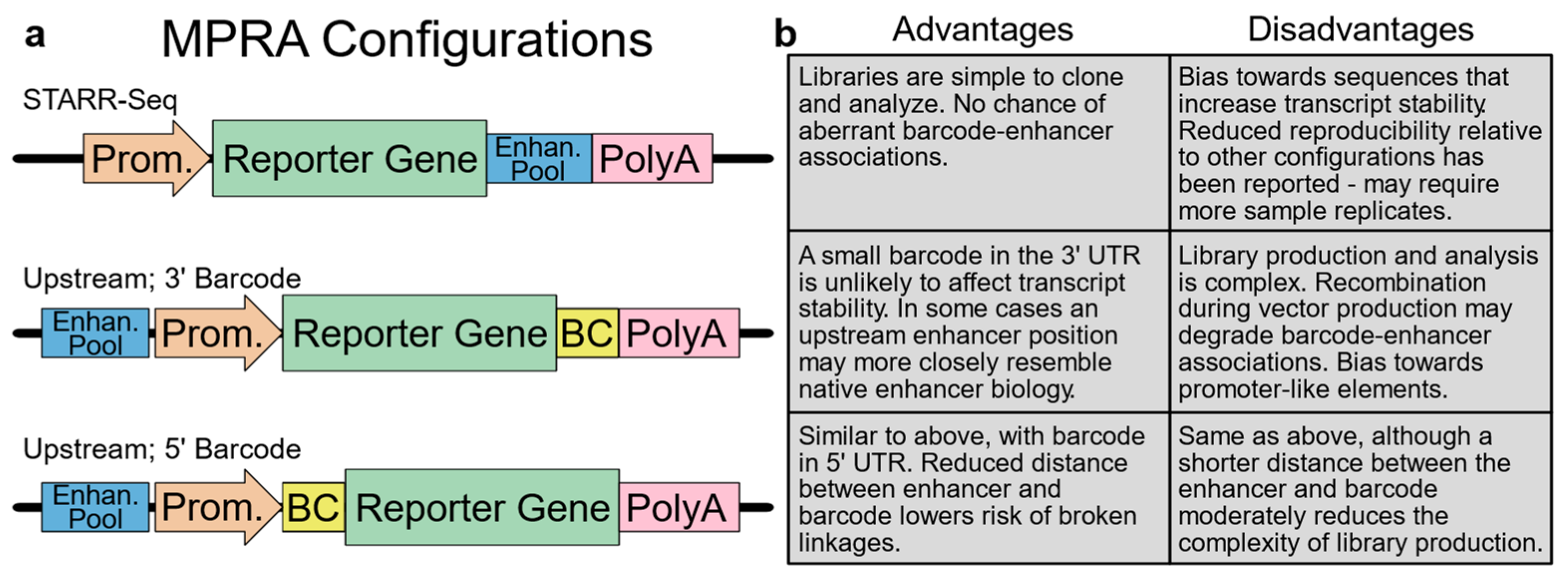
2.1. Experimental Design: Assay Configuration and Context
2.2. Experimental Design: Library Construction, Data Collection, and Analysis
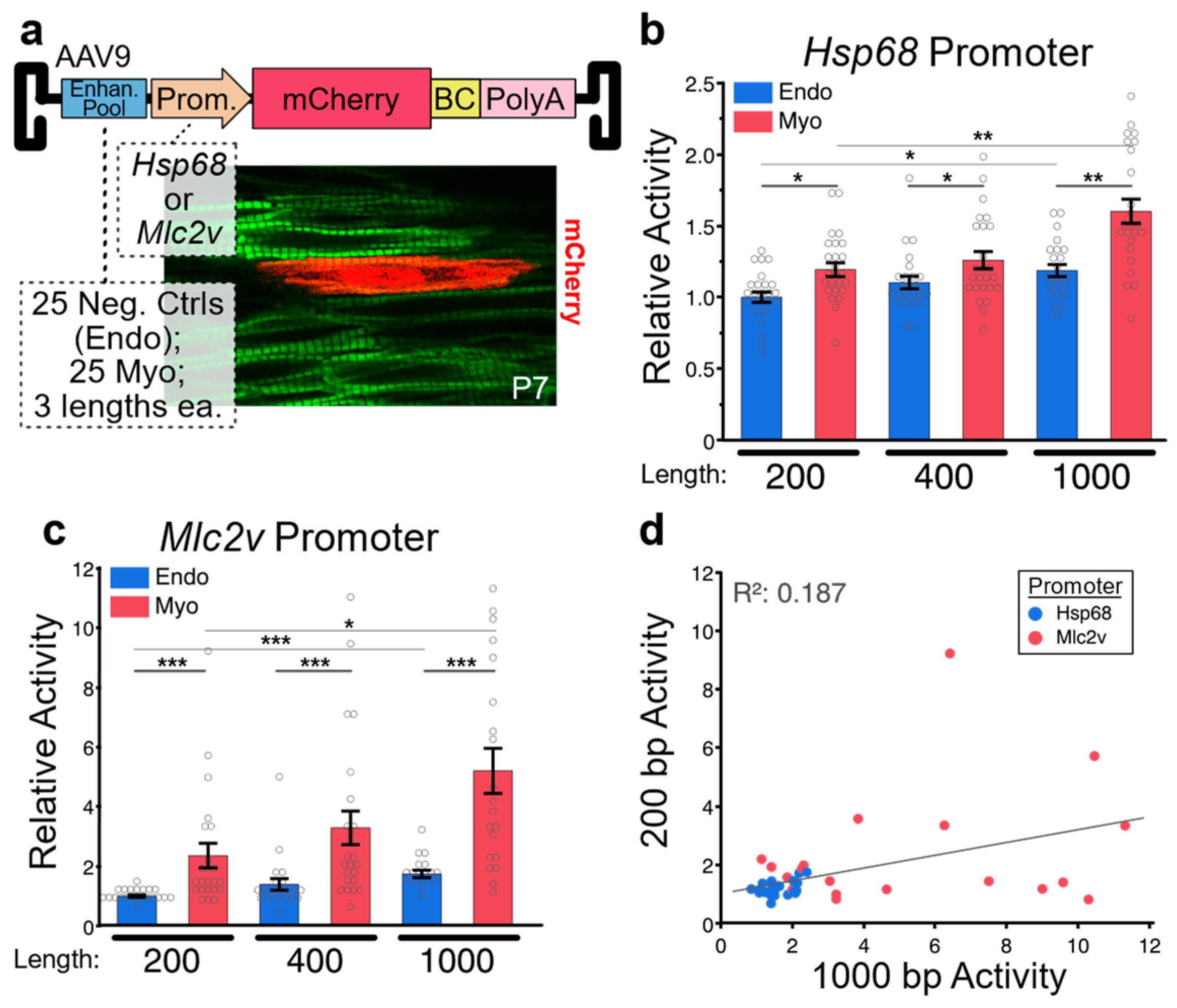
2.3. In Vivo Applications
3. Discussion and Perspectives on Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guo, Y.; Pu, W.T. Cardiomyocyte Maturation: New Phase in Development. Circ. Res. 2020, 126, 1086–1106. [Google Scholar] [CrossRef] [PubMed]
- Christoffels, V.; Jensen, B. Cardiac Morphogenesis: Specification of the Four-Chambered Heart. Cold Spring Harb. Perspect. Biol. 2020, 12, a037143. [Google Scholar] [CrossRef] [PubMed]
- Paige, S.L.; Plonowska, K.; Xu, A.; Wu, S.M. Molecular regulation of cardiomyocyte differentiation. Circ. Res. 2015, 116, 341–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, S.U.; Quiat, D.; Seidman, J.G.; Seidman, C.E. Genomic frontiers in congenital heart disease. Nat. Rev. Cardiol. 2022, 19, 26–42. [Google Scholar] [CrossRef] [PubMed]
- McKenna, W.J.; Judge, D.P. Epidemiology of the inherited cardiomyopathies. Nat. Rev. Cardiol. 2021, 18, 22–36. [Google Scholar] [CrossRef]
- Choudhury, T.Z.; Garg, V. Molecular genetic mechanisms of congenital heart disease. Curr. Opin. Genet. Dev. 2022, 75, 101949. [Google Scholar] [CrossRef]
- Liu, N.; Olson, E.N. CRISPR Modeling and Correction of Cardiovascular Disease. Circ. Res. 2022, 130, 1827–1850. [Google Scholar] [CrossRef]
- Schultheiss, H.P.; Fairweather, D.; Caforio, A.L.P.; Escher, F.; Hershberger, R.E.; Lipshultz, S.E.; Liu, P.P.; Matsumori, A.; Mazzanti, A.; McMurray, J.; et al. Dilated cardiomyopathy. Nat. Rev. Dis. Prim. 2019, 5, 32. [Google Scholar] [CrossRef]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef] [PubMed]
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.T. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2019, 16, 519–537. [Google Scholar] [CrossRef]
- Hall, B.; Limaye, A.; Kulkarni, A.B. Overview: Generation of gene knockout mice. Curr. Protoc. Cell Biol. 2009, 44, 12–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouabe, H.; Okkenhaug, K. Gene targeting in mice: A review. Methods Mol. Biol. 2013, 1064, 315–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Kim, M.; Im, S.K.; Fang, S. Mouse Cre-LoxP system: General principles to determine tissue-specific roles of target genes. Lab. Anim. Res. 2018, 34, 147–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, A.; Haruyama, N.; Kulkarni, A.B. Generation of transgenic mice. Curr. Protoc. Cell Biol. 2009, 42, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welter, D.; MacArthur, J.; Morales, J.; Burdett, T.; Hall, P.; Junkins, H.; Klemm, A.; Flicek, P.; Manolio, T.; Hindorff, L.; et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014, 42, D1001–D1006. [Google Scholar] [CrossRef] [PubMed]
