
The Importance of Telomere Shortening for Atherosclerosis and Mortality


Abstract
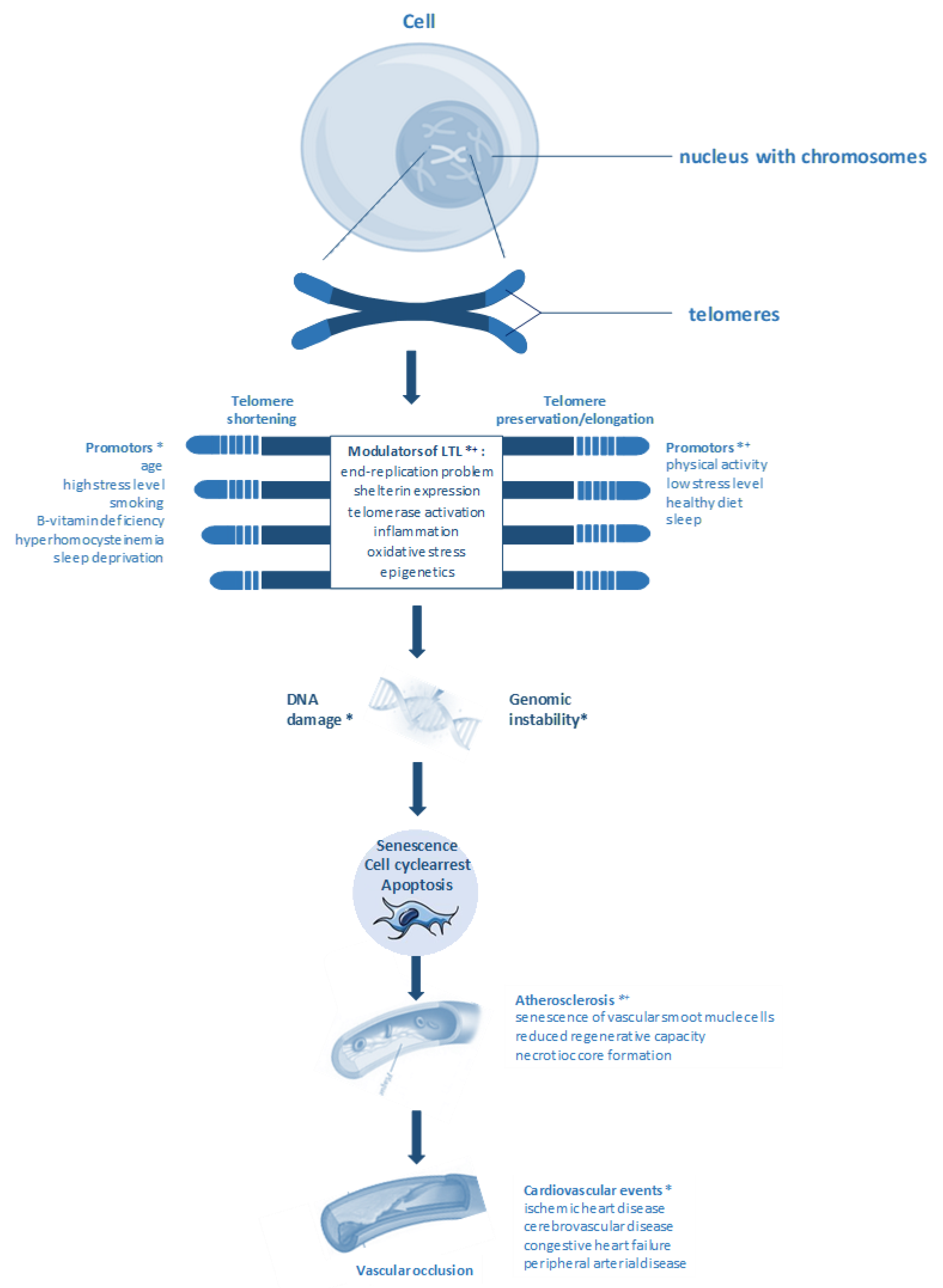
:1. Introduction
2. Telomere Length and Age
3. Telomere Length, Cardiovascular Disease (CVD), and Mortality
4. Telomere Length and Atherosclerosis
5. Telomere Shortening and CVD in Pre-Clinical Models
6. B-vitamins, Homocysteine, Telomere Length, and CVD
7. Markers of Inflammation, Oxidative Stress, HCY, LTL, and Mortality
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vijg, J.; Suh, Y. Genome instability and aging. Annu. Rev. Physiol. 2013, 75, 645–668. [Google Scholar] [CrossRef]
- Lombard, D.B.; Chua, K.F.; Mostoslavsky, R.; Franco, S.; Gostissa, M.; Alt, F.W. DNA repair, genome stability, and aging. Cell 2005, 120, 497–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasco, M.A. Telomere length, stem cells and aging. Nat. Chem. Biol. 2007, 3, 640–649. [Google Scholar] [CrossRef]
- Blasco, M.A. Telomeres and human disease: Ageing, cancer and beyond. Nat. Rev. Genet. 2005, 6, 611–622. [Google Scholar] [CrossRef]
- Muller, H. The remaking of chromosomes. Collect. Net 1938, 13, 181–198. [Google Scholar]
- McClintock, B. The stability of broken ends of chromosomes in Zea mays. Genetics 1941, 26, 234. [Google Scholar] [PubMed]
- de Lange, T. Shelterin-mediated telomere protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef] [PubMed]
- de Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, U.; Griese, E.-U.; Schwab, M.; Fritz, P.; Thon, K.-P.; Klotz, U. Telomere length in different tissues of elderly patients. Mech. Ageing Dev. 2000, 119, 89–99. [Google Scholar] [CrossRef]
- Yeh, J.-K.; Wang, C.-Y. Telomeres and telomerase in cardiovascular diseases. Genes 2016, 7, 58. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick, A.L.; Kronmal, R.A.; Kimura, M.; Gardner, J.P.; Psaty, B.M.; Jenny, N.S.; Tracy, R.P.; Hardikar, S.; Aviv, A. Leukocyte telomere length and mortality in the cardiovascular health study. J. Gerontol. Ser. Biomed. Sci. Med. Sci. 2011, 66, 421–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dlouha, D.; Maluskova, J.; Lesna, I.K.; Lanska, V.; Hubacek, J. Comparison of the relative telomere length measured in leukocytes and eleven different human tissues. Physiol. Res. 2014, 63 (Suppl. 3), S343–350. [Google Scholar]
- Wilson, W.R.; Herbert, K.E.; Mistry, Y.; Stevens, S.E.; Patel, H.R.; Hastings, R.A.; Thompson, M.M.; Williams, B. Blood leucocyte telomere DNA content predicts vascular telomere DNA content in humans with and without vascular disease. Eur. Heart J. 2008, 29, 2689–2694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willeit, P.; Willeit, J.; Brandstätter, A.; Ehrlenbach, S.; Mayr, A.; Gasperi, A.; Weger, S.; Oberhollenzer, F.; Reindl, M.; Kronenberg, F. Cellular aging reflected by leukocyte telomere length predicts advanced atherosclerosis and cardiovascular disease risk. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1649–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mons, U.; Müezzinler, A.; Schöttker, B.; Dieffenbach, A.K.; Butterbach, K.; Schick, M.; Peasey, A.; De Vivo, I.; Trichopoulou, A.; Boffetta, P.; et al. Leukocyte telomere length and all-cause, cardiovascular disease, and cancer mortality: Results from individual-participant-data meta-analysis of 2 large prospective cohort studies. Am. J. Epidemiol. 2017, 185, 1317–1326. [Google Scholar] [CrossRef] [Green Version]
- Needham, B.L.; Rehkopf, D.; Adler, N.; Gregorich, S.; Lin, J.; Blackburn, E.H.; Epel, E.S. Leukocyte telomere length and mortality in the National Health and Nutrition Examination Survey, 1999–2002. Epidemiology 2015, 26, 528. [Google Scholar] [CrossRef] [Green Version]
- Rode, L.; Nordestgaard, B.G.; Bojesen, S.E. Peripheral blood leukocyte telomere length and mortality among 64 637 individuals from the general population. J. Nat. Cancer Inst. 2015, 107, djv074. [Google Scholar] [CrossRef] [Green Version]
- Cawthon, R.M.; Smith, K.R.; O’Brien, E.; Sivatchenko, A.; Kerber, R.A. Association between telomere length in blood and mortality in people aged 60 years or older. Lancet 2003, 361, 393–395. [Google Scholar] [CrossRef]
- Herrmann, M.; Pusceddu, I.; März, W.; Herrmann, W. Telomere biology and age-related diseases. Clin. Chem. Lab. Med. 2018, 56, 1210–1222. [Google Scholar] [CrossRef]
- Müezzinler, A.; Zaineddin, A.K.; Brenner, H. A systematic review of leukocyte telomere length and age in adults. Ageing Res. Rev. 2013, 12, 509–519. [Google Scholar] [CrossRef]
- Pusceddu, I.; Kleber, M.; Delgado, G.; Herrmann, W.; März, W.; Herrmann, M. Telomere length and mortality in the ludwigshafen risk and cardiovascular health study. PLoS ONE 2018, 13, e0198373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Kimura, M.; Kim, S.; Cao, X.; Srinivasan, S.R.; Berenson, G.; Kark, J.; Aviv, A. Longitudinal versus cross-sectional evaluations of leukocyte telomere length dynamics: Age-dependent telomere shortening is the rule. J. Gerontol. Ser. Biomed. Sci. Med. Sci. 2011, 66, 312–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrlenbach, S.; Willeit, P.; Kiechl, S.; Willeit, J.; Reindl, M.; Schanda, K.; Kronenberg, F.; Brandstätter, A. Influences on the reduction of relative telomere length over 10 years in the population-based bruneck study: Introduction of a well-controlled high-throughput assay. Int. J. Epidemiol. 2009, 38, 1725–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsova, T.; Codd, V.; Brouilette, S.; Thijs, L.; González, A.; Jin, Y.; Richart, T.; van der Harst, P.; Díez, J.; Staessen, J.A.; et al. Association between left ventricular mass and telomere length in a population study. Am. J. Epidemiol. 2010, 172, 440–450. [Google Scholar] [CrossRef] [Green Version]
