
Myocardium-Specific Deletion of Rac1 Causes Ventricular Noncompaction and Outflow Tract Defects



Abstract
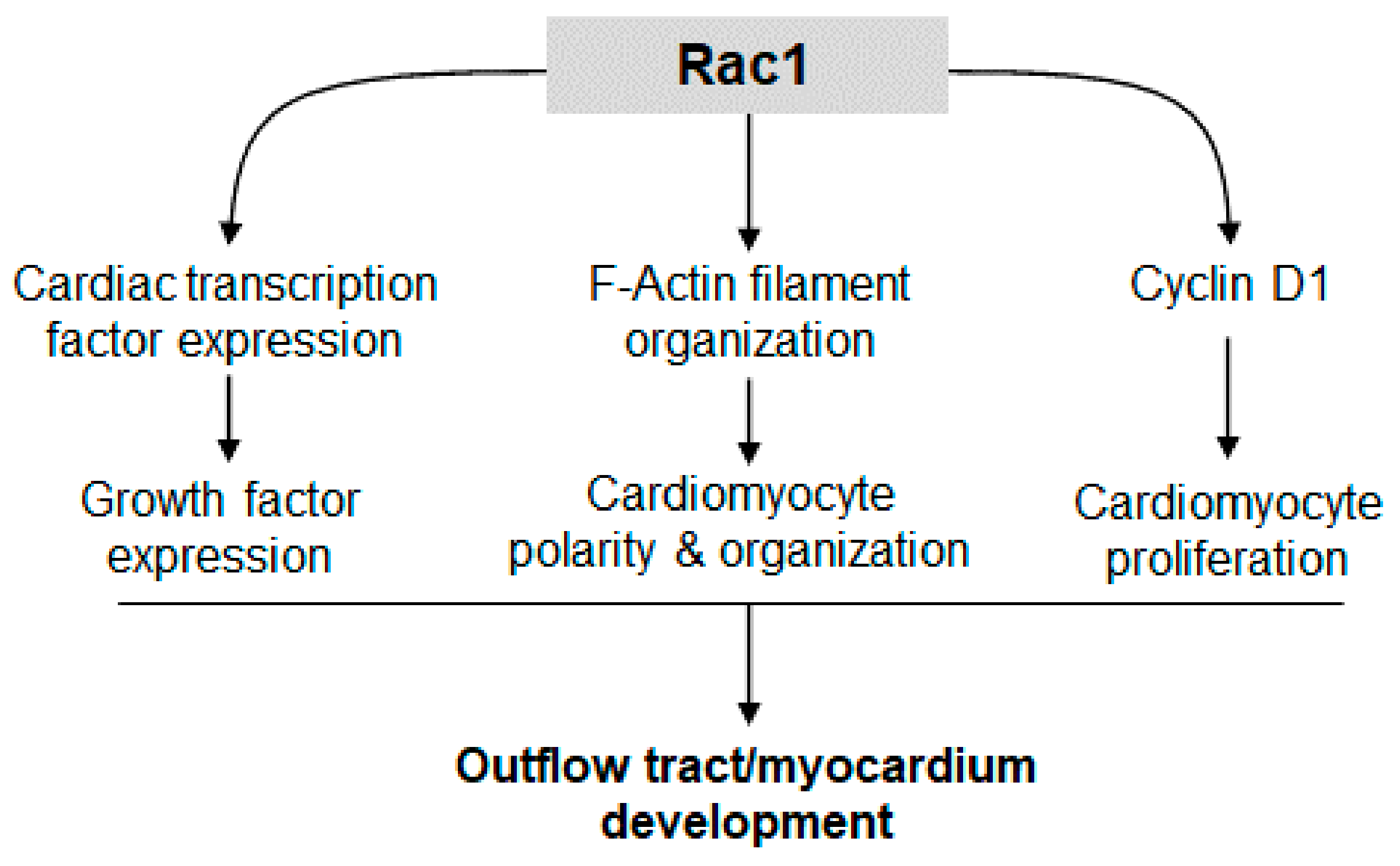
:1. Introduction
2. Methods
2.1. Mice
2.2. Histological Analysis
2.3. Immunohistochemistry
2.4. Quantitative Real Time RT-PCR
2.5. Western Blot Analysis
2.6. Statistical Analysis
3. Results
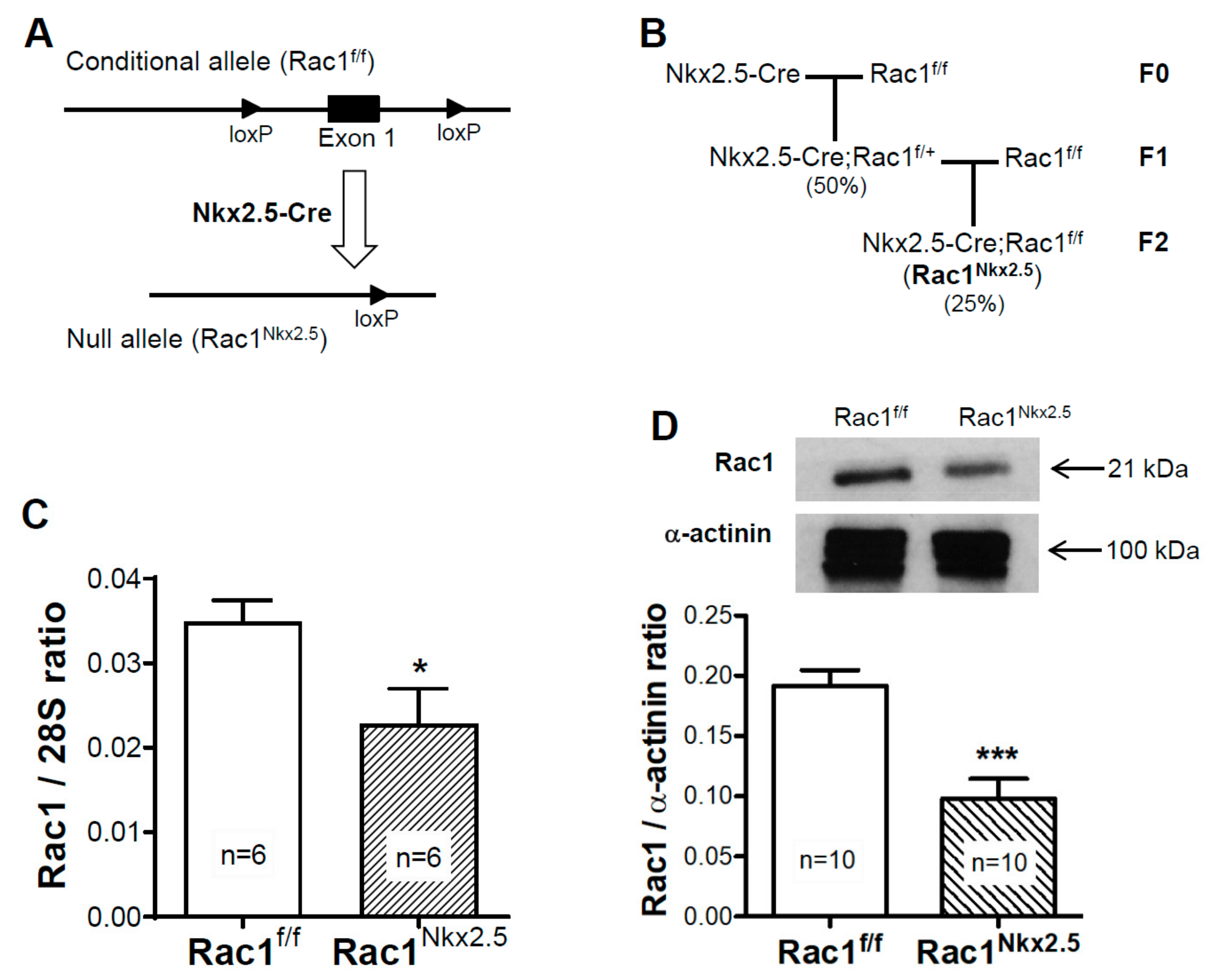
3.1. Generation of a Cardiomyocyte Specific Rac1 Knockout Mouse
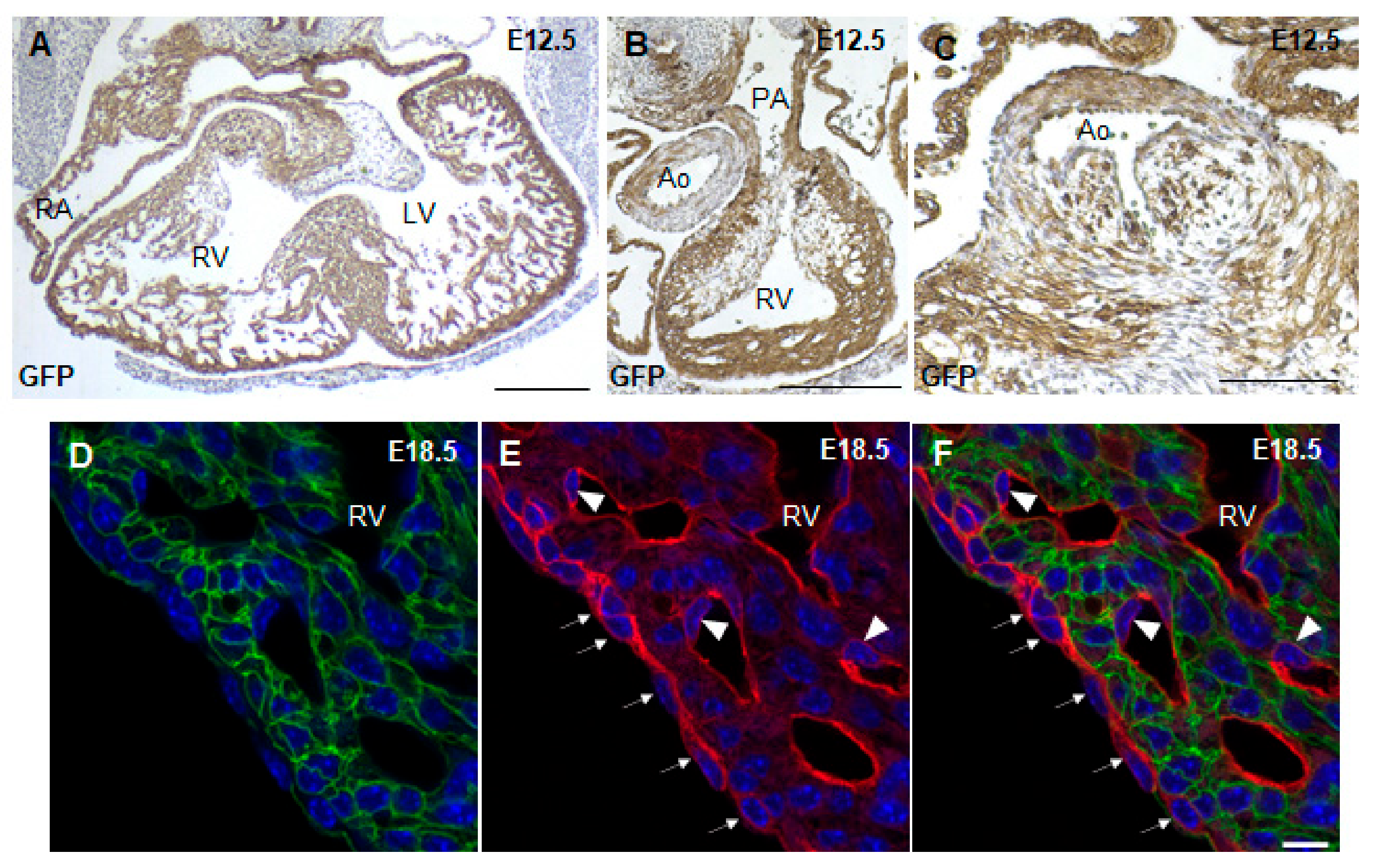
3.2. Lineage Tracing of Nkx2.5-Cre Transgenic Mouse
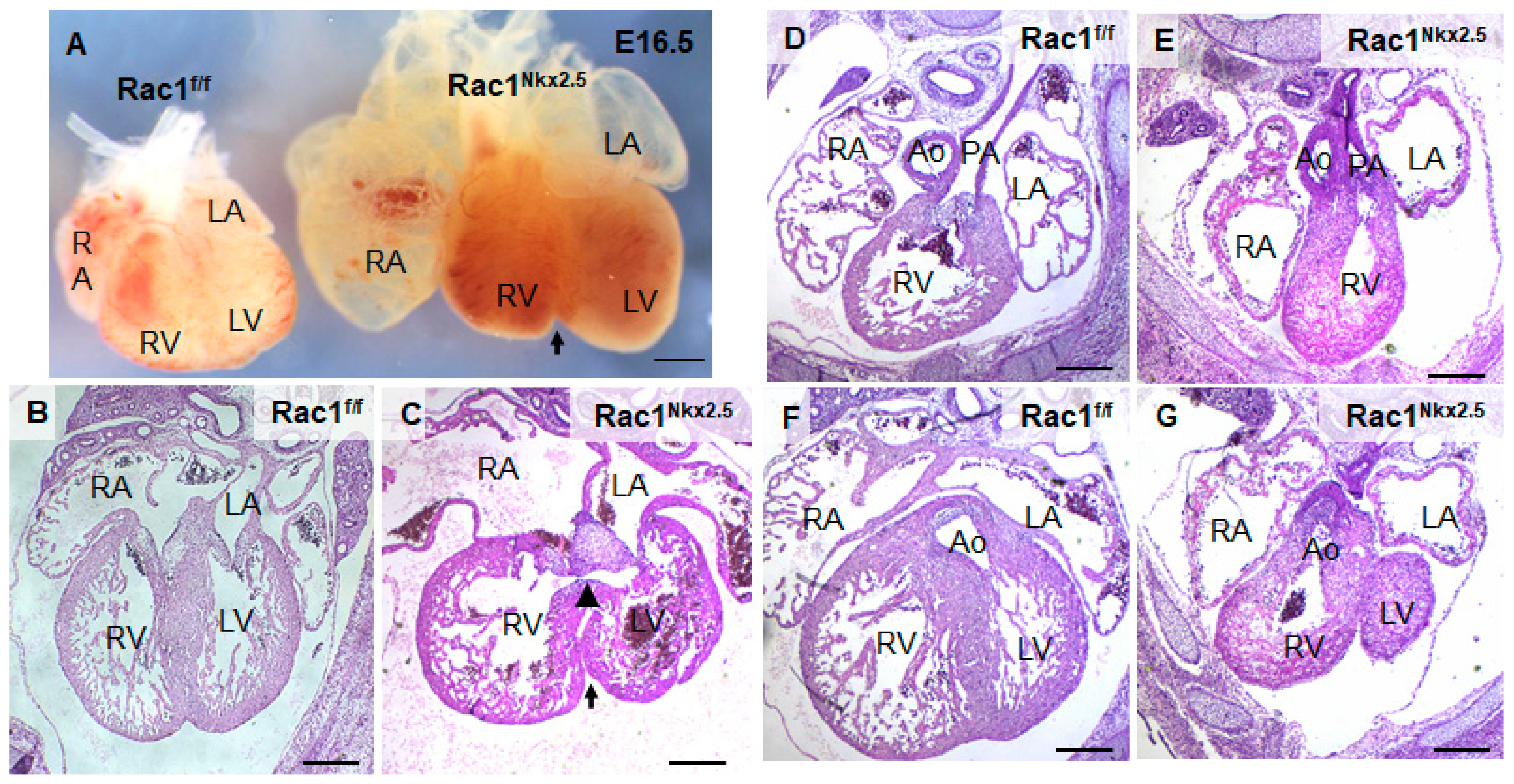
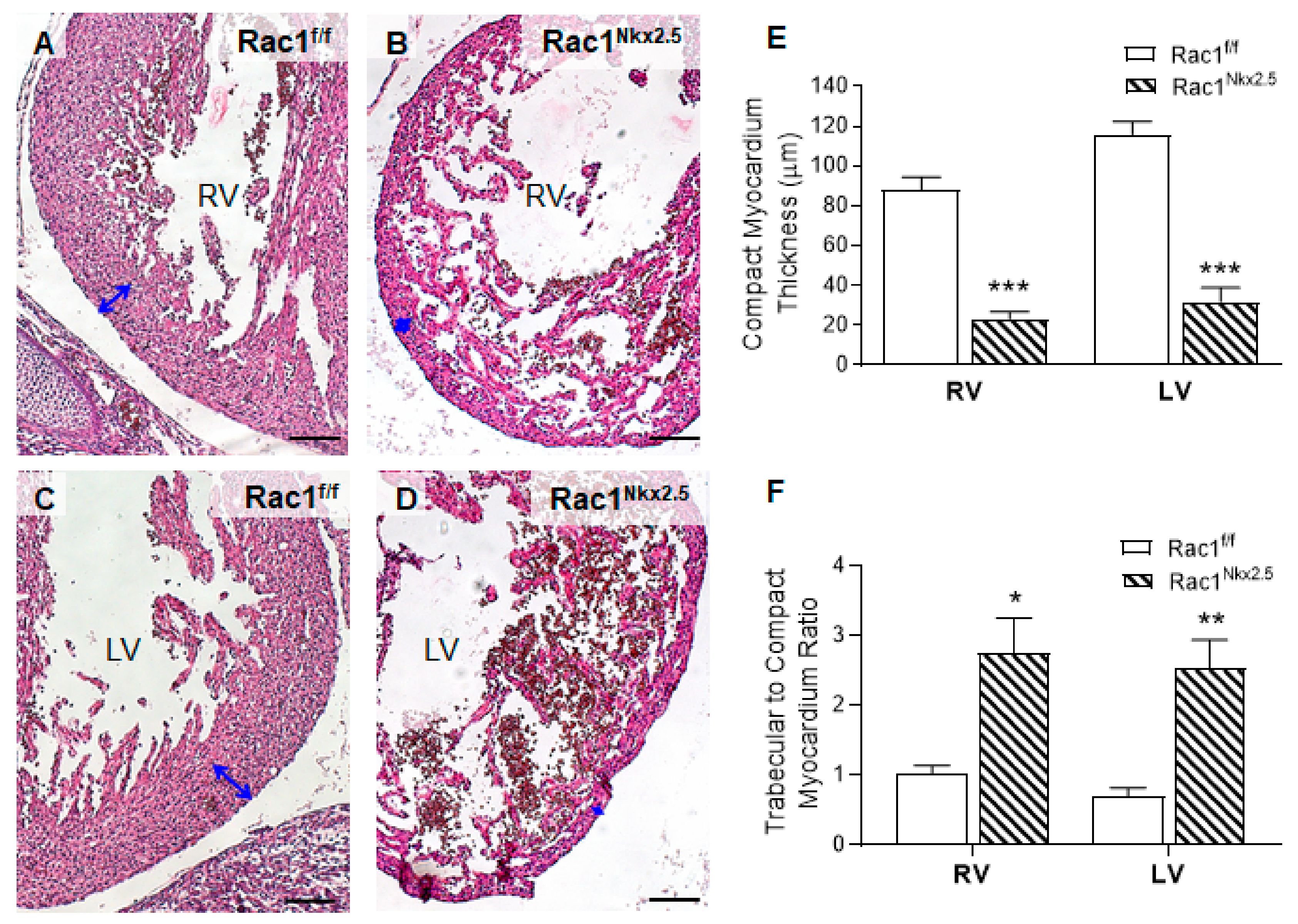
3.3. Congenital Heart Defects in Rac1Nkx2.5 Mice
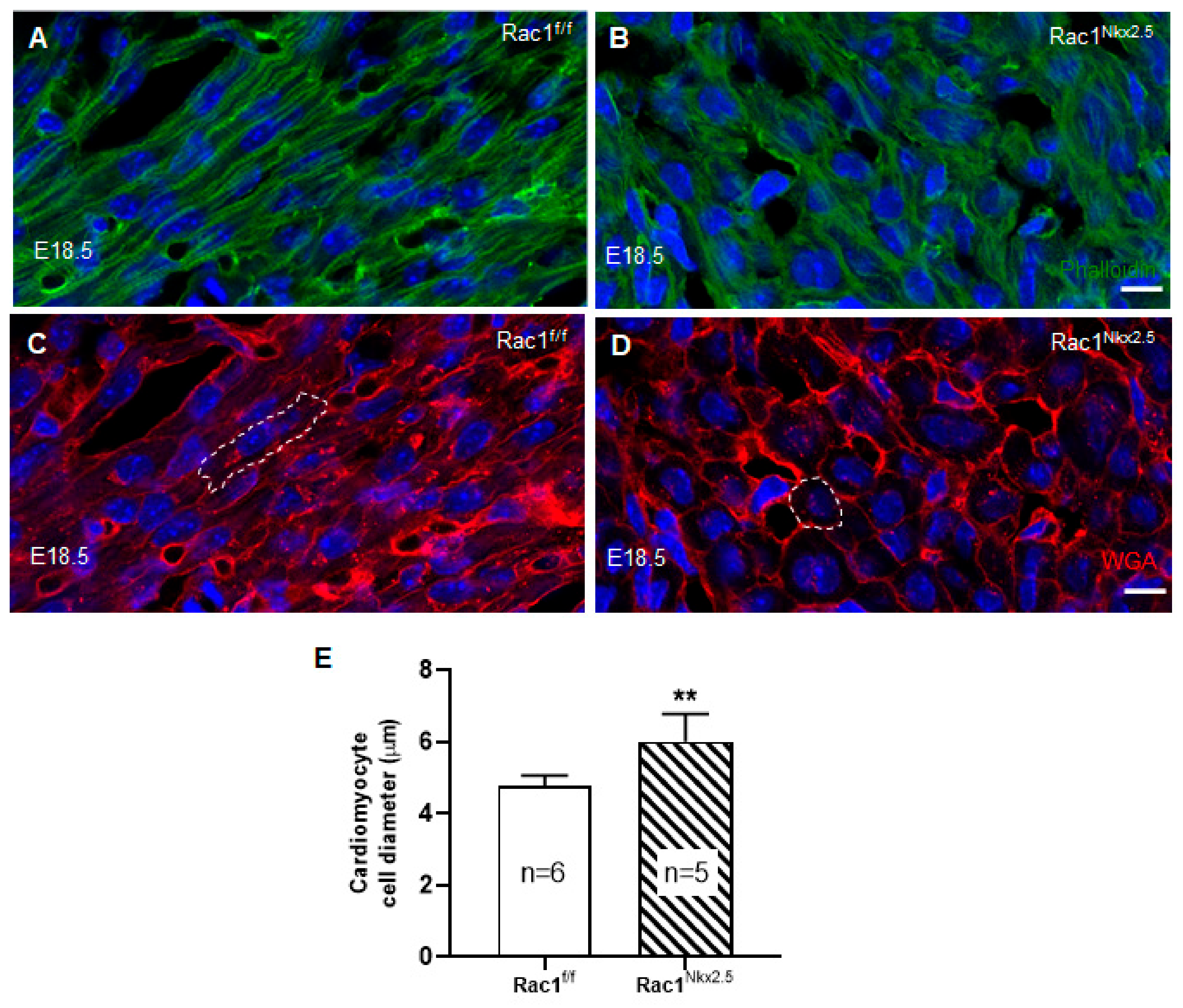
3.4. Loss of F-Actin Filament Organization and Cardiomyocyte Polarity in Rac1Nkx2.5 Hearts
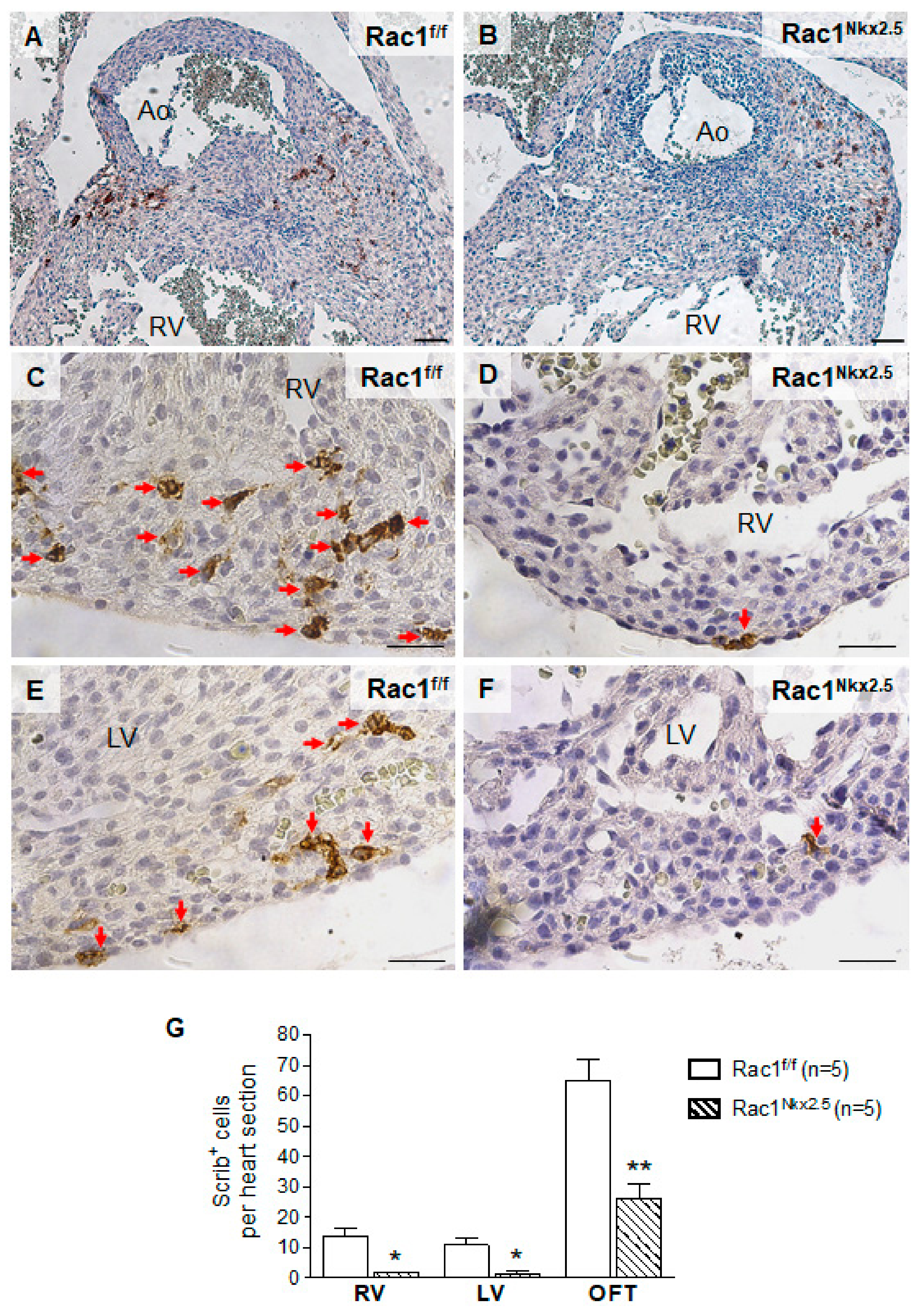