- Winter, D.R.; Song, L.; Mukherjee, S.; Furey, T.S.; Crawford, G.E. DNase-seq predicts regions of rotational nucleosome stability across diverse human cell types. Genome Res. 2013, 23, 1118–1129. [Google Scholar] [CrossRef] [Green Version]
- Grandi, F.C.; Modi, H.; Kampman, L.; Corces, M.R. Chromatin accessibility profiling by ATAC-seq. Nat. Protoc. 2022, 17, 1518–1552. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Li, W. Enhancer RNA: What we know and what we can achieve. Cell Prolif. 2022, 55, e13202. [Google Scholar] [CrossRef] [PubMed]
- Park, P.J. ChIP-seq: Advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, B.E.; Stamatoyannopoulos, J.A.; Costello, J.F.; Ren, B.; Milosavljevic, A.; Meissner, A.; Kellis, M.; Marra, M.A.; Beaudet, A.L.; Ecker, J.R.; et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat. Biotechnol. 2010, 28, 1045–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [Green Version]
- Abell, N.S.; DeGorter, M.K.; Gloudemans, M.J.; Greenwald, E.; Smith, K.S.; He, Z.; Montgomery, S.B. Multiple causal variants underlie genetic associations in humans. Science 2022, 375, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Maricque, B.B.; Dougherty, J.D.; Cohen, B.A. A genome-integrated massively parallel reporter assay reveals DNA sequence determinants of cis-regulatory activity in neural cells. Nucleic Acids Res. 2017, 45, e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, J.C.; Agarwal, V.; Inoue, F.; Keith, A.; Martin, B.; Kircher, M.; Ahituv, N.; Shendure, J. A systematic evaluation of the design and context dependencies of massively parallel reporter assays. Nat. Methods 2020, 17, 1083–1091. [Google Scholar] [CrossRef]
- Ghazi, A.R.; Chen, E.S.; Henke, D.M.; Madan, N.; Edelstein, L.C.; Shaw, C.A. Design tools for MPRA experiments. Bioinformatics 2018, 34, 2682–2683. [Google Scholar] [CrossRef] [Green Version]
- Georgakopoulos-Soares, I.; Jain, N.; Gray, J.M.; Hemberg, M. MPRAnator: A web-based tool for the design of massively parallel reporter assay experiments. Bioinformatics 2017, 33, 137–138. [Google Scholar] [CrossRef] [Green Version]