- Rius-Ottenheim, N.; Houben, J.M.; Kromhout, D.; Kafatos, A.; van der Mast, R.C.; Zitman, F.G.; Geleijnse, J.M.; Hageman, G.J.; Giltay, E.J. Telomere length and mental well-being in elderly men from the Netherlands and Greece. Behav. Genet. 2012, 42, 278–286. [Google Scholar] [CrossRef] [Green Version]
- Benetos, A.; Verhulst, S.; Labat, C.; Lai, T.P.; Girerd, N.; Toupance, S.; Zannad, F.; Rossignol, P.; Aviv, A. Telomere length tracking in children and their parents: Implications for adult onset diseases. FASEB J. 2019, 33, 14248–14253. [Google Scholar] [CrossRef] [Green Version]
- Aviv, A.; Chen, W.; Gardner, J.P.; Kimura, M.; Brimacombe, M.; Cao, X.; Srinivasan, S.R.; Berenson, G.S. Leukocyte telomere dynamics: Longitudinal findings among young adults in the bogalusa heart study. Am. J. Epidemiol. 2009, 169, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Delgado, B.; Yanowsky, K.; Inglada-Perez, L.; Domingo, S.; Urioste, M.; Osorio, A.; Benitez, J. Genetic anticipation is associated with telomere shortening in hereditary breast cancer. PLoS Genet. 2011, 7, e1002182. [Google Scholar] [CrossRef] [Green Version]
- Benitez-Buelga, C.; Sanchez-Barroso, L.; Gallardo, M.; Apellániz-Ruiz, M.; Inglada-Perez, L.; Yanowski, K.; Carrillo, J.; Garcia-Estevez, L.; Calvo, I.; Perona, R. Impact of chemotherapy on telomere length in sporadic and familial breast cancer patients. Breast Cancer Res. Treat. 2015, 149, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Weischer, M.; Bojesen, S.E.; Nordestgaard, B.G. Telomere shortening unrelated to smoking, body weight, physical activity, and alcohol intake: 4576 general population individuals with repeat measurements 10 years apart. PLoS Genet. 2014, 10, e1004191. [Google Scholar] [CrossRef]
- Farzaneh-Far, R.; Lin, J.; Epel, E.; Lapham, K.; Blackburn, E.; Whooley, M.A. Telomere length trajectory and its determinants in persons with coronary artery disease: Longitudinal findings from the heart and soul study. PLoS ONE 2010, 5, e8612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bischoff, C.; Petersen, H.C.; Graakjaer, J.; Andersen-Ranberg, K.; Vaupel, J.W.; Bohr, V.A.; Kølvraa, S.; Christensen, K. No association between telomere length and survival among the elderly and oldest old. Epidemiology 2006, 17, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Svensson, J.; Karlsson, M.K.; Ljunggren, Ö.; Tivesten, Å.; Mellström, D.; Movérare-Skrtic, S. Leukocyte telomere length is not associated with mortality in older men. Exp. Gerontol. 2014, 57, 6–12. [Google Scholar] [CrossRef]
- Brouilette, S.W.; Moore, J.S.; McMahon, A.D.; Thompson, J.R.; Ford, I.; Shepherd, J.; Packard, C.J.; Samani, N.J. Telomere length, risk of coronary heart disease, and statin treatment in the west of scotland primary prevention study: A nested case-control study. Lancet 2007, 369, 107–114. [Google Scholar] [CrossRef]
- Van Der Harst, P.; van der Steege, G.; de Boer, R.A.; Voors, A.A.; Hall, A.S.; Mulder, M.J.; van Gilst, W.H.; van Veldhuisen, D.J.; Group, M.-H.S. Telomere length of circulating leukocytes is decreased in patients with chronic heart failure. J. Am. Coll. Cardiol. 2007, 49, 1459–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Zhu, Z.; Zhou, L.; Yang, M. The joint effects of frailty and telomere length for predicting mortality in older adults: The national health and nutrition examination survey 1999–2002. Aging Clin. Exp. Res. 2019, 1–9. [Google Scholar] [CrossRef]
- Margaritis, M.; Sanna, F.; Lazaros, G.; Akoumianakis, I.; Patel, S.; Antonopoulos, A.S.; Duke, C.; Herdman, L.; Psarros, C.; Oikonomou, E.K. Predictive value of telomere length on outcome following acute myocardial infarction: Evidence for contrasting effects of vascular vs. blood oxidative stress. Eur. Heart J. 2017, 38, 3094–3104. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhan, Y.; Pedersen, N.L.; Fang, F.; Hägg, S. Telomere length and all-cause mortality: A meta-analysis. Ageing Res. Rev. 2018, 48, 11–20. [Google Scholar] [CrossRef]
- Martínez, P.; Blasco, M.A. Heart-breaking telomeres. Circ. Res. 2018, 123, 787–802. [Google Scholar] [CrossRef]
- Calado, R.T.; Yewdell, W.T.; Wilkerson, K.L.; Regal, J.A.; Kajigaya, S.; Stratakis, C.A.; Young, N.S. Sex hormones, acting on the TERT gene, increase telomerase activity in human primary hematopoietic cells. Blood 2009, 114, 2236–2243. [Google Scholar] [CrossRef]
- Bär, C.; Huber, N.; Beier, F.; Blasco, M.A. Therapeutic effect of androgen therapy in a mouse model of aplastic anemia produced by short telomeres. Haematologica 2015, 100, 1267–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townsley, D.M.; Dumitriu, B.; Liu, D.; Biancotto, A.; Weinstein, B.; Chen, C.; Hardy, N.; Mihalek, A.D.; Lingala, S.; Kim, Y.J. Danazol treatment for telomere diseases. N. Engl. J. Med. 2016, 374, 1922–1931. [Google Scholar] [CrossRef] [PubMed]
- Bär, C.; Blasco, M.A. Telomeres and telomerase as therapeutic targets to prevent and treat age-related diseases. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bär, C.; Povedano, J.M.; Serrano, R.; Benitez-Buelga, C.; Popkes, M.; Formentini, I.; Bobadilla, M.; Bosch, F.; Blasco, M.A. Telomerase gene therapy rescues telomere length, bone marrow aplasia, and survival in mice with aplastic anemia. Blood J. Am. Soc. Hematol. 2016, 127, 1770–1779. [Google Scholar] [CrossRef] [Green Version]