3.5. Decreased Scrib Protein Expression in Rac1Nkx2.5 Hearts
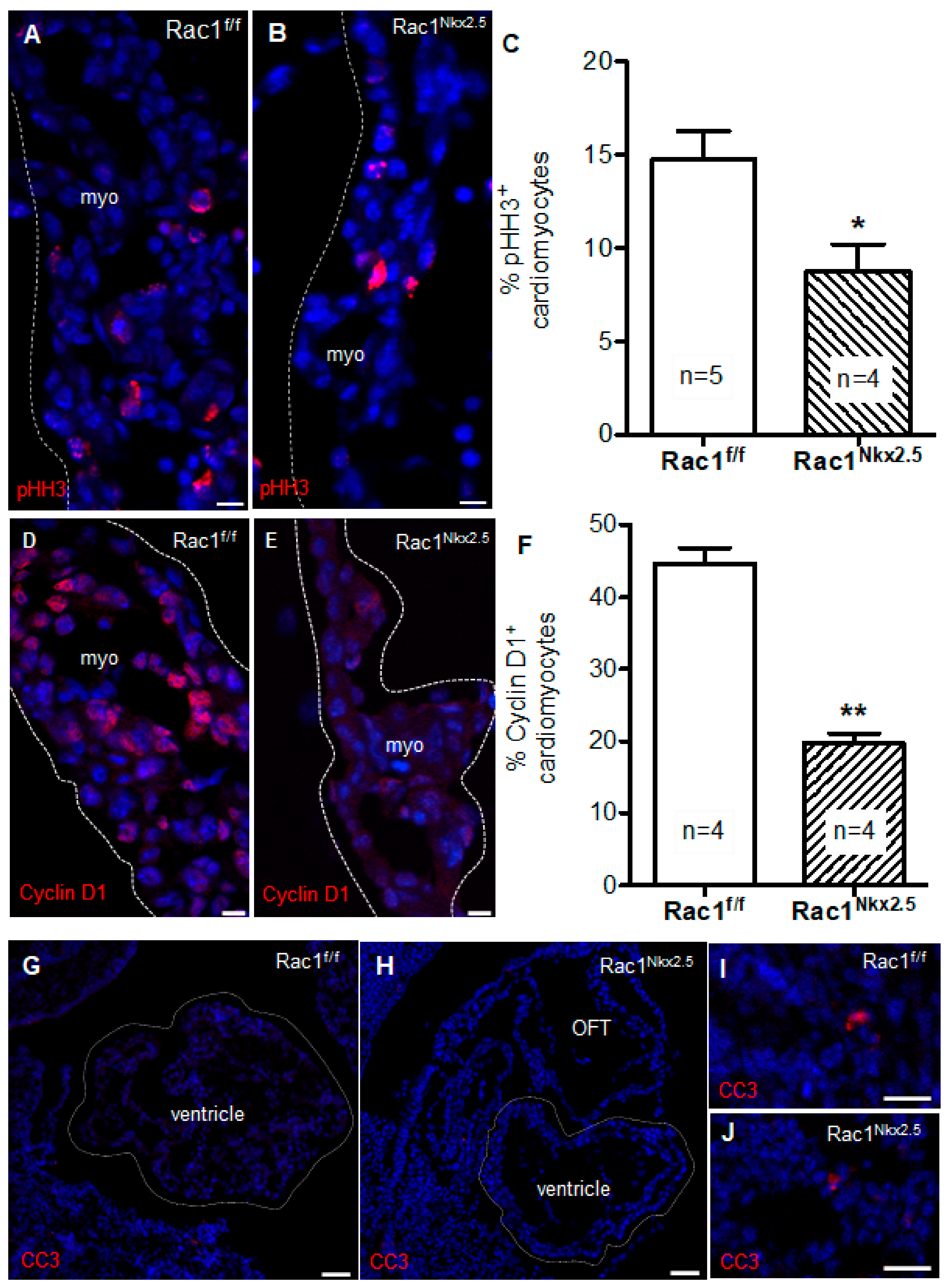
3.6. Decreased Cell Proliferation in Rac1Nkx2.5 Hearts
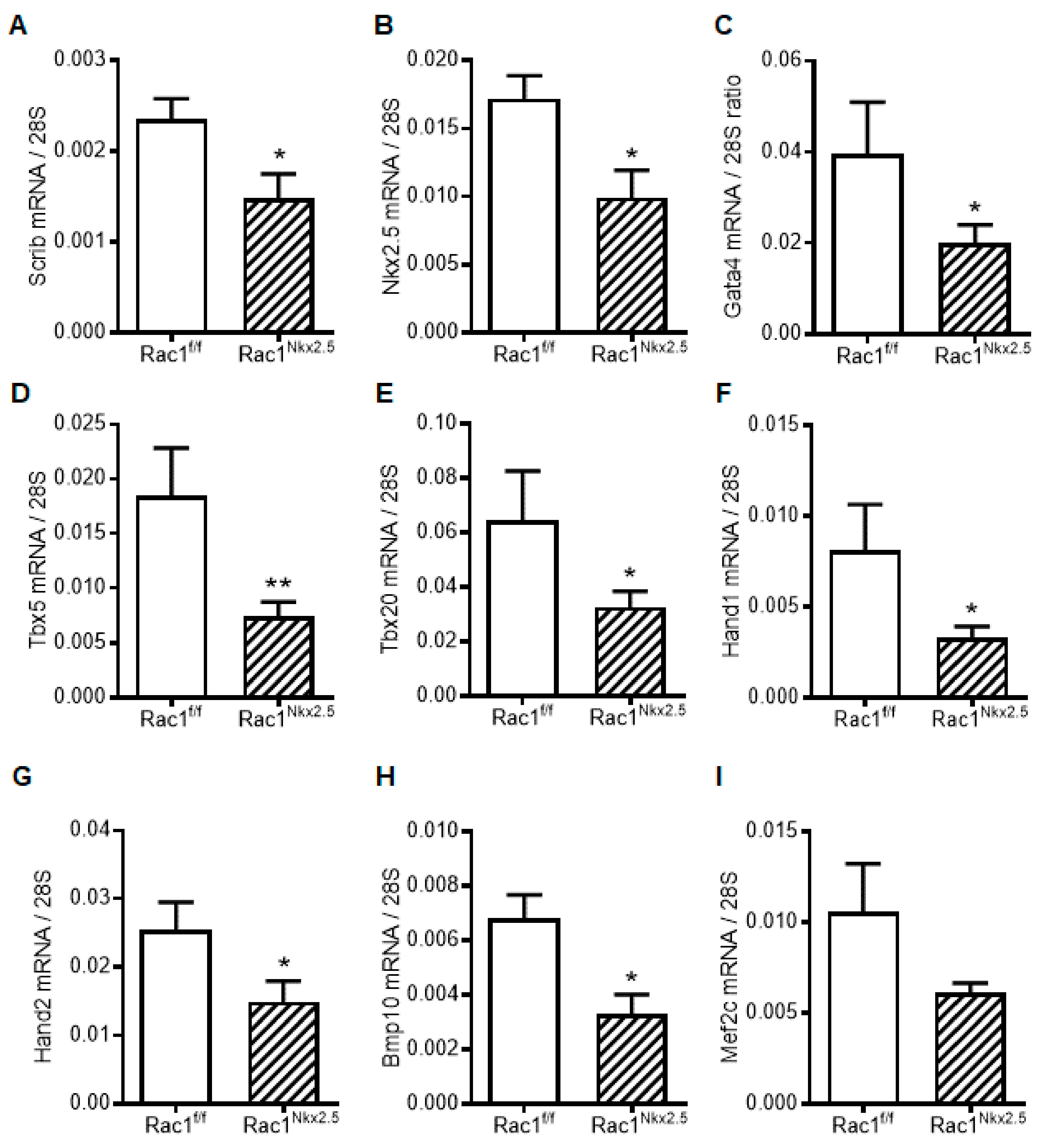
3.7. Decreased Scrib and Cardiac Transcription Factor Expression in Rac1Nkx2.5 Hearts
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pierpont, M.E.; Basson, C.T.; Benson, D.W.; Gelb, B.D.; Giglia, T.M.; Goldmuntz, E.; McGee, G.; Sable, C.A.; Srivastava, D.; Webb, C.L. Genetic Basis for Congenital Heart Defects: Current Knowledge: A Scientific Statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: Endorsed by the American Academy of Pediatrics. Circulation 2007, 115, 3015–3038. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, J.I.; Kaplan, S.; Liberthson, R.R. Prevalence of Congenital Heart Disease. Am. Heart J. 2004, 147, 425–439. [Google Scholar] [CrossRef]