- Niroula, A.; Ajore, R.; Nilsson, B. MPRAscore: Robust and non-parametric analysis of massively parallel reporter assays. Bioinformatics 2019, 35, 5351–5353. [Google Scholar] [CrossRef]
- Ashuach, T.; Fischer, D.S.; Kreimer, A.; Ahituv, N.; Theis, F.J.; Yosef, N. MPRAnalyze: Statistical framework for massively parallel reporter assays. Genome Biol. 2019, 20, 183. [Google Scholar] [CrossRef] [Green Version]
- Arnold, C.D.; Gerlach, D.; Stelzer, C.; Boryn, L.M.; Rath, M.; Stark, A. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 2013, 339, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, A.; Murugan, A.; Zhang, X.; Tesileanu, T.; Wang, L.; Rogov, P.; Feizi, S.; Gnirke, A.; Callan, C.G., Jr.; Kinney, J.B.; et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol. 2012, 30, 271–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kheradpour, P.; Ernst, J.; Melnikov, A.; Rogov, P.; Wang, L.; Zhang, X.; Alston, J.; Mikkelsen, T.S.; Kellis, M. Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay. Genome Res. 2013, 23, 800–811. [Google Scholar] [CrossRef] [Green Version]
- Patwardhan, R.P.; Hiatt, J.B.; Witten, D.M.; Kim, M.J.; Smith, R.P.; May, D.; Lee, C.; Andrie, J.M.; Lee, S.I.; Cooper, G.M.; et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat. Biotechnol. 2012, 30, 265–270. [Google Scholar] [CrossRef] [Green Version]
- Hegde, M.; Strand, C.; Hanna, R.E.; Doench, J.G. Uncoupling of sgRNAs from their associated barcodes during PCR amplification of combinatorial CRISPR screens. PLoS ONE 2018, 13, e0197547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, S.; Cooley, A.; Armendariz, D.; Zhou, P.; Hon, G.C. Frequent sgRNA-barcode recombination in single-cell perturbation assays. PLoS ONE 2018, 13, e0198635. [Google Scholar] [CrossRef] [Green Version]
- Davidsson, M.; Diaz-Fernandez, P.; Schwich, O.D.; Torroba, M.; Wang, G.; Bjorklund, T. A novel process of viral vector barcoding and library preparation enables high-diversity library generation and recombination-free paired-end sequencing. Sci. Rep. 2016, 6, 37563. [Google Scholar] [CrossRef]
- Sack, L.M.; Davoli, T.; Xu, Q.; Li, M.Z.; Elledge, S.J. Sources of Error in Mammalian Genetic Screens. G3 (Bethesda) 2016, 6, 2781–2790. [Google Scholar] [CrossRef] [Green Version]
