
Effects of Serum Estradiol on Proprotein Convertase Subtilisin/Kexin Type 9 Levels and Lipid Profiles in Women Undergoing In Vitro Fertilization

 ,
,
Abstract
:1. Introduction
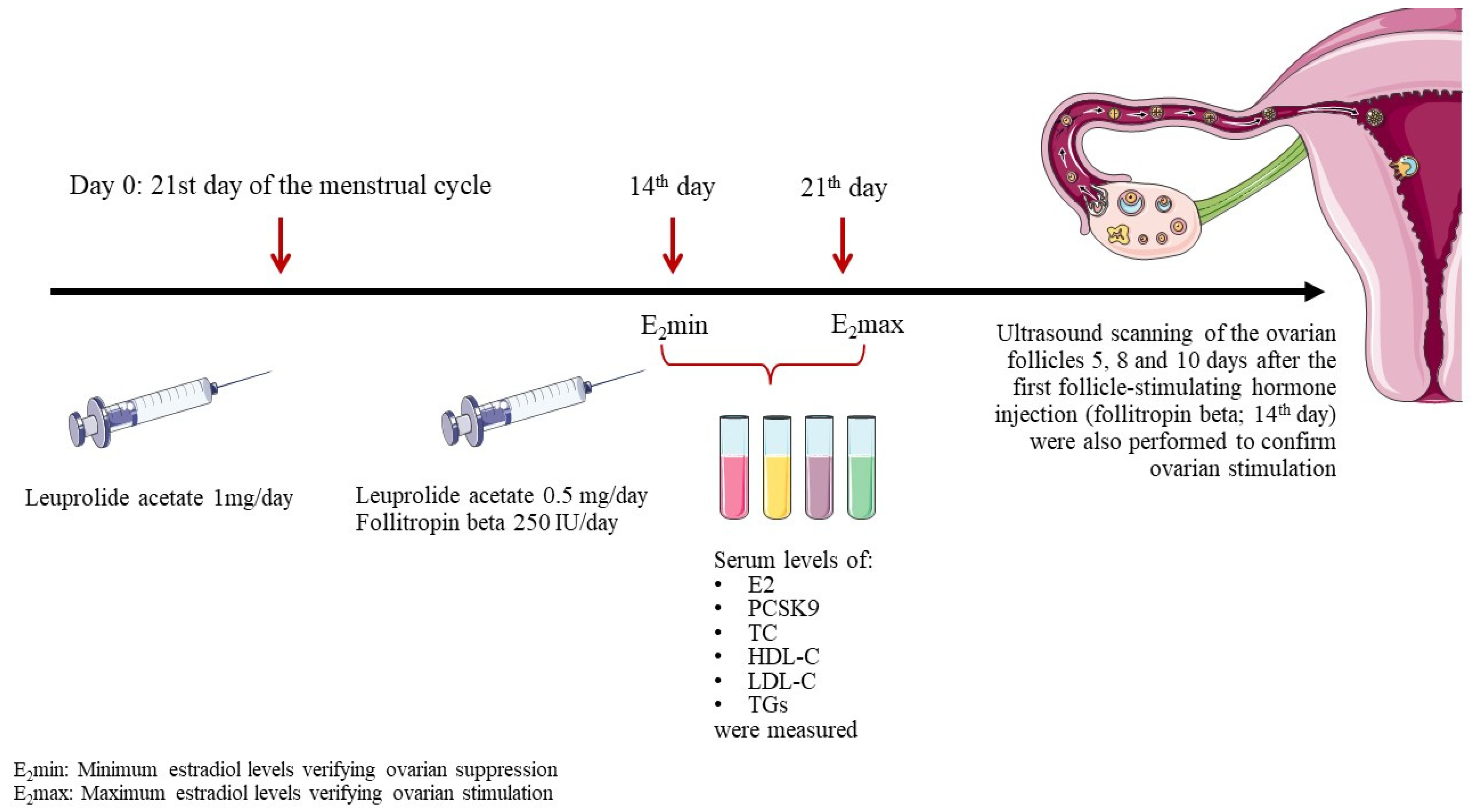
2. Materials and Methods
2.1. Study Design
2.2. Collection and Analysis of Blood Samples
2.3. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heiss, G.; Tamir, I.; Davis, C.E.; Tyroler, H.A.; Rifkand, B.M.; Schonfeld, G.; Jacobs, D.; Frantz, I.D., Jr. Lipoprotein-cholesterol distributions in selected North American populations: The lipid research clinics program prevalence study. Circulation 1980, 61, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.M.; Zhang, D.W. Hypercholesterolemia, low density lipoprotein receptor and proprotein convertase subtilisin/kexin-type 9. J. Biomed. Res. 2015, 29, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Poornima, S.; Subramanyam, K.; Khan, I.A.; Sumanlatha, G.; Hasan, Q. Role of SREBP2 gene polymorphism on knee osteoarthritis in the South Indian Hyderabad Population: A hospital based study with G595C variant. J. Orthop. 2019, 16, 293–297. [Google Scholar] [CrossRef]
- Norata, G.D.; Tavori, H.; Pirillo, A.; Fazio, S.; Catapano, A.L. Biology of proprotein convertase subtilisin kexin 9: Beyond low-density lipoprotein cholesterol lowering. Cardiovasc. Res. 2016, 112, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Parini, P.; Angelin, B.; Rudling, M. Importance of estrogen receptors in hepatic LDL receptor regulation. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1800–1805. [Google Scholar] [CrossRef] [PubMed]
- Heine, P.A.; Taylor, J.A.; Iwamoto, G.A.; Lubahn, D.B.; Cooke, P.S. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc. Natl. Acad. Sci. USA 2000, 97, 12729–12734. [Google Scholar] [CrossRef]
- Ohlsson, C.; Hellberg, N.; Parini, P.; Vidal, O.; Bohlooly, Y.M.; Rudling, M.; Lindberg, M.K.; Warner, M.; Angelin, B.; Gustafsson, J.A. Obesity and disturbed lipoprotein profile in estrogen receptor-alpha-deficient male mice. Biochem. Biophys. Res. Commun. 2000, 278, 640–645. [Google Scholar] [CrossRef]
- Geary, N.; Asarian, L.; Korach, K.S.; Pfaff, D.W.; Ogawa, S. Deficits in E2-dependent control of feeding, weight gain, and cholecystokinin satiation in ER-alpha null mice. Endocrinology 2001, 142, 4751–4757. [Google Scholar] [CrossRef]
- Ribas, V.; Nguyen, M.T.; Henstridge, D.C.; Nguyen, A.K.; Beaven, S.W.; Watt, M.J.; Hevener, A.L. Impaired oxidative metabolism and inflammation are associated with insulin resistance in ERalpha-deficient mice. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E304–E319. [Google Scholar] [CrossRef]