- Povedano, J.M.; Martinez, P.; Serrano, R.; Tejera, Á.; Gómez-López, G.; Bobadilla, M.; Flores, J.M.; Bosch, F.; Blasco, M.A. Therapeutic effects of telomerase in mice with pulmonary fibrosis induced by damage to the lungs and short telomeres. Elife 2018, 7, e31299. [Google Scholar] [CrossRef]
- Martínez, P.; Blasco, M.A. Telomere-driven diseases and telomere-targeting therapies. J. Cell Biol. 2017, 216, 875–887. [Google Scholar] [CrossRef]
- González-Suárez, E.; Samper, E.; Ramírez, A.; Flores, J.M.; Martín-Caballero, J.; Jorcano, J.L.; Blasco, M.A. Increased epidermal tumors and increased skin wound healing in transgenic mice overexpressing the catalytic subunit of telomerase, mTERT, in basal keratinocytes. EMBO J. 2001, 20, 2619–2630. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Lorente, M.A.; Martínez, P.; Tejera, Á.; Whittemore, K.; Moisés-Silva, A.C.; Bosch, F.; Blasco, M.A. AAV9-mediated telomerase activation does not accelerate tumorigenesis in the context of oncogenic K-Ras-induced lung cancer. PLoS Genet. 2018, 14, e1007562. [Google Scholar] [CrossRef]
- Bernardes de Jesus, B.; Schneeberger, K.; Vera, E.; Tejera, A.; Harley, C.B.; Blasco, M.A. The telomerase activator TA-65 elongates short telomeres and increases health span of adult/old mice without increasing cancer incidence. Aging Cell 2011, 10, 604–621. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Lin, J.; Matsuguchi, T.; Blackburn, E.; Yeh, F.; Best, L.G.; Devereux, R.B.; Lee, E.T.; Howard, B.V.; Roman, M.J.; et al. Short leukocyte telomere length predicts incidence and progression of carotid atherosclerosis in American Indians: The strong heart family study. Aging 2014, 6, 414–427. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, C.J.; Demissie, S.; Kimura, M.; Levy, D.; Gardner, J.P.; White, C.; D’Agostino, R.B.; Wolf, P.A.; Polak, J.; Cupples, L.A.; et al. Leukocyte telomere length and carotid artery intimal medial thickness. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1165–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Meyer, T.; Rietzschel, E.R.; De Buyzere, M.L.; Langlois, M.R.; De Bacquer, D.; Segers, P.; Van Damme, P.; De Backer, G.G.; Van Oostveldt, P.; Van Criekinge, W.; et al. Systemic telomere length and preclinical atherosclerosis: The Asklepios study. Eur. Heart J. 2009, 30, 3074–3081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvert, P.A.; Liew, T.V.; Gorenne, I.; Clarke, M.; Costopoulos, C.; Obaid, D.R.; O’Sullivan, M.; Shapiro, L.M.; McNab, D.C.; Densem, C.G.; et al. Leukocyte telomere length is associated with high-risk plaques on virtual histology intravascular ultrasound and increased proinflammatory activity. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2157–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huzen, J.; Peeters, W.; Boer, R.A.d.; Moll, F.L.; Wong, L.S.M.; Codd, V.; Kleijn, D.P.V.d.; Smet, B.J.G.L.d.; Veldhuisen, D.J.v.; Samani, N.J.; et al. Circulating leukocyte and carotid atherosclerotic plaque telomere length. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1219–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panayiotou, A.G.; Nicolaides, A.N.; Griffin, M.; Tyllis, T.; Georgiou, N.; Bond, D.; Martin, R.M.; Hoppensteadt, D.; Fareed, J.; Humphries, S.E. Leukocyte telomere length is associated with measures of subclinical atherosclerosis. Atherosclerosis 2010, 211, 176–181. [Google Scholar] [CrossRef]
- Benetos, A.; Gardner, J.P.; Zureik, M.; Labat, C.; Xiaobin, L.; Adamopoulos, C.; Temmar, M.; Bean, K.E.; Thomas, F.; Aviv, A. Short telomeres are associated with increased carotid atherosclerosis in hypertensive subjects. Hypertension 2004, 43, 182–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mainous, A.G., 3rd; Codd, V.; Diaz, V.A.; Schoepf, U.J.; Everett, C.J.; Player, M.S.; Samani, N.J. Leukocyte telomere length and coronary artery calcification. Atherosclerosis 2010, 210, 262–267. [Google Scholar] [CrossRef]
- Fernández-Alvira, J.M.; Fuster, V.; Dorado, B.; Soberón, N.; Flores, I.; Gallardo, M.; Pocock, S.; Blasco, M.A.; Andrés, V. Short telomere load, telomere length, and subclinical atherosclerosis: The PESA study. J. Am. Coll. Cardiol. 2016, 67, 2467–2476. [Google Scholar] [CrossRef] [Green Version]
- Farzaneh-Far, R.; Cawthon, R.M.; Na, B.; Browner, W.S.; Schiller, N.B.; Whooley, M.A. Prognostic value of leukocyte telomere length in patients with stable coronary artery disease: Data from the heart and soul study. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1379–1384. [Google Scholar] [CrossRef]
- Astrup, A.S.; Tarnow, L.; Jorsal, A.; Lajer, M.; Nzietchueng, R.; Benetos, A.; Rossing, P.; Parving, H.H. Telomere length predicts all-cause mortality in patients with type 1 diabetes. Diabetologia 2010, 53, 45–48. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, L.L.; Fu, R.; Rogers, K.; Freeman, M.; Helfand, M. Homocysteine level and coronary heart disease incidence: A systematic review and meta-analysis. Mayo Clin. Proc. 2008, 83, 1203–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, J.B.; Valdes, A.M.; Gardner, J.P.; Kato, B.S.; Siva, A.; Kimura, M.; Lu, X.; Brown, M.J.; Aviv, A.; Spector, T.D. Homocysteine levels and leukocyte telomere length. Atherosclerosis 2008, 200, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Prescott, J.; Giovannucci, E.; Hankinson, S.E.; Rosner, B.; De Vivo, I. One-carbon metabolism factors and leukocyte telomere length. Am. J. Clin. Nutr. 2013, 97, 794–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masi, S.; D’Aiuto, F.; Cooper, J.; Salpea, K.; Stephens, J.W.; Hurel, S.J.; Deanfield, J.E.; Humphries, S.E. Telomere length, antioxidant status and incidence of ischaemic heart disease in type 2 diabetes. Int. J. Cardiol. 2016, 216, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Steer, S.E.; Williams, F.M.; Kato, B.; Gardner, J.P.; Norman, P.J.; Hall, M.A.; Kimura, M.; Vaughan, R.; Aviv, A.; Spector, T.D. Reduced telomere length in rheumatoid arthritis is independent of disease activity and duration. Ann. Rheum. Dis 2007, 66, 476–480. [Google Scholar] [CrossRef] [Green Version]
