
Failing Heart Transplants and Rejection—A Cellular Perspective


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Heart Transplantation—No Improvements in Long Term Survival
2. The Normal Cellular Composition of the Heart
2.1. Cardiomyocytes
2.2. Endothelial Cells
2.3. Stromal Cells
2.3.1. Fibroblasts
2.3.2. Other Stromal Cells
2.4. Immune Cells
2.5. Neurons
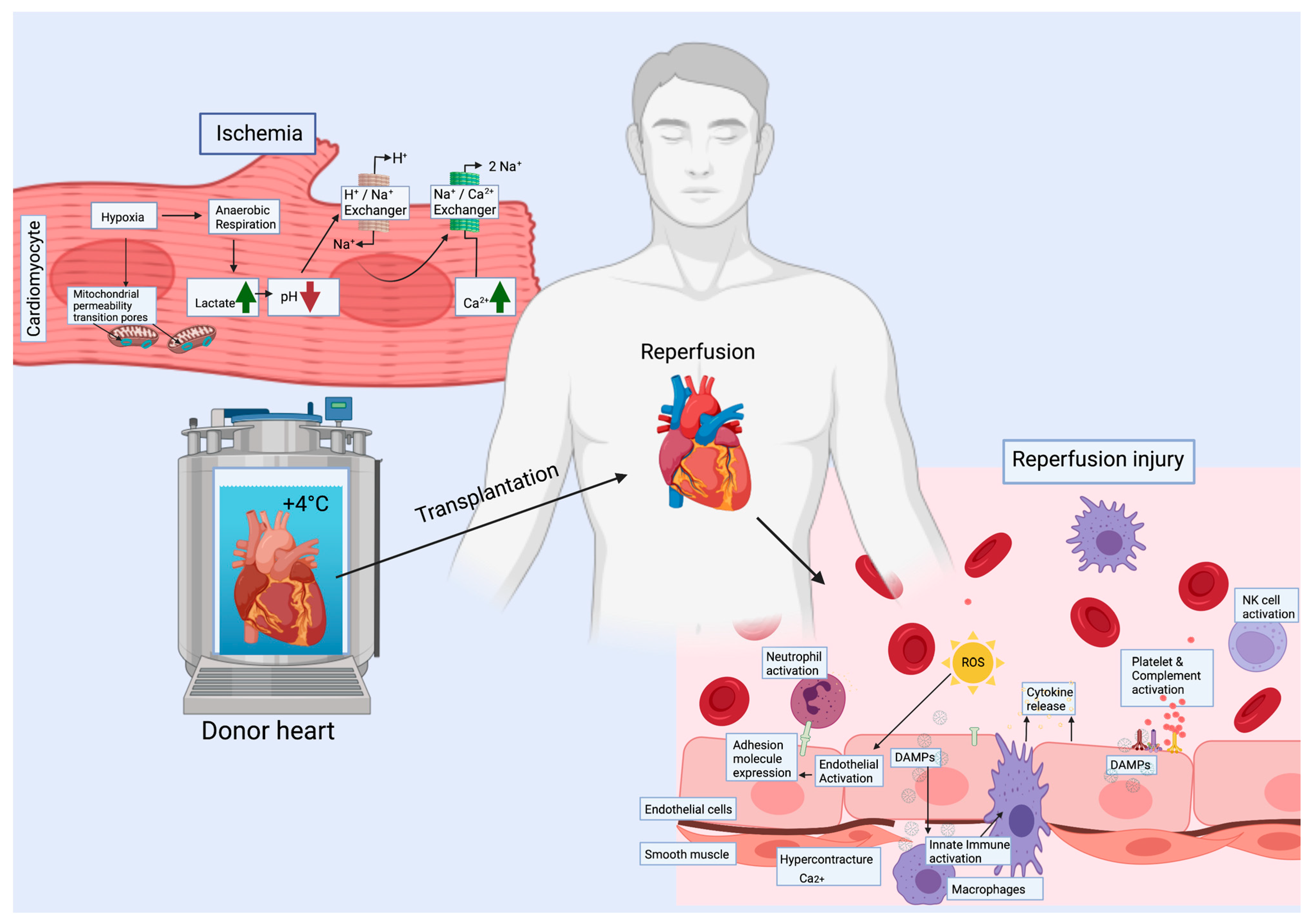
3. Ischemia-Reperfusion Injury—Early Event, Long-Term Consequences
3.1. Cardiomyocytes (CMs)
3.2. Endothelial Cells (ECs)
3.3. Stromal Cells
3.3.1. Fibroblasts and Myofibroblasts
3.3.2. Pericytes
3.4. Immune Cells
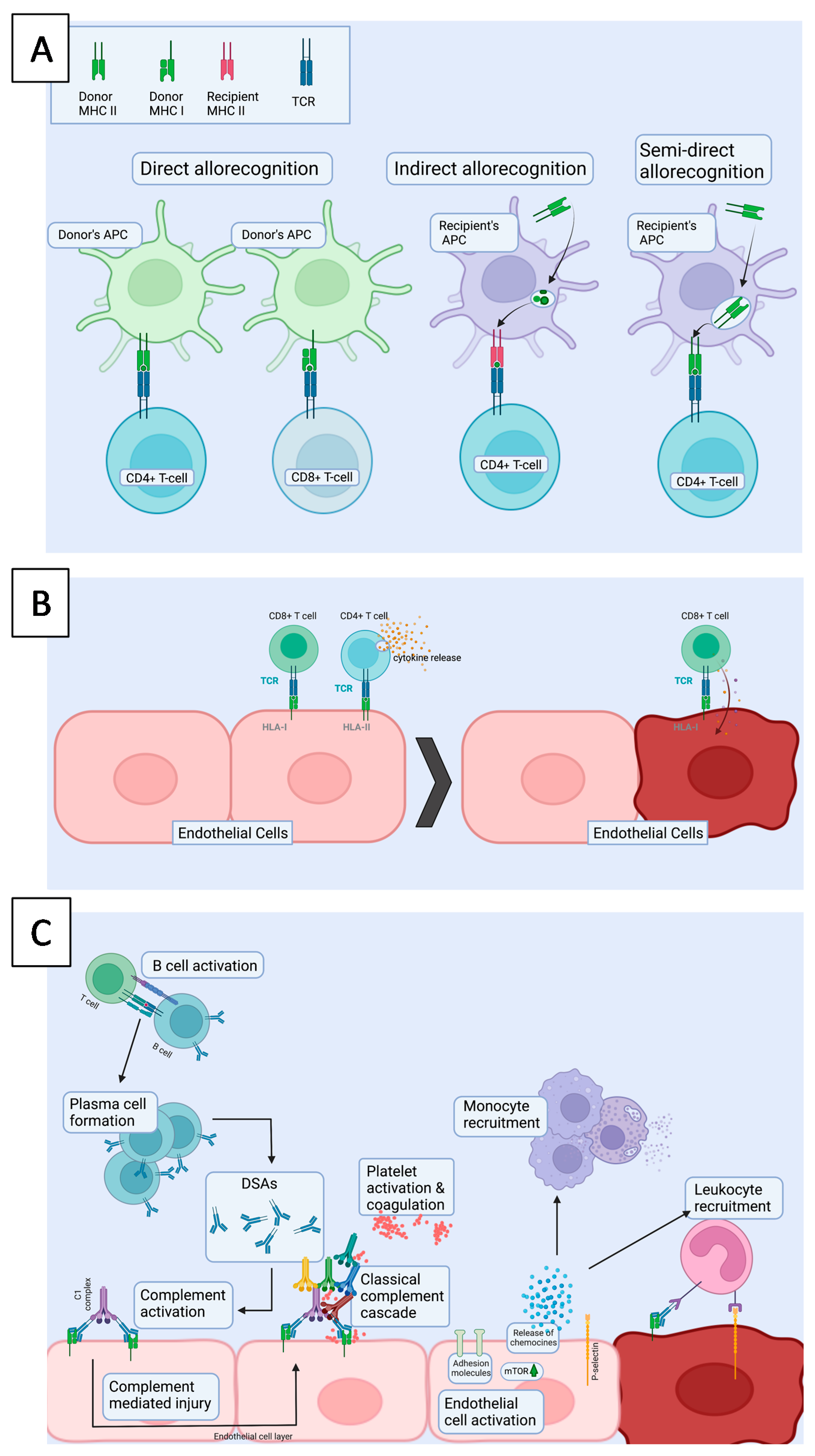
4. Acute Rejection—Better Immunosuppression, Declining Morbidity
4.1. Cellular Rejection
4.2. Antibody-Mediated Rejection
4.2.1. Endothelial Cells
4.2.2. NK Cells
4.2.3. Monocytes and Macrophages
4.2.4. Cardiomyocytes
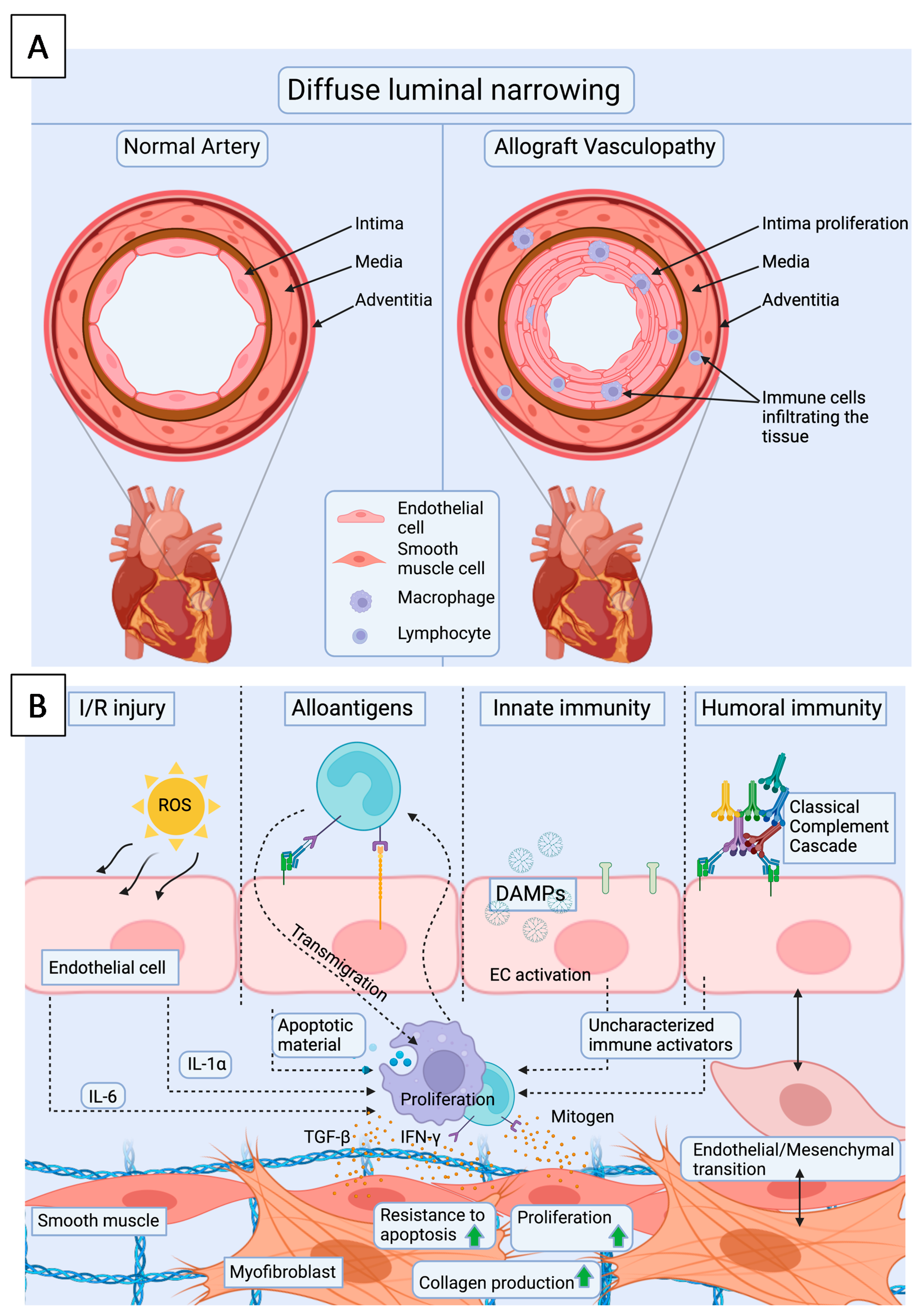
5. Cardiac Allograft Vasculopathy—Endothelium as a Key Player
5.1. Endothelial Cells
5.2. Smooth Muscle Cells
5.3. Immune Cells
5.3.1. T Cells
5.3.2. B Cells
5.3.3. NK Cells
5.3.4. Macrophages
5.3.5. Mast Cells
5.4. Cardiomyocytes
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Sabatino, M.E.; Williams, M.L.; Okwuosa, I.S.; Akhabue, E.; Kim, J.H.; Russo, M.J.; Setoguchi, S. 30-Year Trends in Graft Survival After Heart Transplant: Modeled Analyses of a Transplant Registry. Ann. Thorac. Surg. 2021. [Google Scholar] [CrossRef] [PubMed]
- Khush, K.K.; Cherikh, W.S.; Chambers, D.C.; Harhay, M.O.; Hayes, D.; Hsich, E.; Meiser, B.; Potena, L.; Robinson, A.; Rossano, J.W.; et al. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: Thirty-sixth adult heart transplantation report—2019, focus theme: Donor and recipient size match. J. Hear. Lung Transplant. 2019, 38, 1056–1066. [Google Scholar] [CrossRef]
- Krausgruber, T.; Fortelny, N.; Fife-Gernedl, V.; Senekowitsch, M.; Schuster, L.C.; Lercher, A.; Nemc, A.; Schmidl, C.; Rendeiro, A.F.; Bergthaler, A.; et al. Structural cells are key regulators of organ-specific immune responses. Nature 2020, 583, 296–302. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Gracia Villacampa, E.; Larsson, L.; Kvastad, L.; Andersson, A.; Carlson, J.; Lundeberg, J. Genome-wide Spatial Expression Profiling in FFPE Tissues. BioRxiv Prepr. 2020. [Google Scholar] [CrossRef]
- Merritt, C.R.; Ong, G.T.; Church, S.E.; Barker, K.; Danaher, P.; Geiss, G.; Hoang, M.; Jung, J.; Liang, Y.; McKay-Fleisch, J.; et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol. 2020, 38, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Marín-Sedeño, E.; de Morentin, X.M.; Pérez-Pomares, J.M.; Gómez-Cabrero, D.; Ruiz-Villalba, A. Understanding the Adult Mammalian Heart at Single-Cell RNA-Seq Resolution. Front. Cell Dev. Biol. 2021, 9, 645276. [Google Scholar] [CrossRef] [PubMed]
- Paik, D.T.; Cho, S.; Tian, L.; Chang, H.Y.; Wu, J.C. Single-cell RNA sequencing in cardiovascular development, disease and medicine. Nat. Rev. Cardiol. 2020, 17, 457–473. [Google Scholar] [CrossRef]
- Miao, Z.; Humphreys, B.D.; McMahon, A.P.; Kim, J. Multi-omics integration in the age of million single-cell data. Nat. Rev. Nephrol. 2021, 17, 710–724. [Google Scholar] [CrossRef]
- Wang, L.; Yu, P.; Zhou, B.; Song, J.; Li, Z.; Zhang, M.; Guo, G.; Wang, Y.; Chen, X.; Han, L.; et al. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat. Cell Biol. 2020, 22, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Litviňuková, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef]