- Li, C.; Briggs, M.R.; Ahlborn, T.E.; Kraemer, F.B.; Liu, J. Requirement of Sp1 and estrogen receptor alpha interaction in 17beta-estradiol-mediated transcriptional activation of the low density lipoprotein receptor gene expression. Endocrinology 2001, 142, 1546–1553. [Google Scholar] [CrossRef]
- Horton, J.D.; Cohen, J.C.; Hobbs, H.H. PCSK9: A convertase that coordinates LDL catabolism. J. Lipid Res. 2009, 50, S172–S177. [Google Scholar] [CrossRef] [PubMed]
- Hussain, Y.; Ding, Q.; Connelly, P.W.; Brunt, J.H.; Ban, M.R.; McIntyre, A.D.; Huff, M.W.; Gros, R.; Hegele, R.A.; Feldman, R.D. G-protein estrogen receptor as a regulator of low-density lipoprotein cholesterol metabolism: Cellular and population genetic studies. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 213–221. [Google Scholar] [CrossRef]
- Ghosh, M.; Galman, C.; Rudling, M.; Angelin, B. Influence of physiological changes in endogenous estrogen on circulating PCSK9 and LDL cholesterol. J. Lipid Res. 2015, 56, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Persson, L.; Galman, C.; Angelin, B.; Rudling, M. Importance of proprotein convertase subtilisin/kexin type 9 in the hormonal and dietary regulation of rat liver low-density lipoprotein receptors. Endocrinology 2009, 150, 1140–1146. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Prat, A. The biology and therapeutic targeting of the proprotein convertases. Nat. Rev. Drug. Discov. 2012, 11, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Lakoski, S.G.; Lagace, T.A.; Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Genetic and metabolic determinants of plasma PCSK9 levels. J. Clin. Endocrinol. Metab. 2009, 94, 2537–2543. [Google Scholar] [CrossRef]
- Pyo Kim, H.; Young Lee, J.; Kim Jeong, J.; Won Bae, S.; Kyu Lee, H.; Jo, I. Nongenomic Stimulation of Nitric Oxide Release by Estrogen Is Mediated by Estrogen Receptor α Localized in Caveolae. Biochem. Biophys. Res. Commun. 1999, 263, 257–262. [Google Scholar] [CrossRef]
- Chambliss, K.L.; Shaul, P.W. Rapid activation of endothelial NO synthase by estrogen: Evidence for a steroid receptor fast-action complex (SRFC) in caveolae. Steroids 2002, 67, 413–419. [Google Scholar] [CrossRef]
- Fu, W.; Gao, X.P.; Zhang, S.; Dai, Y.P.; Zou, W.J.; Yue, L.M. 17beta-Estradiol Inhibits PCSK9-Mediated LDLR Degradation Through GPER/PLC Activation in HepG2 Cells. Front. Endocrinol. 2019, 10, 930. [Google Scholar] [CrossRef]
- Starr, A.E.; Lemieux, V.; Noad, J.; Moore, J.I.; Dewpura, T.; Raymond, A.; Chrétien, M.; Figeys, D.; Mayne, J. β-Estradiol results in a proprotein convertase subtilisin/kexin type 9-dependent increase in low-density lipoprotein receptor levels in human hepatic HuH7 cells. FEBS J. 2015, 282, 2682–2696. [Google Scholar] [CrossRef]