- Pusceddu, I.; Herrmann, W.; Kleber, M.E.; Scharnagl, H.; Hoffmann, M.M.; Winklhofer-Roob, B.M.; März, W.; Herrmann, M. Subclinical inflammation, telomere shortening, homocysteine, vitamin B6, and mortality: The ludwigshafen risk and cardiovascular health study. Eur. J. Nutr. 2020, 59, 1399–1411. [Google Scholar] [CrossRef] [Green Version]
- O’Donovan, A.; Pantell, M.S.; Puterman, E.; Dhabhar, F.S.; Blackburn, E.H.; Yaffe, K.; Cawthon, R.M.; Opresko, P.L.; Hsueh, W.C.; Satterfield, S.; et al. Cumulative inflammatory load is associated with short leukocyte telomere length in the health, aging and body composition study. PLoS ONE 2011, 6, e19687. [Google Scholar] [CrossRef] [Green Version]
- Werner, C.M.; Furster, T.; Widmann, T.; Poss, J.; Roggia, C.; Hanhoun, M.; Scharhag, J.; Buchner, N.; Meyer, T.; Kindermann, W.; et al. Physical exercise prevents cellular senescence in circulating leukocytes and in the vessel wall. Circulation 2009, 120, 2438–2447. [Google Scholar] [CrossRef]
- Werner, C.; Hanhoun, M.; Widmann, T.; Kazakov, A.; Semenov, A.; Pöss, J.; Bauersachs, J.; Thum, T.; Pfreundschuh, M.; Müller, P.; et al. Effects of physical exercise on myocardial telomere-regulating proteins, survival pathways, and apoptosis. J. Am. Coll. Cardiol. 2008, 52, 470. [Google Scholar] [CrossRef] [Green Version]
- Ludlow, A.T.; Witkowski, S.; Marshall, M.R.; Wang, J.; Lima, L.C.; Guth, L.M.; Spangenburg, E.E.; Roth, S.M. Chronic exercise modifies age-related telomere dynamics in a tissue-specific fashion. J. Gerontol. Biol. Sci. Med. Sci. 2012, 67, 911–926. [Google Scholar] [CrossRef] [Green Version]
- Ludlow, A.T.; Lima, L.C.J.; Wang, J.; Hanson, E.D.; Guth, L.M.; Spangenburg, E.E.; Roth, S.M. Exercise alters mRNA expression of telomere-repeat binding factor 1 in skeletal muscle via p38 MAPK. J. Appl. Physiol. 2012, 113, 1737–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomás-Loba, A.; Flores, I.; Fernández-Marcos, P.J.; Cayuela, M.L.; Maraver, A.; Tejera, A.; Borrás, C.; Matheu, A.; Klatt, P.; Flores, J.M.; et al. Telomerase reverse transcriptase delays aging in cancer-resistant mice. Cell 2008, 135, 609–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardes de Jesus, B.; Blasco, M.A. Aging by telomere loss can be reversed. Cell Stem Cell 2011, 8, 3–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaskelioff, M.; Muller, F.L.; Paik, J.-H.; Thomas, E.; Jiang, S.; Adams, A.C.; Sahin, E.; Kost-Alimova, M.; Protopopov, A.; Cadiñanos, J.; et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 2011, 469, 102–106. [Google Scholar] [CrossRef] [Green Version]
- Ludlow, A.T.; Gratidao, L.; Ludlow, L.W.; Spangenburg, E.E.; Roth, S.M. Acute exercise activates p38 MAPK and increases the expression of telomere-protective genes in cardiac muscle. Exp. Physiol. 2017, 102, 397–410. [Google Scholar] [CrossRef] [Green Version]
- Tie, G.; Messina, K.E.; Yan, J.; Messina, J.A.; Messina, L.M. Hypercholesterolemia induces oxidant stress that accelerates the ageing of hematopoietic stem cells. J. Am. Heart Assoc. 2014, 3, e000241. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Uryga, A.K.; Reinhold, J.; Figg, N.; Baker, L.; Finigan, A.; Gray, K.; Kumar, S.; Clarke, M.; Bennett, M. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation 2015, 132, 1909–1919. [Google Scholar] [CrossRef]
- Fan, R.; Zhang, A.; Zhong, F. Association between homocysteine levels and all-cause mortality: A dose-response meta-analysis of prospective studies. Sci. Rep. 2017, 7, 4769. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Huang, Y.; Hu, Y.; Zhong, J.; He, Z.; Li, W.; Yang, Y.; Xu, D.; Wu, S. Hyperhomocysteinemia is an independent risk factor in young patients with coronary artery disease in southern China. Herz 2013, 38, 779–784. [Google Scholar] [CrossRef]
- Alam, N.; Khan, H.I.; Chowdhury, A.W.; Haque, M.S.; Ali, M.S.; Sabah, K.M.; Amin, M.G. Elevated serum homocysteine level has a positive correlation with serum cardiac troponin I in patients with acute myocardial infarction. Bangladesh Med. Res. Counc. Bull. 2012, 38, 9–13. [Google Scholar] [CrossRef]
- Wei, M.; Wang, L.; Liu, Y.-S.; Zheng, M.-Q.; Ma, F.-F.; Qi, Y.-C.; Liu, G. Homocysteine as a potential predictive factor for high major adverse cardiovascular events risk in female patients with premature acute coronary syndrome. Medicine 2019, 98, e18019. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Li, L.; Shang, X.-M.; Tan, Z.; Geng, X.-B.; Zhao, B.-Q.; Tian, M.-R. Analysis of factors related to short-term prognosis in patients undergoing percutaneous coronary intervention for acute myocardial infarction. Exp. Ther. Med. 2013, 5, 1206–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.; Qian, G.; Xue, H.; Guo, J.; Chen, L.; Yang, X.; Shen, M.; Dong, W.; Chen, Y. Hyperhomocysteinemia is an independent predictor of long-term clinical outcomes in Chinese octogenarians with acute coronary syndrome. Clin. Interv. Aging 2015, 10, 1467–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganguly, P.; Alam, S.F. Role of homocysteine in the development of cardiovascular disease. Nutr. J. 2015, 14, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marković Boras, M.; Čaušević, A.; Brizić, I.; Mikulić, I.; Vasilj, M.; Jelić Knezović, N. A relation of serum homocysteine, uric acid and C-reactive protein level in patients with acute myocardial infarction. Med. Glas. 2018, 15, 101–108. [Google Scholar] [CrossRef]