- Yang, Q.; He, G.-W.; Underwood, M.J.; Yu, C.-M. Cellular and molecular mechanisms of endothelial ischemia/reperfusion injury: Perspectives and implications for postischemic myocardial protection. Am. J. Transl. Res. 2016, 8, 765–777. [Google Scholar] [PubMed]
- Zhang, H.; Lui, K.O.; Zhou, B. Endocardial Cell Plasticity in Cardiac Development, Diseases and Regeneration. Circ. Res. 2018, 122, 774–789. [Google Scholar] [CrossRef]
- Furtado, M.B.; Nim, H.T.; Boyd, S.E.; Rosenthal, N.A. View from the heart: Cardiac fibroblasts in development, scarring and regeneration. Development 2016, 143, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Moore-Morris, T.; Guimarães-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident fibroblast lineages mediate pressure overload–induced cardiac fibrosis. J. Clin. Investig. 2014, 124, 2921–2934. [Google Scholar] [CrossRef] [Green Version]
- McLellan, M.A.; Skelly, D.A.; Dona, M.S.I.; Squiers, G.T.; Farrugia, G.E.; Gaynor, T.L.; Cohen, C.D.; Pandey, R.; Diep, H.; Vinh, A.; et al. High-Resolution Transcriptomic Profiling of the Heart During Chronic Stress Reveals Cellular Drivers of Cardiac Fibrosis and Hypertrophy. Circulation 2020, 142, 1448–1463. [Google Scholar] [CrossRef] [PubMed]
- Farbehi, N.; Patrick, R.; Dorison, A.; Xaymardan, M.; Janbandhu, V.; Wystub-Lis, K.; Ho, J.W.; Nordon, R.E.; Harvey, R.P. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife 2019, 8, 43882. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Villalba, A.; Romero, J.P.; Hernández, S.C.; Vilas-Zornoza, A.; Fortelny, N.; Castro-Labrador, L.; San Martin-Uriz, P.; Lorenzo-Vivas, E.; García-Olloqui, P.; Palacio, M.; et al. Single-Cell RNA Sequencing Analysis Reveals a Crucial Role for CTHRC1 (Collagen Triple Helix Repeat Containing 1) Cardiac Fibroblasts After Myocardial Infarction. Circulation 2020, 142, 1831–1847. [Google Scholar] [CrossRef]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef]
- Cai, C.-L.; Martin, J.C.; Sun, Y.; Cui, L.; Wang, L.; Ouyang, K.; Yang, L.; Bu, L.; Liang, X.; Zhang, X.; et al. A myocardial lineage derives from Tbx18 epicardial cells. Nature 2008, 454, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Swirski, F.K.; Nahrendorf, M. Cardioimmunology: The immune system in cardiac homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 733–744. [Google Scholar] [CrossRef]
- Sun, K.; Li, Y.; Jin, J. A double-edged sword of immuno-microenvironment in cardiac homeostasis and injury repair. Signal Transduct. Target. Ther. 2021, 6, 79. [Google Scholar] [CrossRef]
- Epelman, S.; Liu, P.P.; Mann, D.L. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat. Rev. Immunol. 2015, 15, 117–129. [Google Scholar] [CrossRef]
- Chu, C.; Artis, D.; Chiu, I.M. Neuro-immune Interactions in the Tissues. Immunity 2020, 52, 464–474. [Google Scholar] [CrossRef]
- Tracey, K.J. The inflammatory reflex. Nature 2002, 420, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Scalco, A.; Moro, N.; Mongillo, M.; Zaglia, T. Neurohumoral Cardiac Regulation: Optogenetics Gets Into the Groove. Front. Physiol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Pratschke, J.; Tullius, S.G.; Neuhaus, P. Brain death associated ischemia/reperfusion injury. Ann. Transplant. 2004, 9, 78–80. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.; Rega, F.; Meyns, B. Current preservation technology and future prospects of thoracic organs. Part 2: Heart. Curr. Opin. Organ Transplant. 2010, 15, 156–159. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.M.; Connolly, M.R.; Coe, T.M.; Calhoun, A.; Pollok, F.; Markmann, J.F.; Burdorf, L.; Azimzadeh, A.; Madsen, J.C.; Pierson, R.N. Minimizing Ischemia Reperfusion Injury in Xenotransplantation. Front. Immunol. 2021, 12, 681504. [Google Scholar] [CrossRef]