- Sun, R.; Liu, M.; Lu, L.; Zheng, Y.; Zhang, P. Congenital Heart Disease: Causes, Diagnosis, Symptoms, and Treatments. Cell Biochem. Biophys. 2015, 72, 857–860. [Google Scholar] [CrossRef]
- Warnes, C.A. The Adult with Congenital Heart Disease: Born to Be Bad? J. Am. Coll. Cardiol. 2005, 46, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Guha, K.; Treibel, T.A.; Roussin, I.; Prasad, S.K.; Duncan, A.M.; Brookes, C.; McDonagh, T.A.; Sharma, R. Treatment of Left Ventricular Non-Compaction with Cardiac Resynchronization Therapy. QJM 2013, 106, 575–579. [Google Scholar] [CrossRef]
- Chin, T.K.; Perloff, J.K.; Williams, R.G.; Jue, K.; Mohrmann, R. Isolated Noncompaction of Left Ventricular Myocardium. A Study of Eight Cases. Circulation 1990, 82, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Finsterer, J. Left Ventricular Non-Compaction and Its Cardiac and Neurologic Implications. Heart Fail. Rev. 2010, 15, 589–603. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, E.; Marshalko, S.J.; Jaffe, C.C.; Hui, P. Isolated Noncompaction of the Ventricular Myocardium: Clinical and Molecular Aspects of a Rare Cardiomyopathy. Lab. Investig. 2002, 82, 117–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritter, M.; Oechslin, E.; Sutsch, G.; Attenhofer, C.; Schneider, J.; Jenni, R. Isolated Noncompaction of the Myocardium in Adults. Mayo Clin. Proc. 1997, 72, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Samsa, L.A.; Yang, B.; Liu, J. Embryonic Cardiac Chamber Maturation: Trabeculation, Conduction, and Cardiomyocyte Proliferation. Am. J. Med. Genet. C Semin. Med. Genet. 