- Chambers, J.C.; McGregor, A.; Jean-Marie, J.; Obeid, O.A.; Kooner, J.S. Demonstration of rapid onset vascular endothelial dysfunction after hyperhomocysteinemia: An effect reversible with vitamin C therapy. Circulation 1999, 99, 1156–1160. [Google Scholar] [CrossRef] [Green Version]
- Eberhardt, R.T.; Forgione, M.A.; Cap, A.; Leopold, J.A.; Rudd, M.A.; Trolliet, M.; Heydrick, S.; Stark, R.; Klings, E.S.; Moldovan, N.I.; et al. Endothelial dysfunction in a murine model of mild hyperhomocyst(e)inemia. J. Clin. Invest. 2000, 106, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Weiss, N.; Heydrick, S.; Zhang, Y.Y.; Bierl, C.; Cap, A.; Loscalzo, J. Cellular redox state and endothelial dysfunction in mildly hyperhomocysteinemic cystathionine beta-synthase-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 34–41. [Google Scholar] [CrossRef]
- Topal, G.; Brunet, A.; Millanvoye, E.; Boucher, J.L.; Rendu, F.; Devynck, M.A.; David-Dufilho, M. Homocysteine induces oxidative stress by uncoupling of no synthase activity through reduction of tetrahydrobiopterin. Free Radic. Biol. Med. 2004, 36, 1532–1541. [Google Scholar] [CrossRef]
- Papatheodorou, L.; Weiss, N. Vascular oxidant stress and inflammation in hyperhomocysteinemia. Antioxid. Redox Signal. 2007, 9, 1941–1958. [Google Scholar] [CrossRef]
- Kim, C.S.; Kim, Y.R.; Naqvi, A.; Kumar, S.; Hoffman, T.A.; Jung, S.B.; Kumar, A.; Jeon, B.H.; McNamara, D.M.; Irani, K. Homocysteine promotes human endothelial cell dysfunction via site-specific epigenetic regulation of p66shc. Cardiovasc. Res. 2011, 92, 466–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Cui, L.; Joseph, J.; Jiang, B.; Pimental, D.; Handy, D.E.; Liao, R.; Loscalzo, J. Homocysteine induces cardiomyocyte dysfunction and apoptosis through p38 MAPK-mediated increase in oxidant stress. J. Mol. Cell Cardiol. 2012, 52, 753–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, P.N.; Liang, Y.R.; Lin, W.B.; Yao, Z.R.; Chen, D.B.; Chen, P.S.; Ouyang, J. Homocysteine induced oxidative stress in human umbilical vein endothelial cells via regulating methylation of SORBS1. Eur. Rev. Med. Pharm. Sci. 2018, 22, 6948–6958. [Google Scholar] [CrossRef]
- Rane, G.; Koh, W.P.; Kanchi, M.M.; Wang, R.; Yuan, J.M.; Wang, X. Association between leukocyte telomere length and plasma homocysteine in a singapore chinese population. Rejuvenation Res. 2015, 18, 203–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, C.; Baik, I. Leukocyte telomere length is associated with serum vitamin B12 and homocysteine levels in older adults with the presence of systemic inflammation. Clin. Nutr. Res. 2016, 5, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Milne, E.; O’Callaghan, N.; Ramankutty, P.; de Klerk, N.H.; Greenop, K.R.; Armstrong, B.K.; Miller, M.; Fenech, M. Plasma micronutrient levels and telomere length in children. Nutrition 2015, 31, 331–336. [Google Scholar] [CrossRef] [Green Version]
- Nomura, S.J.; Robien, K.; Zota, A.R. Serum folate, vitamin B-12, vitamin A, γ-tocopherol, α-tocopherol, and carotenoids do not modify associations between cadmium exposure and leukocyte telomere length in the general US adult population. J. Nutr. 2017, 147, 538–548. [Google Scholar] [CrossRef] [Green Version]
- Stanger, O.; Herrmann, W.; Pietrzik, K.; Fowler, B.; Geisel, J.; Dierkes, J.; Weger, M. DACH-LIGA homocystein (german, austrian and swiss homocysteine society): Consensus paper on the rational clinical use of homocysteine, folic acid and B-vitamins in cardiovascular and thrombotic diseases: Guidelines and recommendations. Clin. Chem. Lab. Med. 2003, 41, 1392–1403. [Google Scholar] [CrossRef]
- Stabler, S.P. Clinical practice. Vitamin B12 deficiency. N. Engl. J. Med. 2013, 368, 149–160. [Google Scholar] [CrossRef]
- Herrmann, W.; Obeid, R. Utility and limitations of biochemical markers of vitamin B12 deficiency. Eur. J. Clin. Invest. 2013, 43, 231–237. [Google Scholar] [CrossRef]
- Herrmann, W.; Obeid, R. Vitamins in the Prevention of Human Diseases, 1st ed.; De Gruyter: Berlin, Germany, 2011. [Google Scholar]
- Herrmann, W.; Obeid, R. Hyperhomocysteinemia and response of methionine cycle intermediates to vitamin treatment in renal patients. Clin. Chem. Lab. Med. 2005, 43, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Friso, S.; Udali, S.; De Santis, D.; Choi, S.W. One-carbon metabolism and epigenetics. Mol. Asp. Med. 2017, 54, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Mendioroz, M.; Puebla-Guedea, M.; Montero-Marín, J.; Urdánoz-Casado, A.; Blanco-Luquin, I.; Roldán, M.; Labarga, A.; García-Campayo, J. Telomere length correlates with subtelomeric DNA methylation in long-term mindfulness practitioners. Sci. Rep. 2020, 10, 4564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novakovic, B.; Napier, C.E.; Vryer, R.; Dimitriadis, E.; Manuelpillai, U.; Sharkey, A.; Craig, J.M.; Reddel, R.R.; Saffery, R. DNA methylation mediated up-regulation of TERRA non-coding RNA is coincident with elongated telomeres in the human placenta. Mol. Hum. Reprod. 2016, 22, 791–799. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Li, B.; Duan, S. The alteration of subtelomeric DNA methylation in aging-related diseases. Front. Genet. 2018, 9, 697. [Google Scholar] [CrossRef] [Green Version]