- Inoue, F.; Kircher, M.; Martin, B.; Cooper, G.M.; Witten, D.M.; McManus, M.T.; Ahituv, N.; Shendure, J. A systematic comparison reveals substantial differences in chromosomal versus episomal encoding of enhancer activity. Genome Res. 2017, 27, 38–52. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Dai, X.; Berry, L.D.; Cogan, J.D.; Liu, Q.; Shyr, Y. HACER: An atlas of human active enhancers to interpret regulatory variants. Nucleic Acids Res. 2019, 47, D106–D112. [Google Scholar] [CrossRef]
- Gorkin, D.U.; Barozzi, I.; Zhao, Y.; Zhang, Y.; Huang, H.; Lee, A.Y.; Li, B.; Chiou, J.; Wildberg, A.; Ding, B.; et al. Author Correction: An atlas of dynamic chromatin landscapes in mouse fetal development. Nature 2021, 589, E4. [Google Scholar] [CrossRef] [PubMed]
- Akerberg, B.N.; Gu, F.; VanDusen, N.J.; Zhang, X.; Dong, R.; Li, K.; Zhang, B.; Zhou, B.; Sethi, I.; Ma, Q.; et al. A reference map of murine cardiac transcription factor chromatin occupancy identifies dynamic and conserved enhancers. Nat. Commun. 2019, 10, 4907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markenscoff-Papadimitriou, E.; Whalen, S.; Przytycki, P.; Thomas, R.; Binyameen, F.; Nowakowski, T.J.; Kriegstein, A.R.; Sanders, S.J.; State, M.W.; Pollard, K.S.; et al. A Chromatin Accessibility Atlas of the Developing Human Telencephalon. Cell 2020, 182, 754–769.e18. [Google Scholar] [CrossRef]
- Cao, Y.; Zhang, X.; Akerberg, B.N.; Yuan, H.; Sakamoto, T.; Xiao, F.; VanDusen, N.J.; Zhou, P.; Sweat, M.E.; Wang, Y.; et al. In Vivo Dissection of Chamber-Selective Enhancers Reveals Estrogen-Related Receptor as a Regulator of Ventricular Cardiomyocyte Identity. Circulation 2023, 147, 881–896. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.Q.; Myers, C.A.; Hughes, A.E.; Byrne, L.C.; Flannery, J.G.; Corbo, J.C. Massively parallel cis-regulatory analysis in the mammalian central nervous system. Genome Res. 2016, 26, 238–255. [Google Scholar] [CrossRef] [Green Version]
- Kuiper, B.P.; Prins, R.C.; Billerbeck, S. Oligo Pools as an Affordable Source of Synthetic DNA for Cost-Effective Library Construction in Protein- and Metabolic Pathway Engineering. Chembiochem 2022, 23, e202100507. [Google Scholar] [CrossRef]
- Williams, R.; Peisajovich, S.G.; Miller, O.J.; Magdassi, S.; Tawfik, D.S.; Griffiths, A.D. Amplification of complex gene libraries by emulsion PCR. Nat. Methods 2006, 3, 545–550. [Google Scholar] [CrossRef]
- Joung, J.; Konermann, S.; Gootenberg, J.S.; Abudayyeh, O.O.; Platt, R.J.; Brigham, M.D.; Sanjana, N.E.; Zhang, F. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 2017, 12, 828–863. [Google Scholar] [CrossRef] [Green Version]