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; Althagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 2019, 20, 29–39. [Google Scholar] [CrossRef]
- Molenaar, B.; Timmer, L.T.; Droog, M.; Perini, I.; Versteeg, D.; Kooijman, L.; Monshouwer-Kloots, J.; de Ruiter, H.; Gladka, M.M.; van Rooij, E. Single-cell transcriptomics following ischemic injury identifies a role for B2M in cardiac repair. Commun. Biol. 2021, 4, 146. [Google Scholar] [CrossRef]
- Tombor, L.S.; John, D.; Glaser, S.F.; Luxán, G.; Forte, E.; Furtado, M.; Rosenthal, N.; Baumgarten, N.; Schulz, M.H.; Wittig, J.; et al. Single cell sequencing reveals endothelial plasticity with transient mesenchymal activation after myocardial infarction. Nat. Commun. 2021, 12, 681. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Solomonidis, E.G.; Meloni, M.; Taylor, R.S.; Duffin, R.; Dobie, R.; Magalhaes, M.S.; Henderson, B.E.P.; Louwe, P.A.; D’Amico, G.; et al. Single-cell transcriptome analyses reveal novel targets modulating cardiac neovascularization by resident endothelial cells following myocardial infarction. Eur. Heart J. 2019, 40, 2507–2520. [Google Scholar] [CrossRef] [PubMed]
- Forte, E.; Skelly, D.A.; Chen, M.; Daigle, S.; Morelli, K.A.; Hon, O.; Philip, V.M.; Costa, M.W.; Rosenthal, N.A.; Furtado, M.B. Dynamic Interstitial Cell Response during Myocardial Infarction Predicts Resilience to Rupture in Genetically Diverse Mice. Cell Rep. 2020, 30, 3149–3163.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gladka, M.M.; Kohela, A.; Molenaar, B.; Versteeg, D.; Kooijman, L.; Monshouwer-Kloots, J.; Kremer, V.; Vos, H.R.; Huibers, M.M.H.; Haigh, J.J.; et al. Cardiomyocytes stimulate angiogenesis after ischemic injury in a ZEB2-dependent manner. Nat. Commun. 2021, 12, 84. [Google Scholar] [CrossRef]
- Piper, H. The first minutes of reperfusion: A window of opportunity for cardioprotection. Cardiovasc. Res. 2004, 61, 365–371. [Google Scholar] [CrossRef]
- Huang, J.; Li, R.; Wang, C. The Role of Mitochondrial Quality Control in Cardiac Ischemia/Reperfusion Injury. Oxid. Med. Cell. Longev. 2021, 2021, 5543452. [Google Scholar] [CrossRef]
- Silvis, M.J.M.; Kaffka genaamd Dengler, S.E.; Odille, C.A.; Mishra, M.; van der Kaaij, N.P.; Doevendans, P.A.; Sluijter, J.P.G.; de Kleijn, D.P.V.; de Jager, S.C.A.; Bosch, L.; et al. Damage-Associated Molecular Patterns in Myocardial Infarction and Heart Transplantation: The Road to Translational Success. Front. Immunol. 2020, 11, 599511. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef]
- Kumar, S.; Kasseckert, S.; Kostin, S.; Abdallah, Y.; Schafer, C.; Kaminski, A.; Reusch, H.; Piper, H.; Steinhoff, G.; Ladilov, Y. Ischemic acidosis causes apoptosis in coronary endothelial cells through activation of caspase-12☆. Cardiovasc. Res. 2007, 73, 172–180. [Google Scholar] [CrossRef] [Green Version]
- Seal, J.B.; Gewertz, B.L. Vascular Dysfunction in Ischemia-Reperfusion Injury. Ann. Vasc. Surg. 2005, 19, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tan, X.; Tampe, B.; Sanchez, E.; Zeisberg, M.; Zeisberg, E.M. Snail Is a Direct Target of Hypoxia-inducible Factor 1α (HIF1α) in Hypoxia-induced Endothelial to Mesenchymal Transition of Human Coronary Endothelial Cells. J. Biol. Chem. 2015, 290, 16653–16664. [Google Scholar] [CrossRef] [Green Version]