- Sibrian-Vazquez, M.; Escobedo, J.O.; Lim, S.; Samoei, G.K.; Strongin, R.M. Homocystamides promote free-radical and oxidative damage to proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 551–554. [Google Scholar] [CrossRef] [Green Version]
- Pusceddu, I.; Herrmann, W.; Kleber, M.E.; Scharnagl, H.; März, W.; Herrmann, M. Telomere length, vitamin B12 and mortality in persons undergoing coronary angiography: The ludwigshafen risk and cardiovascular health study. Aging 2019, 11, 7083–7097. [Google Scholar] [CrossRef]
- Chiriacò, M.; Georgiopoulos, G.; Duranti, E.; Antonioli, L.; Puxeddu, I.; Nannipieri, M.; Rosada, J.; Blandizzi, C.; Taddei, S.; Virdis, A.; et al. Inflammation and vascular ageing: From telomeres to novel emerging mechanisms. High. Blood Press. Cardiovasc. Prev. 2019, 26, 321–329. [Google Scholar] [CrossRef]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Flores, I.; Canela, A.; Vera, E.; Tejera, A.; Cotsarelis, G.; Blasco, M.A. The longest telomeres: A general signature of adult stem cell compartments. Genes Dev. 2008, 22, 654–667. [Google Scholar] [CrossRef] [Green Version]
- Yui, J.; Chiu, C.P.; Lansdorp, P.M. Telomerase activity in candidate stem cells from fetal liver and adult bone marrow. Blood 1998, 91, 3255–3262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Zglinicki, T. Role of oxidative stress in telomere length regulation and replicative senescence. Ann. N. Y. Acad. Sci. 2000, 908, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Bekaert, S.; De Meyer, T.; Rietzschel, E.R.; De Buyzere, M.L.; De Bacquer, D.; Langlois, M.; Segers, P.; Cooman, L.; Van Damme, P.; Cassiman, P.; et al. Telomere length and cardiovascular risk factors in a middle-aged population free of overt cardiovascular disease. Aging Cell 2007, 6, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Fouquerel, E.; Lormand, J.; Bose, A.; Lee, H.T.; Kim, G.S.; Li, J.; Sobol, R.W.; Freudenthal, B.D.; Myong, S.; Opresko, P.L. Oxidative guanine base damage regulates human telomerase activity. Nat. Struct. Mol. Biol. 2016, 23, 1092–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindhardsen, J.; Ahlehoff, O.; Gislason, G.H.; Madsen, O.R.; Olesen, J.B.; Torp-Pedersen, C.; Hansen, P.R. The risk of myocardial infarction in rheumatoid arthritis and diabetes mellitus: A Danish nationwide cohort study. Ann. Rheum. Dis. 2011, 70, 929–934. [Google Scholar] [CrossRef] [Green Version]
- van Halm, V.P.; Peters, M.J.; Voskuyl, A.E.; Boers, M.; Lems, W.F.; Visser, M.; Stehouwer, C.D.; Spijkerman, A.M.; Dekker, J.M.; Nijpels, G.; et al. Rheumatoid arthritis versus diabetes as a risk factor for cardiovascular disease: A cross-sectional study, the CARRE investigation. Ann. Rheum. Dis. 2009, 68, 1395–1400. [Google Scholar] [CrossRef] [Green Version]
- Gamal, R.M.; Hammam, N.; Zakary, M.M.; Abdelaziz, M.M.; Razek, M.R.A.; Mohamed, M.S.E.; Emad, Y.; Elnaggar, M.G.; Furst, D.E. Telomere dysfunction-related serological markers and oxidative stress markers in rheumatoid arthritis patients: Correlation with diseases activity. Clin. Rheumatol. 2018, 37, 3239–3246. [Google Scholar] [CrossRef]
- Sampson, M.J.; Winterbone, M.S.; Hughes, J.C.; Dozio, N.; Hughes, D.A. Monocyte telomere shortening and oxidative DNA damage in type 2 diabetes. Diabetes Care 2006, 29, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Masi, S.; Gkranias, N.; Li, K.; Salpea, K.D.; Parkar, M.; Orlandi, M.; Suvan, J.E.; Eng, H.L.; Taddei, S.; Patel, K.; et al. Association between short leukocyte telomere length, endotoxemia, and severe periodontitis in people with diabetes: A cross-sectional survey. Diabetes Care 2014, 37, 1140–1147. [Google Scholar] [CrossRef] [Green Version]
- García-Redondo, A.B.; Aguado, A.; Briones, A.M.; Salaices, M. NADPH oxidases and vascular remodeling in cardiovascular diseases. Pharm. Res. 2016, 114, 110–120. [Google Scholar] [CrossRef]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH oxidases in vascular pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [Green Version]
- Sorescu, D.; Weiss, D.; Lassègue, B.; Clempus, R.E.; Szöcs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar] [CrossRef] [Green Version]
- Kalinina, N.; Agrotis, A.; Tararak, E.; Antropova, Y.; Kanellakis, P.; Ilyinskaya, O.; Quinn, M.T.; Smirnov, V.; Bobik, A. Cytochrome b558-dependent NAD(P)H oxidase-phox units in smooth muscle and macrophages of atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 2037–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madrigal-Matute, J.; Fernandez-Laso, V.; Sastre, C.; Llamas-Granda, P.; Egido, J.; Martin-Ventura, J.L.; Zalba, G.; Blanco-Colio, L.M. TWEAK/Fn14 interaction promotes oxidative stress through NADPH oxidase activation in macrophages. Cardiovasc Res. 2015, 108, 139–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pejenaute, Á.; Cortés, A.; Marqués, J.; Montero, L.; Beloqui, Ó.; Fortuño, A.; Martí, A.; Orbe, J.; Zalba, G. NADPH oxidase overactivity underlies telomere shortening in human atherosclerosis. Int. J. Mol. Sci. 2020, 21, 1434. [Google Scholar] [CrossRef] [Green Version]
- Billard, P.; Poncet, D.A. Replication stress at telomeric and mitochondrial DNA: Common origins and consequences on ageing. Int. J. Mol. Sci. 2019, 20, 4959. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Role of metabolic H2O2 generation: Redox signaling and oxidative stress. J. Biol. Chem. 2014, 289, 8735–8741. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, S.L.; Simon, A.K. In aged primary T cells, mitochondrial stress contributes to telomere attrition measured by a novel imaging flow cytometry assay. Aging Cell 2017, 16, 1234–1243. [Google Scholar] [CrossRef]