2013, 163C, 157–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedmera, D.; Pexieder, T.; Vuillemin, M.; Thompson, R.P.; Anderson, R.H. Developmental Patterning of the Myocardium. Anat. Rec. 2000, 258, 319–337. [Google Scholar] [CrossRef]
- Risebro, C.A.; Riley, P.R. Formation of the Ventricles. Sci. World J. 2006, 6, 1862–1880. [Google Scholar] [CrossRef]
- Pasumarthi, K.B.; Field, L.J. Cardiomyocyte Cell Cycle Regulation. Circ. Res. 2002, 90, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, H.; Qu, X.; Chang, C.P.; Shou, W. Molecular Mechanism of Ventricular Trabeculation/Compaction and the Pathogenesis of the Left Ventricular Noncompaction Cardiomyopathy (LVNC). Am. J. Med. Genet. C Semin. Med. Genet. 2013, 163C, 144–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oechslin, E.; Jenni, R. Left Ventricular Non-Compaction Revisited: A Distinct Phenotype with Genetic Heterogeneity? Eur. Heart J. 2011, 32, 1446–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luedde, M.; Ehlermann, P.; Weichenhan, D.; Will, R.; Zeller, R.; Rupp, S.; Muller, A.; Steen, H.; Ivandic, B.T.; Ulmer, H.E.; et al. Severe Familial Left Ventricular Non-Compaction Cardiomyopathy Due to a Novel Troponin T (TNNT2) Mutation. Cardiovasc. Res. 2010, 86, 452–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duquette, P.M.; Lamarche-Vane, N. Rho GTPases in Embryonic Development. Small GTPases 2014, 5, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, C.; Lu, X.; Liu, M.; Feng, Q. Rac1 Signaling Is Critical to Cardiomyocyte Polarity and Embryonic Heart Development. J. Am. Heart Assoc. 