- Gogiraju, R.; Bochenek, M.L.; Schäfer, K. Angiogenic Endothelial Cell Signaling in Cardiac Hypertrophy and Heart Failure. Front. Cardiovasc. Med. 2019, 6. [Google Scholar] [CrossRef] [Green Version]
- Burke, R.M.; Burgos Villar, K.N.; Small, E.M. Fibroblast contributions to ischemic cardiac remodeling. Cell. Signal. 2021, 77, 109824. [Google Scholar] [CrossRef]
- van den Borne, S.W.M.; Diez, J.; Blankesteijn, W.M.; Verjans, J.; Hofstra, L.; Narula, J. Myocardial remodeling after infarction: The role of myofibroblasts. Nat. Rev. Cardiol. 2010, 7, 30–37. [Google Scholar] [CrossRef]
- Hurley, J.R.; Balaji, S.; Narmoneva, D.A. Complex temporal regulation of capillary morphogenesis by fibroblasts. Am. J. Physiol. Physiol. 2010, 299, C444–C453. [Google Scholar] [CrossRef] [PubMed]
- Chintalgattu, V. Cardiac myofibroblasts: A novel source of vascular endothelial growth factor (VEGF) and its receptors Flt-1 and KDR. J. Mol. Cell. Cardiol. 2003, 35, 277–286. [Google Scholar] [CrossRef]
- Anzai, A.; Choi, J.L.; He, S.; Fenn, A.M.; Nairz, M.; Rattik, S.; McAlpine, C.S.; Mindur, J.E.; Chan, C.T.; Iwamoto, Y.; et al. The infarcted myocardium solicits GM-CSF for the detrimental oversupply of inflammatory leukocytes. J. Exp. Med. 2017, 214, 3293–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yemisci, M.; Gursoy-Ozdemir, Y.; Vural, A.; Can, A.; Topalkara, K.; Dalkara, T. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat. Med. 2009, 15, 1031–1037. [Google Scholar] [CrossRef]
- O’Farrell, F.M.; Mastitskaya, S.; Hammond-Haley, M.; Freitas, F.; Wah, W.R.; Attwell, D. Capillary pericytes mediate coronary no-reflow after myocardial ischaemia. Elife 2017, 6, 29280. [Google Scholar] [CrossRef] [PubMed]
- Slegtenhorst, B.R.; Dor, F.J.M.F.; Rodriguez, H.; Voskuil, F.J.; Tullius, S.G. Ischemia/Reperfusion Injury and its Consequences on Immunity and Inflammation. Curr. Transplant. Rep. 2014, 1, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamada, S.; Dubois, V.; Koenig, A.; Thaunat, O. Allograft recognition by recipient’s natural killer cells: Molecular mechanisms and role in transplant rejection. HLA 2021, 98, 191–199. [Google Scholar] [CrossRef]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2016, 38, ehw002. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, P.; Rajasingh, J.; Lambers, E.; Qin, G.; Losordo, D.W.; Kishore, R. IL-10 Inhibits Inflammation and Attenuates Left Ventricular Remodeling After Myocardial Infarction via Activation of STAT3 and Suppression of HuR. Circ. Res. 2009, 104, e9–e18. [Google Scholar] [CrossRef]
- Hilgendorf, I.; Gerhardt, L.M.S.; Tan, T.C.; Winter, C.; Holderried, T.A.W.; Chousterman, B.G.; Iwamoto, Y.; Liao, R.; Zirlik, A.; Scherer-Crosbie, M.; et al. Ly-6C high Monocytes Depend on Nr4a1 to Balance Both Inflammatory and Reparative Phases in the Infarcted Myocardium. Circ. Res. 2014, 114, 1611–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, N.; Lu, Y.; Gu, M.; Li, N.; Liu, M.; Jiao, J.; Zhu, Z.; Li, J.; Li, D.; Tang, T.; et al. A Unique Population of Regulatory T Cells in Heart Potentiates Cardiac Protection From Myocardial Infarction. Circulation 2020, 142, 1956–1973. [Google Scholar] [CrossRef] [PubMed]
