
RNA-Binding Proteins in Cardiomyopathies


Abstract
:1. Introduction
2. RBPs Associated with Cardiomyopathies
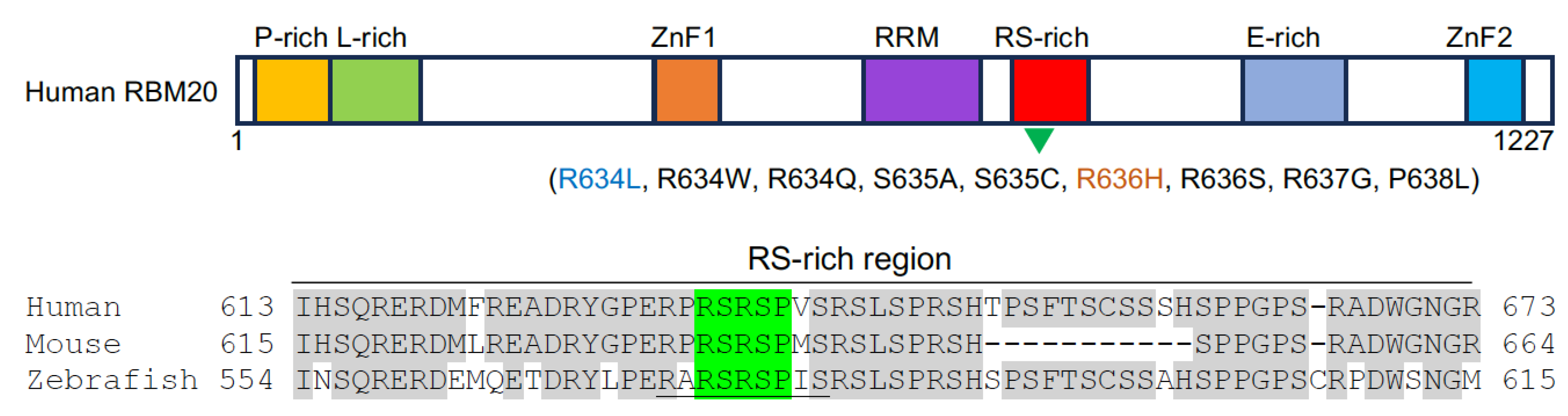
2.1. RBM20 Mutations Disrupt Cardiac-Specific Alternative Splicing in Cardiomyopathies
2.2. RBM24 Is Associated with DCM
2.3. Loss of RBPMS and RBPMS2 Causes DCM or HCM
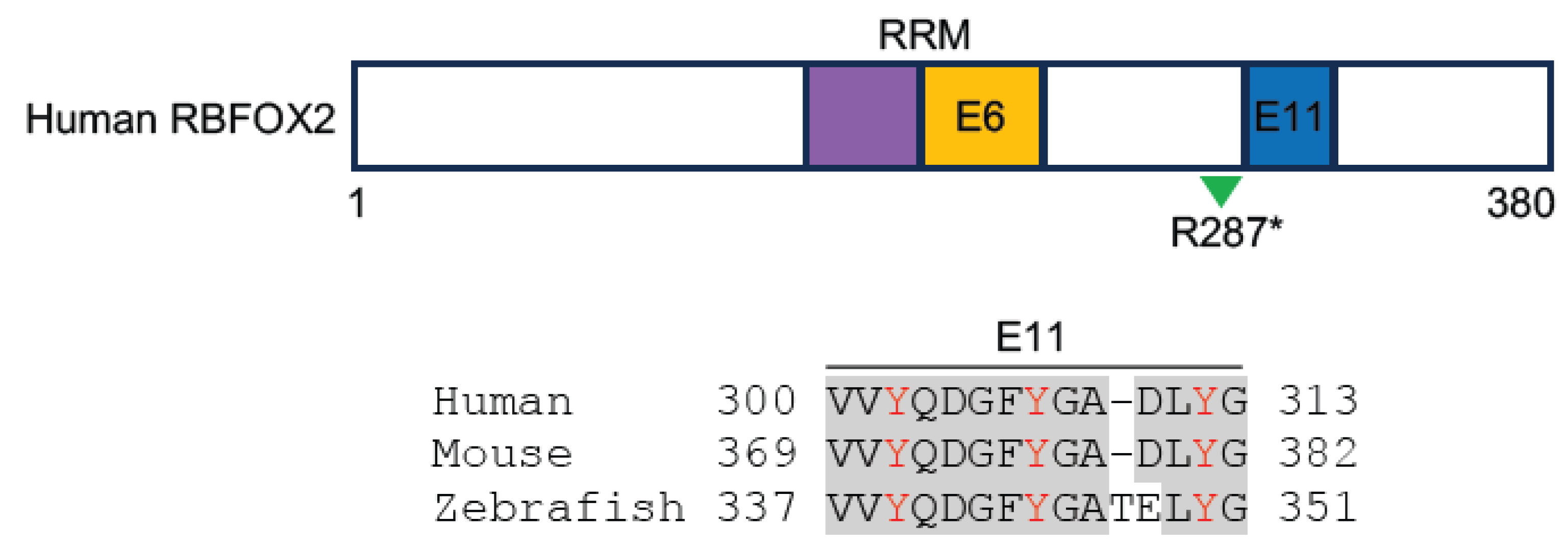
2.4. Rbfox1 and Rbfox2 Are Involved in the Pathogenesis of Heart Disease

2.5. RNA-Binding Proteins Associated with RNA Methylation in Heart Disease
3. Discussion
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Braunwald, E. Cardiomyopathies: An overview. Circ. Res. 2017, 121, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Brieler, J.; Breeden, M.A.; Tucker, J. Cardiomyopathy: An overview. Am. Fam. Physician 2017, 96, 640–646. [Google Scholar] [PubMed]
- Ciarambino, T.; Menna, G.; Sansone, G.; Giordano, M. Cardiomyopathies: An overview. Int. J. Mol. Sci. 2021, 22, 7722. [Google Scholar] [CrossRef] [PubMed]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef] [PubMed]
- Seferović, P.M.; Polovina, M.; Bauersachs, J.; Arad, M.; Ben Gal, T.; Lund, L.H.; Felix, S.B.; Arbustini, E.; Caforio, A.L.P.; Farmakis, D.; et al. Heart failure in cardiomyopathies: A position paper from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 553–576. [Google Scholar] [CrossRef] [PubMed]
- Ziaeian, B.; Fonarow, G.C. Epidemiology and aetiology of heart failure. Nat. Rev. Cardiol. 2016, 13, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Eldemire, R.; Tharp, C.A.; Taylor, M.R.G.; Sbaizero, O.; Mestroni, L. The sarcomeric spring protein Titin: Biophysical properties, molecular mechanisms, and genetic mutations associated with heart failure and cardiomyopathy. Curr. Cardiol. Rep. 2021, 23, 121. [Google Scholar] [CrossRef]
- Fomin, A.; Gärtner, A.; Cyganek, L.; Tiburcy, M.; Tuleta, I.; Wellers, L.; Folsche, L.; Hobbach, A.J.; von Frieling-Salewsky, M.; Unger, A.; et al. Truncated titin proteins and titin haploinsufficiency are targets for functional recovery in human cardiomyopathy due to TTN mutations. Sci. Transl. Med. 2021, 13, eabd3079. [Google Scholar] [CrossRef]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef]
- Loescher, C.M.; Hobbach, A.J.; Linke, W.A. Titin (TTN): From molecule to modifications, mechanics, and medical significance. Cardiovasc. Res. 2022, 118, 2903–2918. [Google Scholar] [CrossRef]
- McAfee, Q.; Chen, C.Y.; Yang, Y.; Caporizzo, M.A.; Morley, M.; Babu, A.; Jeong, S.; Brandimarto, J.; Bedi, K.C., Jr.; Flam, E.; et al. Truncated titin proteins in dilated cardiomyopathy. Sci. Transl. Med. 2021, 13, eabd7287. [Google Scholar] [CrossRef] [PubMed]
- Vikhorev, P.G.; Vikhoreva, N.N.; Yeung, W.; Li, A.; Lal, S.; Dos Remedios, C.G.; Blair, C.A.; Guglin, M.; Campbell, K.S.; Yacoub, M.H.; et al. Titin-truncating mutations associated with dilated cardiomyopathy alter length-dependent activation and its modulation via phosphorylation. Cardiovasc. Res. 2022, 118, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Schäfer, S.; Greaser, M.L.; Radke, M.H.; Liss, M.; Govindarajan, T.; Maatz, H.; Schulz, H.; Li, S.; Parrish, A.M.; et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012, 18, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Beqqali, A. Alternative splicing in cardiomyopathy. Biophys. Rev. 2018, 10, 1061–1071. [Google Scholar] [CrossRef]
- Hasimbegovic, E.; Schweiger, V.; Kastner, N.; Spannbauer, A.; Traxler, D.; Lukovic, D.; Gyöngyösi, M.; Mester-Tonczar, J. Alternative splicing in cardiovascular disease-A survey of recent findings. Genes 2021, 12, 1457. [Google Scholar] [CrossRef] [PubMed]
- Weeland, C.J.; van den Hoogenhof, M.M.; Beqqali, A.; Creemers, E.E. Insights into alternative splicing of sarcomeric genes in the heart. J. Mol. Cell. Cardiol. 2015, 81, 107–113. [Google Scholar] [CrossRef]