- Emmer, B.T.; Sherman, E.J.; Lascuna, P.J.; Graham, S.E.; Willer, C.J.; Ginsburg, D. Genome-scale CRISPR screening for modifiers of cellular LDL uptake. PLoS Genet. 2021, 17, e1009285. [Google Scholar] [CrossRef]
- Alkass, K.; Panula, J.; Westman, M.; Wu, T.D.; Guerquin-Kern, J.L.; Bergmann, O. No Evidence for Cardiomyocyte Number Expansion in Preadolescent Mice. Cell 2015, 163, 1026–1036. [Google Scholar] [CrossRef] [Green Version]
- Clement, K.; Farouni, R.; Bauer, D.E.; Pinello, L. AmpUMI: Design and analysis of unique molecular identifiers for deep amplicon sequencing. Bioinformatics 2018, 34, i202–i210. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Wu, P.H.; Beane, T.; Zamore, P.D.; Weng, Z. Elimination of PCR duplicates in RNA-seq and small RNA-seq using unique molecular identifiers. BMC Genom. 2018, 19, 531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwasnieski, J.C.; Mogno, I.; Myers, C.A.; Corbo, J.C.; Cohen, B.A. Complex effects of nucleotide variants in a mammalian cis-regulatory element. Proc. Natl. Acad. Sci. USA 2012, 109, 19498–19503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, M.A.; Myers, C.A.; Corbo, J.C.; Cohen, B.A. Massively parallel in vivo enhancer assay reveals that highly local features determine the cis-regulatory function of ChIP-seq peaks. Proc. Natl. Acad. Sci. USA 2013, 110, 11952–11957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, A.E.O.; Myers, C.A.; Corbo, J.C. A massively parallel reporter assay reveals context-dependent activity of homeodomain binding sites in vivo. Genome Res. 2018, 28, 1520–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Hong, C.K.Y.; Myers, C.A.; Granas, D.M.; White, M.A.; Corbo, J.C.; Cohen, B.A. A single-cell massively parallel reporter assay detects cell-type-specific gene regulation. Nat. Genet. 2023, 55, 346–354. [Google Scholar] [CrossRef]
- Hrvatin, S.; Tzeng, C.P.; Nagy, M.A.; Stroud, H.; Koutsioumpa, C.; Wilcox, O.F.; Assad, E.G.; Green, J.; Harvey, C.D.; Griffith, E.C.; et al. A scalable platform for the development of cell-type-specific viral drivers. eLife 2019, 8, e48089. [Google Scholar] [CrossRef]
- Lambert, J.T.; Su-Feher, L.; Cichewicz, K.; Warren, T.L.; Zdilar, I.; Wang, Y.; Lim, K.J.; Haigh, J.L.; Morse, S.J.; Canales, C.P.; et al. Parallel functional testing identifies enhancers active in early postnatal mouse brain. eLife 2021, 10, e69479. [Google Scholar] [CrossRef]