- Lakkis, F.G.; Lechler, R.I. Origin and Biology of the Allogeneic Response. Cold Spring Harb. Perspect. Med. 2013, 3, a014993. [Google Scholar] [CrossRef] [Green Version]
- El-Sawy, T.; Miura, M.; Fairchild, R. Early T Cell Response to Allografts Occuring Prior to Alloantigen Priming Up-Regulates Innate-Mediated Inflammation and Graft Necrosis. Am. J. Pathol. 2004, 165, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Merola, J.; Jane-wit, D.D.; Pober, J.S. Recent advances in allograft vasculopathy. Curr. Opin. Organ Transplant. 2017, 22, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, G.J.; Burke, M.M.; Andersen, C.; Bruneval, P.; Fedrigo, M.; Fishbein, M.C.; Goddard, M.; Hammond, E.H.; Leone, O.; Marboe, C.; et al. The 2013 International Society for Heart and Lung Transplantation Working Formulation for the standardization of nomenclature in the pathologic diagnosis of antibody-mediated rejection in heart transplantation. J. Hear. Lung Transplant. 2013, 32, 1147–1162. [Google Scholar] [CrossRef] [PubMed]
- Bruneval, P.; Angelini, A.; Miller, D.; Potena, L.; Loupy, A.; Zeevi, A.; Reed, E.F.; Dragun, D.; Reinsmoen, N.; Smith, R.N.; et al. The XIIIth Banff Conference on Allograft Pathology: The Banff 2015 Heart Meeting Report: Improving Antibody-Mediated Rejection Diagnostics: Strengths, Unmet Needs, and Future Directions. Am. J. Transplant. 2017, 17, 42–53. [Google Scholar] [CrossRef]
- Cross, A.R.; Glotz, D.; Mooney, N. The Role of the Endothelium during Antibody-Mediated Rejection: From Victim to Accomplice. Front. Immunol. 2018, 9, 106. [Google Scholar] [CrossRef] [Green Version]
- Khayyamian, S.; Hutloff, A.; Buchner, K.; Grafe, M.; Henn, V.; Kroczek, R.A.; Mages, H.W. ICOS-ligand, expressed on human endothelial cells, costimulates Th1 and Th2 cytokine secretion by memory CD4+ T cells. Proc. Natl. Acad. Sci. USA 2002, 99, 6198–6203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taflin, C.; Favier, B.; Baudhuin, J.; Savenay, A.; Hemon, P.; Bensussan, A.; Charron, D.; Glotz, D.; Mooney, N. Human endothelial cells generate Th17 and regulatory T cells under inflammatory conditions. Proc. Natl. Acad. Sci. USA 2011, 108, 2891–2896. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela, N.M.; Reed, E.F. Antibody-mediated rejection across solid organ transplants: Manifestations, mechanisms, and therapies. J. Clin. Investig. 2017, 127, 2492–2504. [Google Scholar] [CrossRef] [Green Version]
- Abou-Daya, K.I.; Oberbarnscheidt, M.H. Innate allorecognition in transplantation. J. Hear. Lung Transplant. 2021, 40, 557–561. [Google Scholar] [CrossRef]
- Okabe, Y.; Medzhitov, R. Tissue biology perspective on macrophages. Nat. Immunol. 2016, 17, 9–17. [Google Scholar] [CrossRef]
- Valenzuela, N.M.; Hong, L.; Shen, X.-D.; Gao, F.; Young, S.H.; Rozengurt, E.; Kupiec-Weglinski, J.W.; Fishbein, M.C.; Reed, E.F. Blockade of P-Selectin Is Sufficient to Reduce MHC I Antibody-Elicited Monocyte Recruitment In Vitro and In Vivo. Am. J. Transplant. 2013, 13, 299–311. [Google Scholar] [CrossRef] [Green Version]
- Pabois, A.; Pagie, S.; Gérard, N.; Laboisse, C.; Pattier, S.; Hulin, P.; Nedellec, S.; Toquet, C.; Charreau, B. Notch signaling mediates crosstalk between endothelial cells and macrophages via Dll4 and IL6 in cardiac microvascular inflammation. Biochem. Pharmacol. 2016, 104, 95–107. [Google Scholar] [CrossRef] [PubMed]
- PUIG, M. Burden of myocardial damage in cardiac allograft rejection: Scintigraphic evidence of myocardial injury and histologic evidence of myocyte necrosis and apoptosis. J. Nucl. Cardiol. 2000, 7, 132–139. [Google Scholar] [CrossRef]