- Shi, D.L.; Grifone, R. RNA-binding proteins in the post-transcriptional control of skeletal muscle development, regeneration and disease. Front. Cell Dev. Biol. 2021, 9, 738978. [Google Scholar] [CrossRef]
- Gebauer, F.; Schwarzl, T.; Valcárcel, J.; Hentze, M.W. RNA-binding proteins in human genetic disease. Nat. Rev. Genet. 2021, 22, 185–198. [Google Scholar] [CrossRef]
- Glisovic, T.; Bachorik, J.L.; Yong, J.; Dreyfuss, G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008, 582, 1977–1986. [Google Scholar] [CrossRef]
- D’Antonio, M.; Nguyen, J.P.; Arthur, T.D.; Matsui, H.; Donovan, M.K.R.; D’Antonio-Chronowska, A.; Frazer, K.A. In heart failure reactivation of RNA-binding proteins is associated with the expression of 1,523 fetal-specific isoforms. PLoS Comput. Biol. 2022, 18, e1009918. [Google Scholar] [CrossRef]
- Riechert, E.; Kmietczyk, V.; Stein, F.; Schwarzl, T.; Sekaran, T.; Jürgensen, L.; Kamuf-Schenk, V.; Varma, E.; Hofmann, C.; Rettel, M.; et al. Identification of dynamic RNA-binding proteins uncovers a Cpeb4-controlled regulatory cascade during pathological cell growth of cardiomyocytes. Cell Rep. 2021, 35, 109100. [Google Scholar] [CrossRef] [PubMed]
- Blech-Hermoni, Y.; Ladd, A.N. RNA binding proteins in the regulation of heart development. Int. J. Biochem. Cell Biol. 2013, 45, 2467–2478. [Google Scholar] [CrossRef] [PubMed]
- de Bruin, R.G.; Rabelink, T.J.; van Zonneveld, A.J.; van der Veer, E.P. Emerging roles for RNA-binding proteins as effectors and regulators of cardiovascular disease. Eur. Heart J. 2017, 38, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Kelaini, S.; Chan, C.; Cornelius, V.A.; Margariti, A. RNA-binding proteins hold key roles in function, dysfunction, and disease. Biology 2021, 10, 366. [Google Scholar] [CrossRef] [PubMed]
- Ruffenach, G.; Medzikovic, L.; Sun, W.; Hong, J.; Eghbali, M. Functions of RNA-binding proteins in cardiovascular disease. Cells 2023, 12, 2794. [Google Scholar] [CrossRef]
- Völkers, M.; Preiss, T.; Hentze, M.W. RNA-binding proteins in cardiovascular biology and disease: The beat goes on. Nat. Rev. Cardiol. 2024. [Google Scholar] [CrossRef]
- Grosch, M.; Schraft, L.; Chan, A.; Küchenhoff, L.; Rapti, K.; Ferreira, A.M.; Kornienko, J.; Li, S.; Radke, M.H.; Krämer, C.; et al. Striated muscle-specific base editing enables correction of mutations causing dilated cardiomyopathy. Nat. Commun. 2023, 14, 3714. [Google Scholar] [CrossRef]
- Nishiyama, T.; Zhang, Y.; Cui, M.; Li, H.; Sanchez-Ortiz, E.; McAnally, J.R.; Tan, W.; Kim, J.; Chen, K.; Xu, L.; et al. Precise genomic editing of pathogenic mutations in RBM20 rescues dilated cardiomyopathy. Sci. Transl. Med. 2022, 14, eade1633. [Google Scholar] [CrossRef]
- Shi, D.L. RNA-binding proteins as critical post-transcriptional regulators of cardiac regeneration. Int. J. Mol. Sci. 2023, 24, 12004. [Google Scholar] [CrossRef]
- Cornelius, V.A.; Naderi-Meshkin, H.; Kelaini, S.; Margariti, A. RNA-binding proteins: Emerging therapeutics for vascular dysfunction. Cells 2022, 11, 2494. [Google Scholar] [CrossRef]
- Upadhyay, S.K.; Mackereth, C.D. Structural basis of UCUU RNA motif recognition by splicing factor RBM20. Nucleic Acids Res. 2020, 48, 4538–4550. [Google Scholar] [CrossRef] [PubMed]
- Brauch, K.M.; Karst, M.L.; Herron, K.J.; de Andrade, M.; Pellikka, P.A.; Rodeheffer, R.J.; Michels, V.V.; Olson, T.M. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 2009, 54, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Kimura, A.; Kuroyanagi, H. Alternative splicing regulator RBM20 and cardiomyopathy. Front. Mol. Biosci. 2018, 5, 105. [Google Scholar] [CrossRef] [PubMed]
- Koelemen, J.; Gotthardt, M.; Steinmetz, L.M.; Meder, B. RBM20-related cardiomyopathy: Current understanding and future options. J. Clin. Med. 2021, 10, 4101. [Google Scholar] [CrossRef] [PubMed]