- Liu, L.; Trimarchi, J.R.; Smith, P.J.; Keefe, D.L. Mitochondrial dysfunction leads to telomere attrition and genomic instability. Aging Cell 2002, 1, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [Green Version]
- Kolling, J.; Scherer, E.B.; da Cunha, A.A.; da Cunha, M.J.; Wyse, A.T. Homocysteine induces oxidative-nitrative stress in heart of rats: Prevention by folic acid. Cardiovasc. Toxicol. 2011, 11, 67–73. [Google Scholar] [CrossRef]
- Matté, C.; Mackedanz, V.; Stefanello, F.M.; Scherer, E.B.; Andreazza, A.C.; Zanotto, C.; Moro, A.M.; Garcia, S.C.; Gonçalves, C.A.; Erdtmann, B.; et al. Chronic hyperhomocysteinemia alters antioxidant defenses and increases DNA damage in brain and blood of rats: Protective effect of folic acid. Neurochem. Int. 2009, 54, 7–13. [Google Scholar] [CrossRef]
- Li, J.J.; Li, Q.; Du, H.P.; Wang, Y.L.; You, S.J.; Wang, F.; Xu, X.S.; Cheng, J.; Cao, Y.J.; Liu, C.F.; et al. Homocysteine triggers inflammatory responses in macrophages through inhibiting CSE-H2S signaling via DNA hypermethylation of CSE promoter. Int. J. Mol. Sci. 2015, 16, 12560–12577. [Google Scholar] [CrossRef]
- Steele, M.L.; Fuller, S.; Maczurek, A.E.; Kersaitis, C.; Ooi, L.; Münch, G. Chronic inflammation alters production and release of glutathione and related thiols in human U373 astroglial cells. Cell Mol. Neurobiol. 2013, 33, 19–30. [Google Scholar] [CrossRef]
- Ulvik, A.; Midttun, Ø.; Pedersen, E.R.; Eussen, S.J.; Nygård, O.; Ueland, P.M. Evidence for increased catabolism of vitamin B-6 during systemic inflammation. Am. J. Clin. Nutr. 2014, 100, 250–255. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.Y.; De Vivo, I.; Lin, X.; Fang, S.C.; Christiani, D.C. The relationship between inflammatory biomarkers and telomere length in an occupational prospective cohort study. PLoS ONE 2014, 9, e87348. [Google Scholar] [CrossRef] [Green Version]
- Tan, R.; Lan, L. Guarding chromosomes from oxidative DNA damage to the very end. Acta Biochim. Biophys. Sin. 2016, 48, 617–622. [Google Scholar] [CrossRef] [Green Version]
- Opresko, P.L.; Fan, J.; Danzy, S.; Wilson, D.M., 3rd; Bohr, V.A. Oxidative damage in telomeric DNA disrupts recognition by TRF1 and TRF2. Nucleic Acids Res. 2005, 33, 1230–1239. [Google Scholar] [CrossRef]
- Lin, N.; Qin, S.; Luo, S.; Cui, S.; Huang, G.; Zhang, X. Homocysteine induces cytotoxicity and proliferation inhibition in neural stem cells via DNA methylation in vitro. FEBS J. 2014, 281, 2088–2096. [Google Scholar] [CrossRef]
- Zhang, D.; Sun, X.; Liu, J.; Xie, X.; Cui, W.; Zhu, Y. Homocysteine accelerates senescence of endothelial cells via DNA hypomethylation of human telomerase reverse transcriptase. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Farcas, R.; Schneider, E.; Frauenknecht, K.; Kondova, I.; Bontrop, R.; Bohl, J.; Navarro, B.; Metzler, M.; Zischler, H.; Zechner, U.; et al. Differences in DNA methylation patterns and expression of the CCRK gene in human and nonhuman primate cortices. Mol. Biol Evol. 2009, 26, 1379–1389. [Google Scholar] [CrossRef]
- Svenson, U.; Nordfjäll, K.; Baird, D.; Roger, L.; Osterman, P.; Hellenius, M.L.; Roos, G. Blood cell telomere length is a dynamic feature. PLoS ONE 2011, 6, e21485. [Google Scholar] [CrossRef]
- Gutierrez-Rodrigues, F.; Santana-Lemos, B.A.; Scheucher, P.S.; Alves-Paiva, R.M.; Calado, R.T. Direct comparison of flow-FISH and qPCR as diagnostic tests for telomere length measurement in humans. PLoS ONE 2014, 9, e113747. [Google Scholar] [CrossRef] [Green Version]
- Behrens, Y.L.; Thomay, K.; Hagedorn, M.; Ebersold, J.; Henrich, L.; Nustede, R.; Schlegelberger, B.; Göhring, G. Comparison of different methods for telomere length measurement in whole blood and blood cell subsets: Recommendations for telomere length measurement in hematological diseases. Genes Chromosomes Cancer 2017, 56, 700–708. [Google Scholar] [CrossRef]
- Aviv, A.; Hunt, S.C.; Lin, J.; Cao, X.; Kimura, M.; Blackburn, E. Impartial comparative analysis of measurement of leukocyte telomere length/DNA content by Southern blots and qPCR. Nucleic Acids Res. 2011, 39, e134. [Google Scholar] [CrossRef] [Green Version]
- Lai, T.P.; Wright, W.E.; Shay, J.W. Comparison of telomere length measurement methods. Philos. Trans. R. Soc. 2018, 373. [Google Scholar] [CrossRef] [Green Version]
- Montpetit, A.J.; Alhareeri, A.A.; Montpetit, M.; Starkweather, A.R.; Elmore, L.W.; Filler, K.; Mohanraj, L.; Burton, C.W.; Menzies, V.S.; Lyon, D.E.; et al. Telomere length: A review of methods for measurement. Nurs. Res. 2014, 63, 289–299. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Topic | Reference | Type of Study | Participants | Follow-up | Primary Outcome |
| Telomere length & age | [23] | Longitudinal population-based | 510 males and females mean age at baseline 60 y | 10 y | LTL reduction of 45.5 bp/y |
| [24] | Longitudinal | 334 randomly selected flemish males and females, mean age at baseline 51.9 y | average 7.4 y | Significant LTL reduction, results provided as T/S ratio | |
| [22] | Longitudinal | 271 males and females, Caucasian and African Americans, mean age at baseline 31.9 y | average 12.4 y | LTL reduction of 31 bp/y | |
| [25] | Longitudinal | 75 Dutch men, mean age at baseline 77.6 y | 7 y | LTL reduction of 45.5 bp/y | |
| [26] | Longitudinal | 67 children, mean age at baseline 11.4 y, 99 of their parents, mean age at baseline 43.4 y | 14 y | LTL reduction in children 40.7 bp/y LTL reduction in adults 20.3 bp/y | |
| [20] | meta-analysis of 124 cross-sectional studies | 124 cross-sectional studies, age range 0–104 y, participants (range): 23–12,409 | n/a | weighted mean loss rate: 21.9 bp/y weighted median loss rate: 30.3 bp/y | |