- Colvin-Adams, M.; Harcourt, N.; Duprez, D. Endothelial Dysfunction and Cardiac Allograft Vasculopathy. J. Cardiovasc. Transl. Res. 2013, 6, 263–277. [Google Scholar] [CrossRef]
- Angelini, A.; Castellani, C.; Fedrigo, M.; de Boer, O.J.; Meijer-Jorna, L.B.; Li, X.; Valente, M.; Thiene, G.; van der Wal, A.C. Coronary cardiac allograft vasculopathy versus native atherosclerosis: Difficulties in classification. Virchows Arch. 2014, 464, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Rickenbacher, P.R.; Pinto, F.J.; Chenzbraun, A.; Botas, J.; Lewis, N.P.; Alderman, E.L.; Valantine, H.A.; Hunt, S.A.; Schroeder, J.S.; Popp, R.L.; et al. Incidence and severity of transplant coronary artery disease early and up to 15 years after transplantation as detected by intravascular ultrasound. J. Am. Coll. Cardiol. 1995, 25, 171–177. [Google Scholar] [CrossRef] [Green Version]
- Tsutsui, H.; Schoenhagen, P.; Klingensmith, J.D.; Vince, D.G.; Nissen, S.E.; Tuzcu, E.M. Regression of a Donor Atheroma after Cardiac Transplantation. Circulation 2001, 104, 2874. [Google Scholar] [CrossRef] [Green Version]
- Castellani, C.; Angelini, A.; de Boer, O.J.; van der Loos, C.M.; Fedrigo, M.; Frigo, A.C.; Meijer-Jorna, L.B.; Li, X.; Ploegmakers, H.J.P.; Tona, F.; et al. Intraplaque Hemorrhage in Cardiac Allograft Vasculopathy. Am. J. Transplant. 2014, 14, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Pober, J.S.; Jane-wit, D.; Qin, L.; Tellides, G. Interacting Mechanisms in the Pathogenesis of Cardiac Allograft Vasculopathy. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1609–1614. [Google Scholar] [CrossRef] [Green Version]
- Costello, J.P.; Mohanakumar, T.; Nath, D.S. Mechanisms of chronic cardiac allograft rejection. Tex. Hear. Inst. J. 2013, 40, 395–399. [Google Scholar]
- Kfoury, A.G.; Stehlik, J.; Renlund, D.G.; Snow, G.; Seaman, J.T.; Gilbert, E.M.; Stringham, J.S.; Long, J.W.; Hammond, M.E.H. Impact of Repetitive Episodes of Antibody-mediated or Cellular Rejection on Cardiovascular Mortality in Cardiac Transplant Recipients: Defining Rejection Patterns. J. Hear. Lung Transplant. 2006, 25, 1277–1282. [Google Scholar] [CrossRef] [PubMed]
- Fedoseyeva, E.V.; Zhang, F.; Orr, P.L.; Levin, D.; Buncke, H.J.; Benichou, G. De novo autoimmunity to cardiac myosin after heart transplantation and its contribution to the rejection process. J. Immunol. 1999, 162, 6836–6842. [Google Scholar]
- Divanyan, T.; Acosta, E.; Patel, D.; Constantino, D.; Lopez-Soler, R.I. Anti-vimentin antibodies in transplant and disease. Hum. Immunol. 2019, 80, 602–607. [Google Scholar] [CrossRef]
- Mehra, M.R.; Uber, P.A.; Ventura, H.O.; Scott, R.L.; Park, M.H. The impact of mode of donor brain death on cardiac allograft vasculopathy. J. Am. Coll. Cardiol. 2004, 43, 806–810. [Google Scholar] [CrossRef] [Green Version]
- Koskinen, P.K.; Kallio, E.A.; Tikkanen, J.M.; Sihvola, R.K.; Häyry, P.J.; Lemström, K.B. Cytomegalovirus infection and cardiac allograft vasculopathy. Transpl. Infect. Dis. 1999, 1, 115–126. [Google Scholar] [CrossRef]
- Fateh-Moghadam, S.; Bocksch, W.; Wessely, R.; Jäger, G.; Hetzer, R.; Gawaz, M. Cytomegalovirus infection status predicts progression of heart-transplant vasculopathy. Transplantation 2003, 76, 1470–1474. [Google Scholar] [CrossRef] [PubMed]