- Kircher, M.; Xiong, C.; Martin, B.; Schubach, M.; Inoue, F.; Bell, R.J.A.; Costello, J.F.; Shendure, J.; Ahituv, N. Saturation mutagenesis of twenty disease-associated regulatory elements at single base-pair resolution. Nat. Commun. 2019, 10, 3583. [Google Scholar] [CrossRef] [Green Version]
- Shigaki, D.; Adato, O.; Adhikari, A.N.; Dong, S.; Hawkins-Hooker, A.; Inoue, F.; Juven-Gershon, T.; Kenlay, H.; Martin, B.; Patra, A.; et al. Integration of multiple epigenomic marks improves prediction of variant impact in saturation mutagenesis reporter assay. Hum. Mutat. 2019, 40, 1280–1291. [Google Scholar] [CrossRef] [Green Version]
- Patwardhan, R.P.; Lee, C.; Litvin, O.; Young, D.L.; Pe’er, D.; Shendure, J. High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat. Biotechnol. 2009, 27, 1173–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossman, S.R.; Zhang, X.; Wang, L.; Engreitz, J.; Melnikov, A.; Rogov, P.; Tewhey, R.; Isakova, A.; Deplancke, B.; Bernstein, B.E.; et al. Systematic dissection of genomic features determining transcription factor binding and enhancer function. Proc. Natl. Acad. Sci. USA 2017, 114, E1291–E1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvon, E.Z.; Waymack, R.; Gad, M.; Wunderlich, Z. Enhancer redundancy in development and disease. Nat. Rev. Genet. 2021, 22, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Osterwalder, M.; Barozzi, I.; Tissieres, V.; Fukuda-Yuzawa, Y.; Mannion, B.J.; Afzal, S.Y.; Lee, E.A.; Zhu, Y.; Plajzer-Frick, I.; Pickle, C.S.; et al. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature 2018, 554, 239–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankel, N.; Davis, G.K.; Vargas, D.; Wang, S.; Payre, F.; Stern, D.L. Phenotypic robustness conferred by apparently redundant transcriptional enhancers. Nature 2010, 466, 490–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, K.J.; Makarewich, C.A.; McAnally, J.; Anderson, D.M.; Zentilin, L.; Liu, N.; Giacca, M.; Bassel-Duby, R.; Olson, E.N. A mouse model for adult cardiac-specific gene deletion with CRISPR/Cas9. Proc. Natl. Acad. Sci. USA 2016, 113, 338–343. [Google Scholar] [CrossRef] [Green Version]
- Johansen, A.K.; Molenaar, B.; Versteeg, D.; Leitoguinho, A.R.; Demkes, C.; Spanjaard, B.; de Ruiter, H.; Akbari Moqadam, F.; Kooijman, L.; Zentilin, L.; et al. Postnatal Cardiac Gene Editing Using CRISPR/Cas9 With AAV9-Mediated Delivery of Short Guide RNAs Results in Mosaic Gene Disruption. Circ. Res. 2017, 121, 1168–1181. [Google Scholar] [CrossRef]