- Roubtsova, A.; Garçon, D.; Lacoste, S.; Chamberland, A.; Marcinkiewicz, J.; Métivier, R.; Sotin, T.; Paquette, M.; Bernard, S.; Cariou, B.; et al. PCSK9 deficiency results in a specific shedding of excess LDLR in female mice only: Role of hepatic cholesterol. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2022, 1867, 159217. [Google Scholar] [CrossRef] [PubMed]
- Nanda, S.; Gupta, N.; Mehta, H.C.; Sangwan, K. Effect of oestrogen replacement therapy on serum lipid profile. Aust. N. Z. J. Obstet. Gynaecol. 2003, 43, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Barrett-Connor, E.; Wingard, D.L.; Criqui, M.H. Postmenopausal estrogen use and heart disease risk factors in the 1980s. Rancho Bernardo, Calif, revisited. JAMA 1989, 261, 2095–2100. [Google Scholar] [CrossRef] [PubMed]
- Krauss, R.M.; Perlman, J.A.; Ray, R.; Petitti, D. Effects of estrogen dose and smoking on lipid and lipoprotein levels in postmenopausal women. Am. J. Obstet. Gynecol. 1988, 158, 1606–1611. [Google Scholar] [CrossRef]
- Bush, T.L.; Barrett-Connor, E.; Cowan, L.D.; Criqui, M.H.; Wallace, R.B.; Suchindran, C.M.; Tyroler, H.A.; Rifkind, B.M. Cardiovascular mortality and noncontraceptive use of estrogen in women: Results from the Lipid Research Clinics Program Follow-up Study. Circulation 1987, 75, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Metka, M.; Hanes, V.; Heytmanek, G. Hormone replacement therapy: Lipid responses to continuous combined oestrogen and progestogen versus oestrogen monotherapy. Maturitas 1992, 15, 53–59. [Google Scholar] [CrossRef]
- Kim, C.J.; Jang, H.C.; Cho, D.H.; Min, Y.K. Effects of hormone replacement therapy on lipoprotein(a) and lipids in postmenopausal women. Arterioscler. Thromb. J. Vasc. Biol. 1994, 14, 275–281. [Google Scholar] [CrossRef]
- Persson, L.; Henriksson, P.; Westerlund, E.; Hovatta, O.; Angelin, B.; Rudling, M. Endogenous estrogens lower plasma PCSK9 and LDL cholesterol but not Lp(a) or bile acid synthesis in women. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 810–814. [Google Scholar] [CrossRef]
- Davezac, M.; Buscato, M.; Zahreddine, R.; Lacolley, P.; Henrion, D.; Lenfant, F.; Arnal, J.F.; Fontaine, C. Estrogen Receptor and Vascular Aging. Front. Aging 2021, 2, 727380. [Google Scholar] [CrossRef]
- Rivera, F.B.; Cha, S.W.; Aparece, J.P.; Rocimo, A.; Ong, B.A.; Golbin, J.M.; Alfonso, P.G.; Enkhmaa, B.; Khan, S.U.; Cainzos-Achirica, M.; et al. Sex Differences in Cardiovascular Outcomes and Cholesterol-Lowering Efficacy of PCSK9 Inhibitors. JACC Adv. 2023, 2, 100669. [Google Scholar] [CrossRef]