- Caforio, A.L.P.; Tona, F.; Fortina, A.B.; Angelini, A.; Piaserico, S.; Gambino, A.; Feltrin, G.; Ramondo, A.; Valente, M.; Iliceto, S.; et al. Immune and Nonimmune Predictors of Cardiac Allograft Vasculopathy Onset and Severity: Multivariate Risk Factor Analysis and Role of Immunosuppression. Am. J. Transplant. 2004, 4, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis—An Inflammatory Disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Glaser, R.; Lu, M.M.; Narula, N.; Epstein, J.A. Smooth Muscle Cells, But Not Myocytes, of Host Origin in Transplanted Human Hearts. Circulation 2002, 106, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Quaini, F.; Urbanek, K.; Beltrami, A.P.; Finato, N.; Beltrami, C.A.; Nadal-Ginard, B.; Kajstura, J.; Leri, A.; Anversa, P. Chimerism of the Transplanted Heart. N. Engl. J. Med. 2002, 346, 5–15. [Google Scholar] [CrossRef]
- Simper, D.; Wang, S.; Deb, A.; Holmes, D.; McGregor, C.; Frantz, R.; Kushwaha, S.S.; Caplice, N.M. Endothelial Progenitor Cells Are Decreased in Blood of Cardiac Allograft Patients with Vasculopathy and Endothelial Cells of Noncardiac Origin Are Enriched in Transplant Atherosclerosis. Circulation 2003, 108, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Hillebrands, J.-L.; Klatter, F.A.; van den Hurk, B.M.H.; Popa, E.R.; Nieuwenhuis, P.; Rozing, J. Origin of neointimal endothelium and α-actin–positive smooth muscle cells in transplant arteriosclerosis. J. Clin. Investig. 2001, 107, 1411–1422. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Davison, F.; Ludewig, B.; Erdel, M.; Mayr, M.; Url, M.; Dietrich, H.; Xu, Q. Smooth Muscle Cells in Transplant Atherosclerotic Lesions Are Originated From Recipients, but Not Bone Marrow Progenitor Cells. Circulation 2002, 106, 1834–1839. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, C.; Horsley, J.; Rhind-Tutt, S.; Charman, S.; Phillpotts, C.J.; Wallwork, J.; Goddard, M.J. Neointimal smooth muscle cells in human cardiac allograft coronary artery vasculopathy are of donor origin. J. Hear. Lung Transplant. 2004, 23, 427–435. [Google Scholar] [CrossRef]
- Minami, E.; Laflamme, M.A.; Saffitz, J.E.; Murry, C.E. Extracardiac Progenitor Cells Repopulate Most Major Cell Types in the Transplanted Human Heart. Circulation 2005, 112, 2951–2958. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Liu, S.; Song, Z. Mechanism of arterial remodeling in chronic allograft vasculopathy. Front. Med. 2011, 5, 248–253. [Google Scholar] [CrossRef]
- Jansen, M.A.A.; Otten, H.G.; de Weger, R.A.; Huibers, M.M.H. Immunological and Fibrotic Mechanisms in Cardiac Allograft Vasculopathy. Transplantation 2015, 99, 2467–2475. [Google Scholar] [CrossRef] [PubMed]
- EULERTAIMOR, G.; HEGER, J. The complex pattern of SMAD signaling in the cardiovascular system☆. Cardiovasc. Res. 2006, 69, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Huibers, M.; De Jonge, N.; Van Kuik, J.; Koning, E.S.-D.; Van Wichen, D.; Dullens, H.; Schipper, M.; De Weger, R. Intimal fibrosis in human cardiac allograft vasculopathy. Transpl. Immunol. 2011, 25, 124–132. [Google Scholar] [CrossRef]
- Gareau, A.; Hirsch, G.M.; Lee, T.D.G.; Nashan, B. Contribution of B Cells and Antibody to Cardiac Allograft Vasculopathy. Transplantation 2009, 88, 470–477. [Google Scholar] [CrossRef]