- Guo, Y.; VanDusen, N.J.; Zhang, L.; Gu, W.; Sethi, I.; Guatimosim, S.; Ma, Q.; Jardin, B.D.; Ai, Y.; Zhang, D.; et al. Analysis of Cardiac Myocyte Maturation Using CASAAV, a Platform for Rapid Dissection of Cardiac Myocyte Gene Function In Vivo. Circ. Res. 2017, 120, 1874–1888. [Google Scholar] [CrossRef]
- Kremer, L.P.M.; Cerrizuela, S.; Dehler, S.; Stiehl, T.; Weinmann, J.; Abendroth, H.; Kleber, S.; Laure, A.; El Andari, J.; Anders, S.; et al. High throughput screening of novel AAV capsids identifies variants for transduction of adult NSCs within the subventricular zone. Mol. Ther. Methods Clin. Dev. 2021, 23, 33–50. [Google Scholar] [CrossRef]
- Davidsson, M.; Wang, G.; Aldrin-Kirk, P.; Cardoso, T.; Nolbrant, S.; Hartnor, M.; Mudannayake, J.; Parmar, M.; Bjorklund, T. A systematic capsid evolution approach performed in vivo for the design of AAV vectors with tailored properties and tropism. Proc. Natl. Acad. Sci. USA 2019, 116, 27053–27062. [Google Scholar] [CrossRef] [Green Version]
- Flitsch, L.J.; Borner, K.; Stullein, C.; Ziegler, S.; Sonntag-Buck, V.; Wiedtke, E.; Semkova, V.; Au Yeung, S.W.C.; Schlee, J.; Hajo, M.; et al. Identification of adeno-associated virus variants for gene transfer into human neural cell types by parallel capsid screening. Sci. Rep. 2022, 12, 8356. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Lee, J.S.; Wang, L.; Desai, T.; Akache, B.; Storm, T.A.; Kay, M.A. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J. Virol. 2008, 82, 5887–5911. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.B.; Xu, B.C.; Chen, Y.T.; Yuan, X.; Liu, J.Y.; Liu, T.; Du, G.Z.; Jiang, W.; Yang, Y.; Zhu, Y.; et al. Directed evolution of AAV accounting for long-term and enhanced transduction of cardiovascular endothelial cells in vivo. Mol. Ther. Methods Clin. Dev. 2021, 22, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Korbelin, J.; Sieber, T.; Michelfelder, S.; Lunding, L.; Spies, E.; Hunger, A.; Alawi, M.; Rapti, K.; Indenbirken, D.; Muller, O.J.; et al. Pulmonary Targeting of Adeno-associated Viral Vectors by Next-generation Sequencing-guided Screening of Random Capsid Displayed Peptide Libraries. Mol. Ther. 2016, 24, 1050–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurema, A. Adenoviral Gene Therapy and Fertility; Distribution Studies in Reproductive Organs and Risk of Vertical Transmission in Female Rabbits and Rats. Ph.D. Thesis, University of Kuopio, Kuopio, Finland, 2008. Available online: https://erepo.uef.fi/bitstream/handle/123456789/8980/urn_isbn_978-951-27-1107-9.pdf?sequence=1 (accessed on 15 December 2022).