{kind=link}
| Age (years) | 38 (18–48) |
| Weight (kg) | 64 (55–78) |
| BMI (kg/m2) | 22.8 (19.1–26.7) |
| Smokers (%) | 0 |
| E2min | E2max | Change, % | |
|---|---|---|---|
| E2, pg/mL | 108 (47–346) | 464 (241–2471) | +329.6 * |
| ΤC, mg/dL | 164 ± 44 | 163 ± 42 | −0.4 |
| TGs, mg/dL | 69 ± 36 | 67 ± 24 | −2.2 |
| LDL-C, mg/dL | 108 ± 43 | 103 ± 42 | −3.8 |
| HDL-C, mg/dL | 42 ±10 | 45 ± 14 | +5.3 |
| PCSK9, ng/mL | 245 ± 80 | 170 ± 64 | −30.8 * |
| E2min | E2max | Change, % | |
|---|---|---|---|
| E2, pg/mL | 101 (56–327) | 439 (208–794) | +334.6% * |
| ΤC, mg/dL | 155 ± 40 | 155 ± 36 | 0% |
| TGs, mg/dL | 83 ± 34 | 79 ± 24 | −54.9% |
| LDL-C, mg/dL | 102 ± 39 | 100 ± 34 | −1.7% |
| HDL-C, mg/dL | 37 ± 5 | 39 ± 7 | +5.7% |
| PCSK9, ng/mL | 239 ± 60 | 143 ± 25 | −40.1% *^ |
| E2min | E2max | Change, % | |
|---|---|---|---|
| E2, pg/mL | 115 (47–346) | 488 (265–2471) | +324.3% * |
| ΤC, mg/dL | 169 ± 47 | 166 ± 45 | −2.1% |
| TGs, mg/dL | 64 ± 36 | 63 ± 23 | −0.7% |
| LDL-C, mg/dL | 112 ± 46 | 106 ± 45 | −5.5% |
| HDL-C, mg/dL | 45 ± 10 | 47 ± 14 | +4.9% |
| PCSK9, ng/mL | 248 ± 85 | 177 ± 71 | −28.6% * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papanikolaou, A.; Anastasiou, G.; Barkas, F.; Tellis, C.; Zikopoulos, K.; Liberopoulos, E. Effects of Serum Estradiol on Proprotein Convertase Subtilisin/Kexin Type 9 Levels and Lipid Profiles in Women Undergoing In Vitro Fertilization. J. Cardiovasc. Dev. Dis. 2024, 11, 25. https://doi.org/10.3390/jcdd11010025
Papanikolaou A, Anastasiou G, Barkas F, Tellis C, Zikopoulos K, Liberopoulos E. Effects of Serum Estradiol on Proprotein Convertase Subtilisin/Kexin Type 9 Levels and Lipid Profiles in Women Undergoing In Vitro Fertilization. Journal of Cardiovascular Development and Disease. 2024; 11(1):25. https://doi.org/10.3390/jcdd11010025
Chicago/Turabian StylePapanikolaou, Anna, Georgia Anastasiou, Fotios Barkas, Constantinos Tellis, Konstantinos Zikopoulos, and Evangelos Liberopoulos. 2024. "Effects of Serum Estradiol on Proprotein Convertase Subtilisin/Kexin Type 9 Levels and Lipid Profiles in Women Undergoing In Vitro Fertilization" Journal of Cardiovascular Development and Disease 11, no. 1: 25. https://doi.org/10.3390/jcdd11010025
APA StylePapanikolaou, A., Anastasiou, G., Barkas, F., Tellis, C., Zikopoulos, K., & Liberopoulos, E. (2024). Effects of Serum Estradiol on Proprotein Convertase Subtilisin/Kexin Type 9 Levels and Lipid Profiles in Women Undergoing In Vitro Fertilization. Journal of Cardiovascular Development and Disease, 11(1), 25. https://doi.org/10.3390/jcdd11010025