- Lennermann, D.; Backs, J.; van den Hoogenhof, M.M.G. New insights in RBM20 cardiomyopathy. Curr. Heart Fail. Rep. 2020, 17, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Eberl, H.; Rebs, S.; Hoppe, S.; Sedaghat-Hamedani, F.; Kayvanpour, E.; Meder, B.; Streckfuss-Bömeke, K. Generation of an RBM20-mutation-associated left-ventricular non-compaction cardiomyopathy iPSC line (UMGi255-A) into a DCM genetic background to investigate monogenetic cardiomyopathies. Stem Cell Res. 2024, 74, 103290. [Google Scholar] [CrossRef] [PubMed]
- Ihara, K.; Sasano, T.; Hiraoka, Y.; Togo-Ohno, M.; Soejima, Y.; Sawabe, M.; Tsuchiya, M.; Ogawa, H.; Furukawa, T.; Kuroyanagi, H. A missense mutation in the RSRSP stretch of Rbm20 causes dilated cardiomyopathy and atrial fibrillation in mice. Sci. Rep. 2020, 10, 17894. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.W.; Oommen, S.; Qureshi, M.Y.; Goetsch, S.C.; Pease, D.R.; Sundsbak, R.S.; Guo, W.; Sun, M.; Sun, H.; Kuroyanagi, H.; et al. Dysregulated ribonucleoprotein granules promote cardiomyopathy in RBM20 gene-edited pigs. Nat. Med. 2020, 26, 1788–1800. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, Y.; Methawasin, M.; Braz, C.U.; Gao-Hu, J.; Yang, B.; Strom, J.; Gohlke, J.; Hacker, T.; Khatib, H.; et al. RBM20S639G mutation is a high genetic risk factor for premature death through RNA-protein condensates. J. Mol. Cell. Cardiol. 2022, 165, 115–129. [Google Scholar] [CrossRef]
- Kornienko, J.; Rodríguez-Martínez, M.; Fenzl, K.; Hinze, F.; Schraivogel, D.; Grosch, M.; Tunaj, B.; Lindenhofer, D.; Schraft, L.; Kueblbeck, M.; et al. Mislocalization of pathogenic RBM20 variants in dilated cardiomyopathy is caused by loss-of-interaction with Transportin-3. Nat. Commun. 2023, 14, 4312. [Google Scholar] [CrossRef]
- Zhang, Y.; Gregorich, Z.R.; Wang, Y.; Braz, C.U.; Zhang, J.; Liu, Y.; Liu, P.; Shen, J.; Aori, N.; Hacker, T.A.; et al. Disruption of the nuclear localization signal in RBM20 is causative in dilated cardiomyopathy. JCI Insight 2023, 8, e170001. [Google Scholar] [CrossRef] [PubMed]
- van den Hoogenhof, M.M.G.; Beqqali, A.; Amin, A.S.; van der Made, I.; Aufiero, S.; Khan, M.A.F.; Schumacher, C.A.; Jansweijer, J.A.; van Spaendonck-Zwarts, K.Y.; Remme, C.A.; et al. RBM20 mutations induce an arrhythmogenic dilated cardiomyopathy related to disturbed calcium handling. Circulation 2018, 138, 1330–1342. [Google Scholar] [CrossRef] [PubMed]
- Gaertner, A.; Klauke, B.; Felski, E.; Kassner, A.; Brodehl, A.; Gerdes, D.; Stanasiuk, C.; Ebbinghaus, H.; Schulz, U.; Dubowy, K.O.; et al. Cardiomyopathy-associated mutations in the RS domain affect nuclear localization of RBM20. Hum. Mutat. 2020, 41, 1931–1943. [Google Scholar] [CrossRef] [PubMed]
- Murayama, R.; Kimura-Asami, M.; Togo-Ohno, M.; Yamasaki-Kato, Y.; Naruse, T.K.; Yamamoto, T.; Hayashi, T.; Ai, T.; Spoonamore, K.G.; Kovacs, R.J.; et al. Phosphorylation of the RSRSP stretch is critical for splicing regulation by RNA-Binding Motif Protein 20 (RBM20) through nuclear localization. Sci. Rep. 2018, 8, 8970. [Google Scholar] [CrossRef] [PubMed]
- Fenix, A.M.; Miyaoka, Y.; Bertero, A.; Blue, S.M.; Spindler, M.J.; Tan, K.K.B.; Perez-Bermejo, J.A.; Chan, A.H.; Mayerl, S.J.; Nguyen, T.D.; et al. Gain-of-function cardiomyopathic mutations in RBM20 rewire splicing regulation and re-distribute ribonucleoprotein granules within processing bodies. Nat. Commun. 2021, 12, 6324. [Google Scholar] [CrossRef]
- Sedaghat-Hamedani, F.; Haas, J.; Zhu, F.; Geier, C.; Kayvanpour, E.; Liss, M.; Lai, A.; Frese, K.; Pribe-Wolferts, R.; Amr, A.; et al. Clinical genetics and outcome of left ventricular non-compaction cardiomyopathy. Eur. Heart J. 2017, 38, 3449–3460. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, N.; Hayashi, T.; Takei, Y.; Kosuge, H.; Suzuki, S.; Tanimoto, K.; Chikamori, T.; Kimura, A. Pathogenic variant of RBM20 in a multiplex family with hypertrophic cardiomyopathy. Hum. Genome Var. 2022, 9, 6. [Google Scholar] [CrossRef]