- Christensen, G.; Minamisawa, S.; Gruber, P.J.; Wang, Y.; Chien, K.R. High-efficiency, long-term cardiac expression of foreign genes in living mouse embryos and neonates. Circulation 2000, 101, 178–184. [Google Scholar] [CrossRef] [Green Version]
- Lai, L.; Davison, B.B.; Veazey, R.S.; Fisher, K.J.; Baskin, G.B. A preliminary evaluation of recombinant adeno-associated virus biodistribution in rhesus monkeys after intrahepatic inoculation in utero. Hum. Gene Ther. 2002, 13, 2027–2039. [Google Scholar] [CrossRef] [PubMed]
- Dejneka, N.S.; Surace, E.M.; Aleman, T.S.; Cideciyan, A.V.; Lyubarsky, A.; Savchenko, A.; Redmond, T.M.; Tang, W.; Wei, Z.; Rex, T.S.; et al. In utero gene therapy rescues vision in a murine model of congenital blindness. Mol. Ther. 2004, 9, 182–188. [Google Scholar] [CrossRef]
- Sabatino, D.E.; Mackenzie, T.C.; Peranteau, W.; Edmonson, S.; Campagnoli, C.; Liu, Y.L.; Flake, A.W.; High, K.A. Persistent expression of hF.IX After tolerance induction by in utero or neonatal administration of AAV-1-F.IX in hemophilia B mice. Mol. Ther. 2007, 15, 1677–1685. [Google Scholar] [CrossRef]
- Joyeux, L.; Danzer, E.; Limberis, M.P.; Zoltick, P.W.; Radu, A.; Flake, A.W.; Davey, M.G. In utero lung gene transfer using adeno-associated viral and lentiviral vectors in mice. Hum. Gene Ther. Methods 2014, 25, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; VanDusen, N.J.; Butler, C.E.; Ma, Q.; King, J.S.; Pu, W.T. Efficient In Vivo Homology-Directed Repair Within Cardiomyocytes. Circulation 2022, 145, 787–789. [Google Scholar] [CrossRef]
- Penaud-Budloo, M.; Le Guiner, C.; Nowrouzi, A.; Toromanoff, A.; Cherel, Y.; Chenuaud, P.; Schmidt, M.; von Kalle, C.; Rolling, F.; Moullier, P.; et al. Adeno-associated virus vector genomes persist as episomal chromatin in primate muscle. J. Virol. 2008, 82, 7875–7885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, D.; Sharma, P.; Yang, J.; Yue, Y.; Dudus, L.; Zhang, Y.; Fisher, K.J.; Engelhardt, J.F. Circular intermediates of recombinant adeno-associated virus have defined structural characteristics responsible for long-term episomal persistence in muscle tissue. J. Virol. 1998, 72, 8568–8577. [Google Scholar] [CrossRef] [PubMed]
- Kvon, E.Z.; Zhu, Y.; Kelman, G.; Novak, C.S.; Plajzer-Frick, I.; Kato, M.; Garvin, T.H.; Pham, Q.; Harrington, A.N.; Hunter, R.D.; et al. Comprehensive In Vivo Interrogation Reveals Phenotypic Impact of Human Enhancer Variants. Cell 2020, 180, 1262–1271.e15. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, Y.; VanDusen, N.J. Massively Parallel Reporter Assays for High-Throughput In Vivo Analysis of Cis-Regulatory Elements. J. Cardiovasc. Dev. Dis. 2023, 10, 144. https://doi.org/10.3390/jcdd10040144
Zheng Y, VanDusen NJ. Massively Parallel Reporter Assays for High-Throughput In Vivo Analysis of Cis-Regulatory Elements. Journal of Cardiovascular Development and Disease. 2023; 10(4):144. https://doi.org/10.3390/jcdd10040144
Chicago/Turabian StyleZheng, Yanjiang, and Nathan J. VanDusen. 2023. "Massively Parallel Reporter Assays for High-Throughput In Vivo Analysis of Cis-Regulatory Elements" Journal of Cardiovascular Development and Disease 10, no. 4: 144. https://doi.org/10.3390/jcdd10040144
APA StyleZheng, Y., & VanDusen, N. J. (2023). Massively Parallel Reporter Assays for High-Throughput In Vivo Analysis of Cis-Regulatory Elements. Journal of Cardiovascular Development and Disease, 10(4), 144. https://doi.org/10.3390/jcdd10040144