- Zeng, Q.; Ng, Y.-H.; Singh, T.; Jiang, K.; Sheriff, K.A.; Ippolito, R.; Zahalka, S.; Li, Q.; Randhawa, P.; Hoffman, R.A.; et al. B cells mediate chronic allograft rejection independently of antibody production. J. Clin. Investig. 2014, 124, 1052–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirohashi, T.; Chase, C.M.; Della Pelle, P.; Sebastian, D.; Alessandrini, A.; Madsen, J.C.; Russell, P.S.; Colvin, R.B. A Novel Pathway of Chronic Allograft Rejection Mediated by NK Cells and Alloantibody. Am. J. Transplant. 2012, 12, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millington, T.M.; Madsen, J.C. Innate immunity and cardiac allograft rejection. Kidney Int. 2010, 78, S18–S21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabel, E.G.; Shum, L.; Pompili, V.J.; Yang, Z.Y.; San, H.; Shu, H.B.; Liptay, S.; Gold, L.; Gordon, D.; Derynck, R. Direct transfer of transforming growth factor beta 1 gene into arteries stimulates fibrocellular hyperplasia. Proc. Natl. Acad. Sci. USA 1993, 90, 10759–10763. [Google Scholar] [CrossRef] [Green Version]
- Lemström, K.B.; Koskinen, P.K. Expression and Localization of Platelet-Derived Growth Factor Ligand and Receptor Protein During Acute and Chronic Rejection of Rat Cardiac Allografts. Circulation 1997, 96, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Sihvola, R.; Koskinen, P.; Myllärniemi, M.; Loubtchenkov, M.; Häyry, P.; Buchdunger, E.; Lemström, K. Prevention of Cardiac Allograft Arteriosclerosis by Protein Tyrosine Kinase Inhibitor Selective for Platelet-Derived Growth Factor Receptor. Circulation 1999, 99, 2295–2301. [Google Scholar] [CrossRef] [Green Version]
- Koskinen, P.K.; Kovanen, P.T.; Lindstedt, K.A.; Lemström, K.B. mast cells in acute and chronic rejection of rat cardiac allografts—A major source of basic fibroblast growth factor1. Transplantation 2001, 71, 1741–1747. [Google Scholar] [CrossRef]
- Kitchens, W.H.; Chase, C.M.; Uehara, S.; Cornell, L.D.; Colvin, R.B.; Russell, P.S.; Madsen, J.C. Macrophage Depletion Suppresses Cardiac Allograft Vasculopathy in Mice. Am. J. Transplant. 2007, 7, 2675–2682. [Google Scholar] [CrossRef]
- Lemström, K.B.; Krebs, R.; Nykänen, A.I.; Tikkanen, J.M.; Sihvola, R.K.; Aaltola, E.M.; Häyry, P.J.; Wood, J.; Alitalo, K.; Ylä-Herttuala, S.; et al. Vascular Endothelial Growth Factor Enhances Cardiac Allograft Arteriosclerosis. Circulation 2002, 105, 2524–2530. [Google Scholar] [CrossRef] [Green Version]
- Tuuminen, R.; Syrjälä, S.; Krebs, R.; Keränen, M.A.I.; Koli, K.; Abo-Ramadan, U.; Neuvonen, P.J.; Tikkanen, J.M.; Nykänen, A.I.; Lemström, K.B. Donor Simvastatin Treatment Abolishes Rat Cardiac Allograft Ischemia/Reperfusion Injury and Chronic Rejection Through Microvascular Protection. Circulation 2011, 124, 1138–1150. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hurskainen, M.; Ainasoja, O.; Lemström, K.B. Failing Heart Transplants and Rejection—A Cellular Perspective. J. Cardiovasc. Dev. Dis. 2021, 8, 180. https://doi.org/10.3390/jcdd8120180
Hurskainen M, Ainasoja O, Lemström KB. Failing Heart Transplants and Rejection—A Cellular Perspective. Journal of Cardiovascular Development and Disease. 2021; 8(12):180. https://doi.org/10.3390/jcdd8120180
Chicago/Turabian StyleHurskainen, Maria, Olli Ainasoja, and Karl B. Lemström. 2021. "Failing Heart Transplants and Rejection—A Cellular Perspective" Journal of Cardiovascular Development and Disease 8, no. 12: 180. https://doi.org/10.3390/jcdd8120180
APA StyleHurskainen, M., Ainasoja, O., & Lemström, K. B. (2021). Failing Heart Transplants and Rejection—A Cellular Perspective. Journal of Cardiovascular Development and Disease, 8(12), 180. https://doi.org/10.3390/jcdd8120180