- Guo, W.; Bharmal, S.J.; Esbona, K.; Greaser, M.L. Titin diversity--alternative splicing gone wild. J. Biomed. Biotechnol. 2010, 2010, 753675. [Google Scholar] [CrossRef]
- Tharp, C.A.; Haywood, M.E.; Sbaizero, O.; Taylor, M.R.G.; Mestroni, L. The giant protein Titin’s role in cardiomyopathy: Genetic, transcriptional, and post-translational modifications of TTN and their contribution to cardiac disease. Front. Physiol. 2019, 10, 1436. [Google Scholar] [CrossRef]
- Neagoe, C.; Opitz, C.A.; Makarenko, I.; Linke, W.A. Gigantic variety: Expression patterns of titin isoforms in striated muscles and consequences for myofibrillar passive stiffness. J. Muscle Res. Cell Motil. 2003, 24, 175–189. [Google Scholar] [CrossRef]
- Li, S.; Guo, W.; Dewey, C.N.; Greaser, M.L. Rbm20 regulates titin alternative splicing as a splicing repressor. Nucleic Acids Res. 2013, 41, 2659–2672. [Google Scholar] [CrossRef]
- Maatz, H.; Jens, M.; Liss, M.; Schafer, S.; Heinig, M.; Kirchner, M.; Adami, E.; Rintisch, C.; Dauksaite, V.; Radke, M.H.; et al. RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J. Clin. Investig. 2014, 124, 3419–3430. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Li, Z.; Huang, W.; Chen, P.; Sun, Y.; Wang, H.; Wu, D.; Chen, Y.; Li, C.; Xiao, L.; et al. RBM20 Is a candidate gene for hypertrophic cardiomyopathy. Can. J. Cardiol. 2021, 37, 1751–1759. [Google Scholar] [CrossRef]
- Grifone, R.; Shao, M.; Saquet, A.; Shi, D.L. RNA-binding protein Rbm24 as a multifaceted post-transcriptional regulator of embryonic lineage differentiation and cellular homeostasis. Cells 2020, 9, 1891. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.; Kazan, H.; Chan, E.T.; Peña-Castillo, L.; Chaudhry, S.; Talukder, S.; Blencowe, B.J.; Morris, Q.; Hughes, T.R. Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nat. Biotechnol. 2009, 27, 667–670. [Google Scholar] [CrossRef] [PubMed]
- Fetka, I.; Radeghieri, A.; Bouwmeester, T. Expression of the RNA recognition motif-containing protein SEB-4 during Xenopus embryonic development. Mech. Dev. 2000, 94, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Grifone, R.; Xie, X.; Bourgeois, A.; Saquet, A.; Duprez, D.; Shi, D.L. The RNA-binding protein Rbm24 is transiently expressed in myoblasts and is required for myogenic differentiation during vertebrate development. Mech. Dev. 2014, 134, 1–15. [Google Scholar] [CrossRef]
- Maragh, S.; Miller, R.A.; Bessling, S.L.; McGaughey, D.M.; Wessels, M.W.; de Graaf, B.; Stone, E.A.; Bertoli-Avella, A.M.; Gearhart, J.D.; Fisher, S.; et al. Identification of RNA binding motif proteins essential for cardiovascular development. BMC Dev. Biol. 2011, 11, 62. [Google Scholar] [CrossRef]
- Miller, R.A.; Christoforou, N.; Pevsner, J.; McCallion, A.S.; Gearhart, J.D. Efficient array-based identification of novel cardiac genes through differentiation of mouse ESCs. PLoS ONE. 2008, 3, e2176. [Google Scholar] [CrossRef]
- Poon, K.L.; Tan, K.T.; Wei, Y.Y.; Ng, C.P.; Colman, A.; Korzh, V.; Xu, X.Q. RNA-binding protein RBM24 is required for sarcomere assembly and heart contractility. Cardiovasc. Res. 2012, 94, 418–427. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, Y.; Xu, E.; Mohibi, S.; De Anda, D.M.; Jiang, Y.; Zhang, J.; Chen, X. Rbm24, a target of p53, is necessary for proper expression of p53 and heart development. Cell Death Differ. 2018, 25, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Lu, T.; Zhang, C.; Zhang, Y.Z.; Kong, S.H.; Shi, D.L. Rbm24 controls poly(A) tail length and translation efficiency of crystallin mRNAs in the lens via cytoplasmic polyadenylation. Proc. Natl. Acad. Sci. USA 2020, 117, 7245–7254. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hung, L.H.; Licht, T.; Kostin, S.; Looso, M.; Khrameeva, E.E.; Bindereif, A.; Schneider, A.; Braun, T. RBM24 Is a major regulator of muscle-specific alternative splicing. Dev. Cell. 2014, 31, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kong, X.; Zhang, M.; Yang, X.; Xu, X.Q. RNA binding protein 24 deletion disrupts global alternative splicing and causes dilated cardiomyopathy. Protein Cell. 2019, 10, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.H.; Lee, K.Z.; Hsu, P.W.; Su, L.Y.; Yeh, Y.C.; Pan, C.Y.; Tsai, S.Y. Alternative splicing mediated by RNA-binding protein RBM24 facilitates cardiac myofibrillogenesis in a differentiation stage-specific manner. Circ. Res. 2022, 130, 112–129. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, K.; Liu, X.; Pan, L.; Zhou, W.; Huang, J.; Liu, H.; Su, Z.; Xu, X.Q. RBM24 controls cardiac QT interval through CaMKIIδ splicing. Cell. Mol. Life Sci. 2022, 79, 613. [Google Scholar] [CrossRef] [PubMed]
- Grifone, R.; Saquet, A.; Desgres, M.; Sangiorgi, C.; Gargano, C.; Li, Z.; Coletti, D.; Shi, D.L. Rbm24 displays dynamic functions required for myogenic differentiation during muscle regeneration. Sci. Rep. 2021, 11, 9423. [Google Scholar] [CrossRef]
- Akerberg, A.A.; Burns, C.E.; Burns, C.G. Exploring the activities of RBPMS proteins in myocardial biology. Pediatr. Cardiol. 2019, 40, 1410–1418. [Google Scholar] [CrossRef]
- Soufari, H.; Mackereth, C.D. Conserved binding of GCAC motifs by MEC-8, couch potato, and the RBPMS protein family. RNA. 2017, 23, 308–316. [Google Scholar] [CrossRef]
- Gan, P.; Wang, Z.; Morales, M.G.; Zhang, Y.; Bassel-Duby, R.; Liu, N.; Olson, E.N. RBPMS is an RNA-binding protein that mediates cardiomyocyte binucleation and cardiovascular development. Dev. Cell. 2022, 57, 959–973.e7. [Google Scholar] [CrossRef]
- Gan, P.; Wang, Z.; Bezprozvannaya, S.; McAnally, J.R.; Tan, W.; Li, H.; Bassel-Duby, R.; Liu, N.; Olson, E.N. RBPMS regulates cardiomyocyte contraction and cardiac function through RNA alternative splicing. Cardiovasc. Res. 2024, 120, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Akerberg, A.A.; Trembley, M.; Butty, V.; Schwertner, A.; Zhao, L.; Beerens, M.; Liu, X.; Mahamdeh, M.; Yuan, S.; Boyer, L.; et al. RBPMS2 Is a myocardial-enriched splicing regulator required for cardiac function. Circ. Res. 2022, 131, 980–1000. [Google Scholar] [CrossRef] [PubMed]
- Tudurachi, B.S.; Zăvoi, A.; Leonte, A.; Țăpoi, L.; Ureche, C.; Bîrgoan, S.G.; Chiuariu, T.; Anghel, L.; Radu, R.; Sascău, R.A.; et al. An update on MYBPC3 gene mutation in hypertrophic cardiomyopathy. Int. J. Mol. Sci. 2023, 24, 10510. [Google Scholar] [CrossRef] [PubMed]
- Auxerre-Plantié, E.; Nielsen, T.; Grunert, M.; Olejniczak, O.; Perrot, A.; Özcelik, C.; Harries, D.; Matinmehr, F.; Dos Remedios, C.; Mühlfeld, C.; et al. Identification of MYOM2 as a candidate gene in hypertrophic cardiomyopathy and Tetralogy of Fallot, and its functional evaluation in the Drosophila heart. Dis. Model Mech. 2020, 13, dmm045377. [Google Scholar] [CrossRef] [PubMed]
- Conboy, J.G. Developmental regulation of RNA processing by Rbfox proteins. Wiley Interdiscip. Rev. RNA 2017, 8, e1398. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Ren, S.; Lee, J.H.; Qiu, J.; Chapski, D.J.; Rau, C.D.; Zhou, Y.; Abdellatif, M.; Nakano, A.; Vondriska, T.M.; et al. RBFox1-mediated RNA splicing regulates cardiac hypertrophy and heart failure. J. Clin. Investig. 2016, 126, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Lee, J.Z.; Rau, C.D.; Pezhouman, A.; Yokota, T.; Miwa, H.; Feldman, M.; Kong, T.K.; Yang, Z.; Tay, W.T.; et al. Regulation of postnatal cardiomyocyte maturation by an RNA splicing regulator RBFox1. Circulation 2023, 148, 1263–1266. [Google Scholar] [CrossRef] [PubMed]