| Telomere length & all-cause mortality | [21] | prospectice cohort study | 3316 patients hospitalized for elective coronary angiography, mean age 62.7 y | median 9.9 y | LTL quartiles 2–4 vs. 1 (shortest telomeres): HR(95% CI) 0.82 (0.71–0.92) |
| [15] | prospectice cohort study | 8633 females from the Nuses Health study, mean age at baseline 59 y, 3566 males and females from the ESTHER study, mean age at baseline 61.9 y | 18.4 y | shortest vs. longest LTL quintile: HR (95% CI) 1.23 (1.04–1.46) | |
| [17] | prospectice cohort study | 64,637 participants from the Copenhagen City Heart Study, Copenhagen General Population Study | median 7 y | shortest vs. the longest decile of LTL: HR (95% CI) 1.40 (1.25–1.57) | |
| [38] | meta-analysis of 25 prospective cohort studies | 12,083 participants, 2517 deaths | n/a | per 1 SD LTL decrement: HR (95% CI) 1.09 (1.06-1.13); shortest vs longest LTL quartile: HR (95% CI) 1.26 (1.15–1.38) | |
| Telomere length & cardiovascular mortality | [14] | populatin-based prospectice cohort study | 800 males and females, mean age at baseline 62.7 y | 10 y | shortest vs. the longest tertile of LTL: HR 3.04 (95% CI: 1.13–8.19) |
| [21] | prospectice cohort study | 3316 patients hospitalized for elective coronary angiography, mean age 62.7 y | median 9.9 y | LTL quartiles 2–4 vs. 1 (shortest telomeres): HR(95% CI) 0.84; (0.72–0.97) | |
| [15] | prospectice cohort study | 8633 females from the Nuses Health study, mean age at baseline 59 y, 3566 males and females from the ESTHER study, mean age at baseline 61.9 y | 18.4 y | shortest vs. longest LTL quintile: HR (95% CI) 1.10 (0.88–1.37) | |
| [17] | prospectice cohort study | 64,637 participants from the Copenhagen City Heart Study, Copenhagen General Population Study | median 7 y | per 200 bp reduction of LTL: HR(95% CI) 1.02 (1.01–1.03) | |
| Telomere length & atherosclerosis | [50] | prospectice cohort study | 2819 participants, were free of overt CVD, mean age at baseline 38.5 y | average 5.5 y | shortes vs. longest LTL tertile: HR(95% CI) for incident plaque 1.49 (1.09–2.03) HR(95% CI) plaque progression 1.61 (1.26–2.07) |
| [14] | populatin-based prospectice cohort study | 800 males and females, mean age at baseline 62.7 y | 10 y | shortest vs. the longest tertile of LTL: HR 3.18 (95% CI: 1.66–6.08) composite CVD end points (stroke, myocardial infarction, vascular death) | |
| [58] | cross-sectional | 1459 participants without CVD at recruitment, age at baseline 40–54 y | n/a | Average LTL and short telomere load are no significant predictors of total and femoral plaques | |
| [57] | cross-sectional | 325 subjects free of diabetes, coronary artery disease, stroke and cancer, age 40–64 years | n/a | Shortest vs. longest tertile of LTL: OR (95% CI) 2.36 (1.23–4.52) for having coronary artery calcification (after adjustment for age, race, gender, metabolic syndrome) | |
| [52] | cross-sectional | 2509 participants withoutestablished CVD, aged approximately 35–55 | n/a | LTL is neither an independent determinant of intima-media-thickness nor plaque presence | |
| Telomere length, HCY and B-vitamins | [61] | meta-analysis of 26 studies | n/a | n/a | estimated RR (95% CI) for coronary heart disease events associated with each 5-μmol/L increase in homocysteine 1.18 (1.10–1.26) |
| [62] | cross-sectional population-based cohort study | 1319 healthy subjects, mean age 49 y | n/a | adjusted LTL difference in the highest and lowest tertile of HCY was 111 base pairs (corresponding to 6.0 years of telomeric aging) | |
| [63] | cross-sectional cohort study | 1715 females | n/a | no sognificant association between LTL, HCY and B-vitamins | |
| Telomere length, oxidative stress and inflammation | [64] | cross-sectional and prospective cohort-study | 489 type 2 diabetics, mean age 67 y | 10 y | at baseline correltation between LTL and total antioxidant staus (r = 0.106, p = 0.024), lower TAOS and shorter LTL at baseline predicted increased risk of incident ischemic heart disease |
| [65] | cross-sectional | 176 patients with rheumatoid arthritis and 1151 controls | n/a | in rheumatoid arthritis patients significantly lower LTL, LTL unrelated to disease duration, CRP or rheumatoid factor | |
| [66] | cross-sectional | 2968 patients hospitalized for elective coronary angiography, mean age 63.5 y | n/a | Subjects with the longest telomeres had lower concentrations of HCY, IL-6, and hs-CRP | |
| [67] | cross-sectional | 1962 adults; age range: 70–79 y | n/a | OR (95% CI) for LTL in the shortest tertile: 1.3 (1.1–1.7) for subjects with IL-6 in top tertile 1.5 (1.2–1.9) for subjects with TNF-a in top tertile 1.8 (1.3–2.4) for subjects IL-6 + TNF-a in top tertile |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrmann, W.; Herrmann, M. The Importance of Telomere Shortening for Atherosclerosis and Mortality. J. Cardiovasc. Dev. Dis. 2020, 7, 29. https://doi.org/10.3390/jcdd7030029
Herrmann W, Herrmann M. The Importance of Telomere Shortening for Atherosclerosis and Mortality. Journal of Cardiovascular Development and Disease. 2020; 7(3):29. https://doi.org/10.3390/jcdd7030029
Chicago/Turabian StyleHerrmann, Wolfgang, and Markus Herrmann. 2020. "The Importance of Telomere Shortening for Atherosclerosis and Mortality" Journal of Cardiovascular Development and Disease 7, no. 3: 29. https://doi.org/10.3390/jcdd7030029
APA StyleHerrmann, W., & Herrmann, M. (2020). The Importance of Telomere Shortening for Atherosclerosis and Mortality. Journal of Cardiovascular Development and Disease, 7(3), 29. https://doi.org/10.3390/jcdd7030029