- Nutter, C.A.; Jaworski, E.A.; Verma, S.K.; Deshmukh, V.; Wang, Q.; Botvinnik, O.B.; Lozano, M.J.; Abass, I.J.; Ijaz, T.; Brasier, A.R.; et al. Dysregulation of RBFOX2 Is an early event in cardiac pathogenesis of diabetes. Cell Rep. 2016, 15, 2200–2213. [Google Scholar] [CrossRef]
- Damianov, A.; Black, D.L. Autoregulation of Fox protein expression to produce dominant negative splicing factors. RNA 2010, 16, 405–416. [Google Scholar] [CrossRef]
- Glessner, J.T.; Bick, A.G.; Ito, K.; Homsy, J.; Rodriguez-Murillo, L.; Fromer, M.; Mazaika, E.; Vardarajan, B.; Italia, M.; Leipzig, J.; et al. Increased frequency of de novo copy number variants in congenital heart disease by integrative analysis of single nucleotide polymorphism array and exome sequence data. Circ. Res. 2014, 115, 884–896. [Google Scholar] [CrossRef]
- Homsy, J.; Zaidi, S.; Shen, Y.; Ware, J.S.; Samocha, K.E.; Karczewski, K.J.; DePalma, S.R.; McKean, D.; Wakimoto, H.; Gorham, J.; et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 2015, 350, 1262–1266. [Google Scholar] [CrossRef] [PubMed]
- McKean, D.M.; Homsy, J.; Wakimoto, H.; Patel, N.; Gorham, J.; DePalma, S.R.; Ware, J.S.; Zaidi, S.; Ma, W.; Patel, N.; et al. Loss of RNA expression and allele-specific expression associated with congenital heart disease. Nat. Commun. 2016, 7, 12824. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.K.; Deshmukh, V.; Nutter, C.A.; Jaworski, E.; Jin, W.; Wadhwa, L.; Abata, J.; Ricci, M.; Lincoln, J.; Martin, J.F.; et al. Rbfox2 function in RNA metabolism is impaired in hypoplastic left heart syndrome patient hearts. Sci. Rep. 2016, 6, 30896. [Google Scholar] [CrossRef] [PubMed]
- Ying, Y.; Wang, X.J.; Vuong, C.K.; Lin, C.H.; Damianov, A.; Black, D.L. Splicing activation by Rbfox requires self-aggregation through its tyrosine-rich domain. Cell 2017, 170, 312–323.e10. [Google Scholar] [CrossRef]
- Wei, C.; Qiu, J.; Zhou, Y.; Xue, Y.; Hu, J.; Ouyang, K.; Banerjee, I.; Zhang, C.; Chen, B.; Li, H.; et al. Repression of the central splicing regulator RBFox2 is functionally linked to pressure overload-induced heart failure. Cell Rep. 2015, 10, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.K.; Deshmukh, V.; Thatcher, K.; Belanger, K.K.; Rhyner, A.M.; Meng, S.; Holcomb, R.J.; Bressan, M.; Martin, J.F.; Cooke, J.P.; et al. RBFOX2 is required for establishing RNA regulatory networks essential for heart development. Nucleic Acids Res. 2022, 50, 2270–2286. [Google Scholar] [CrossRef] [PubMed]
- Frese, K.S.; Meder, B.; Keller, A.; Just, S.; Haas, J.; Vogel, B.; Fischer, S.; Backes, C.; Matzas, M.; Köhler, D.; et al. RNA splicing regulated by RBFOX1 is essential for cardiac function in zebrafish. J. Cell Sci. 2015, 128, 3030–3040. [Google Scholar] [CrossRef]
- Huang, M.; Akerberg, A.A.; Zhang, X.; Yoon, H.; Joshi, S.; Hallinan, C.; Nguyen, C.; Pu, W.T.; Haigis, M.C.; Burns, C.G.; et al. Intrinsic myocardial defects underlie an Rbfox-deficient zebrafish model of hypoplastic left heart syndrome. Nat. Commun. 2022, 13, 5877. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, B.; Nie, Z.; Duan, L.; Xiong, Q.; Jin, Z.; Yang, C.; Chen, Y. The role of m6A modification in the biological functions and diseases. Signal Transduct. Target. Ther. 2021, 6, 74. [Google Scholar] [CrossRef]
- Wang, S.; Lv, W.; Li, T.; Zhang, S.; Wang, H.; Li, X.; Wang, L.; Ma, D.; Zang, Y.; Shen, J.; et al. Dynamic regulation and functions of mRNA m6A modification. Cancer Cell Int. 2022, 22, 48. [Google Scholar] [CrossRef]
- Sikorski, V.; Vento, A.; Kankuri, E.; IHD-EPITRAN Consortium. Emerging roles of the RNA modifications N6-methyladenosine and adenosine-to-inosine in cardiovascular diseases. Mol. Ther. Nucleic Acids 2022, 29, 426–461. [Google Scholar] [CrossRef] [PubMed]
- Krumbein, M.; Oberman, F.; Cinnamon, Y.; Golomb, M.; May, D.; Vainer, G.; Belzer, V.; Meir, K.; Fridman, I.; Haybaeck, J.; et al. RNA binding protein IGF2BP2 expression is induced by stress in the heart and mediates dilated cardiomyopathy. Commun. Biol. 2023, 6, 1229. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Sun, H.; Chen, K.; Gu, X.; Chen, H.; Jiang, L.; Chen, L.; Zhang, S.; Liu, Y.; Shi, D.; et al. Depletion of m(6) A reader protein YTHDC1 induces dilated cardiomyopathy by abnormal splicing of Titin. J. Cell Mol. Med. 2021, 25, 10879–10891. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, J.; Wu, X.; Lin, X.; Liu, X.M.; Zhou, J. Differential roles of YTHDF1 and YTHDF3 in embryonic stem cell-derived cardiomyocyte differentiation. RNA Biol. 2021, 18, 1354–1363. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, Z.; Chen, M.; Zhao, W.; Tao, T.; Ma, L.; Ni, Y.; Li, W. YTHDF2 alleviates cardiac hypertrophy via regulating Myh7 mRNA decoy. Cell Biosci. 2021, 11, 132. [Google Scholar] [CrossRef] [PubMed]
- Golubeva, V.A.; Dorn, L.E.; Gilbert, C.J.; Rabolli, C.P.; Das, A.S.; Wanasinghe, V.S.; Veress, R.; Terentyev, D.; Accornero, F. Loss of YTHDF2 alters the expression of m6A-modified Myzap and causes adverse cardiac remodeling. JACC Basic Transl. Sci. 2023, 8, 1180–1194. [Google Scholar] [CrossRef] [PubMed]
- Kmietczyk, V.; Oelschläger, J.; Gupta, P.; Varma, E.; Hartl, S.; Furkel, J.; Konstandin, M.; Marx, A.; Loewenthal, Z.; Kamuf-Schenk, V.; et al. Ythdf2 regulates cardiac remodeling through its mRNA target transcripts. J. Mol. Cell. Cardiol. 2023, 181, 57–66. [Google Scholar] [CrossRef]
- Martino, F.; Varadarajan, N.M.; Perestrelo, A.R.; Hejret, V.; Durikova, H.; Vukic, D.; Horvath, V.; Cavalieri, F.; Caruso, F.; Albihlal, W.S.; et al. The mechanical regulation of RNA binding protein hnRNPC in the failing heart. Sci. Transl. Med. 2022, 14, eabo5715. [Google Scholar] [CrossRef]
- Montañés-Agudo, P.; Pinto, Y.M.; Creemers, E.E. Splicing factors in the heart: Uncovering shared and unique targets. J. Mol. Cell. Cardiol. 2023, 179, 72–79. [Google Scholar] [CrossRef]
- Ito, J.; Iijima, M.; Yoshimoto, N.; Niimi, T.; Kuroda, S.; Maturana, A.D. RBM20 and RBM24 cooperatively promote the expression of short enh splice variants. FEBS Lett. 2016, 590, 2262–2274. [Google Scholar] [CrossRef]
- Lee, K.Y.; Seah, C.; Li, C.; Chen, Y.F.; Chen, C.Y.; Wu, C.I.; Liao, P.C.; Shyu, Y.C.; Olafson, H.R.; McKee, K.K.; et al. Mice lacking MBNL1 and MBNL2 exhibit sudden cardiac death and molecular signatures recapitulating myotonic dystrophy. Hum. Mol. Genet. 2022, 31, 3144–3160. [Google Scholar] [CrossRef] [PubMed]
- Montañés-Agudo, P.; Aufiero, S.; Schepers, E.N.; van der Made, I.; Cócera-Ortega, L.; Ernault, A.C.; Richard, S.; Kuster, D.W.D.; Christoffels, V.M.; Pinto, Y.M.; et al. The RNA-binding protein QKI governs a muscle-specific alternative splicing program that shapes the contractile function of cardiomyocytes. Cardiovasc. Res. 2023, 119, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Yu, S.; Liu, X.; Zhang, Y.; Fakadej, T.; Liu, Z.; Yin, C.; Shen, W.; Locasale, J.W.; Taylor, J.M.; et al. Lin28a regulates pathological cardiac hypertrophic growth through Pck2-mediated enhancement of anabolic synthesis. Circulation 2019, 139, 1725–1740. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Tan, K.T.; Liu, J.; Kong, X.; Huang, Z.; Xu, X.Q. Global profiling of Rbm24 bound RNAs uncovers a multi-tasking RNA binding protein. Int. J. Biochem. Cell Biol. 2018, 94, 10–21. [Google Scholar] [CrossRef]
- Bartsch, D.; Kalamkar, K.; Ahuja, G.; Lackmann, J.W.; Hescheler, J.; Weber, T.; Bazzi, H.; Clamer, M.; Mendjan, S.; Papantonis, A.; et al. mRNA translational specialization by RBPMS presets the competence for cardiac commitment in hESCs. Sci. Adv. 2023, 9, eade1792. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Verma, S.K.; Jaworski, E.; Mohan, S.; Nagasawa, C.K.; Rayavara, K.; Sooter, A.; Miller, S.N.; Holcomb, R.J.; Powell, M.J.; et al. RBFOX2 is critical for maintaining alternative polyadenylation patterns and mitochondrial health in rat myoblasts. Cell Rep. 2021, 37, 109910. [Google Scholar] [CrossRef] [PubMed]
- Kemmler, C.L.; Riemslagh, F.W.; Moran, H.R.; Mosimann, C. From stripes to a beating heart: Early cardiac development in zebrafish. J. Cardiovasc. Dev. Dis. 2021, 8, 17. [Google Scholar] [CrossRef]
- Staudt, D.; Stainier, D. Uncovering the molecular and cellular mechanisms of heart development using the zebrafish. Annu. Rev. Genet. 2012, 46, 397–418. [Google Scholar] [CrossRef]
- Bakkers, J. Zebrafish as a model to study cardiac development and human cardiac disease. Cardiovasc. Res. 2011, 91, 279–288. [Google Scholar] [CrossRef]
- González-Rosa, J.M. Zebrafish models of cardiac disease: From fortuitous mutants to precision medicine. Circ. Res. 2022, 130, 1803–1826. [Google Scholar] [CrossRef]
- Miura, G.I.; Yelon, D. A guide to analysis of cardiac phenotypes in the zebrafish embryo. Methods Cell Biol. 2011, 101, 161–180. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| RBPs | Mutations or Knockout | Cardiovascular Phenotypes | Dysregulated Target Genes | References |
|---|---|---|---|---|
| RBM20 | Heterozygous missense mutations in human patients | DCM and likely HCM in humans | Defective splicing of Titin, Camk2d, and many other sarcomeric genes | [13,32,42,47,53] |
| RBM24 | Knockout in mice, zebrafish, and human embryonic stem cells | DCM and prolongation of the QT interval in mice | Defective splicing of Titin, Camk2d, α-actinin 2, and other muscle-specific genes; increased p53 mRNA translation | [61,64,65,66] |
| RBPMS | Knockout in mice and human cardiomyocytes | Non-compaction cardiomyopathy and DCM in mice | Defective splicing of Titin, Pdlim5, and cardiac myofibrillogenesis genes | [70,71] |
| RBPMS2 | Knockout in zebrafish and human cardiomyocytes | Cardiac defects in zebrafish reminiscent of HCM | Defective splicing of Rbfox2, Mybpc3, Slc8a1, and Myom2a | [72] |
| RBFOX1 | Knockout in mice and knockdown in zebrafish | Cardiac hypertrophy, cardiomyoapthy, and heart failure | Defective splicing of Mef2 in mice, and huG, actn3a, ptpla, camk2g1, and ktn1 in zebrafish | [76,87] |
| RBFOX2 | De novo frameshift, nonsense, or splice site mutations in HLHS human patients; knockout in mice and zebrafish | HLHS in humans and zebrafish (rbfox1/2 mutants); DCM in mice | Defective splicing of sarcomere components (tpm1, tpm3, and tnnt3b), MICOS complex components (mic19a and mic19b), and cytoskeletal components (pdlim5b, alcama, and fmnl3) | [80,81,82,83,85,88] |
| IGF2BP2 | Overexpression in mice | DCM in mice | Reduced expression of sarcomeric and mitochondrial proteins (Titin and Mybpc3 unaffected) | [92] |
| YTHDC1 | Knockout in mice | DCM in mice | Defective splicing of Titin | [93] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, D.-L. RNA-Binding Proteins in Cardiomyopathies. J. Cardiovasc. Dev. Dis. 2024, 11, 88. https://doi.org/10.3390/jcdd11030088
Shi D-L. RNA-Binding Proteins in Cardiomyopathies. Journal of Cardiovascular Development and Disease. 2024; 11(3):88. https://doi.org/10.3390/jcdd11030088
Chicago/Turabian StyleShi, De-Li. 2024. "RNA-Binding Proteins in Cardiomyopathies" Journal of Cardiovascular Development and Disease 11, no. 3: 88. https://doi.org/10.3390/jcdd11030088
APA StyleShi, D. -L. (2024). RNA-Binding Proteins in Cardiomyopathies. Journal of Cardiovascular Development and Disease, 11(3), 88. https://doi.org/10.3390/jcdd11030088