2014, 3, e001271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boczonadi, V.; Gillespie, R.; Keenan, I.; Ramsbottom, S.A.; Donald-Wilson, C.; Al Nazer, M.; Humbert, P.; Schwarz, R.J.; Chaudhry, B.; Henderson, D.J. Scrib: Rac1 Interactions Are Required for the Morphogenesis of the Ventricular Myocardium. Cardiovasc. Res. 2014, 104, 103–115. [Google Scholar] [CrossRef] [Green Version]
- McFadden, D.G.; Barbosa, A.C.; Richardson, J.A.; Schneider, M.D.; Srivastava, D.; Olson, E.N. The Hand1 and Hand2 Transcription Factors Regulate Expansion of the Embryonic Cardiac Ventricles in a Gene Dosage-Dependent Manner. Development 2005, 132, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Glogauer, M.; Marchal, C.C.; Zhu, F.; Worku, A.; Clausen, B.E.; Foerster, I.; Marks, P.; Downey, G.P.; Dinauer, M.; Kwiatkowski, D.J. Rac1 Deletion in Mouse Neutrophils Has Selective Effects on Neutrophil Functions. J. Immunol. 2003, 170, 5652–5657. [Google Scholar] [CrossRef] [Green Version]
- Muzumdar, M.D.; Tasic, B.; Miyamichi, K.; Li, L.; Luo, L. A Global Double-Fluorescent Cre Reporter Mouse. Genesis 2007, 45, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Xiang, F.L.; Lu, X.; Hammoud, L.; Zhu, P.; Chidiac, P.; Robbins, J.; Feng, Q. Cardiomyocyte-Specific Overexpression of Human Stem Cell Factor Improves Cardiac Function and Survival Post Myocardial Infarction in Mice. Circulation 2009, 120, 1065–1074. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lu, X.; Xiang, F.L.; Poelmann, R.E.; Gittenberger-de Groot, A.C.; Robbins, J.; Feng, Q. Nitric Oxide Synthase-3 Deficiency Results in Hypoplastic Coronary Arteries and Postnatal Myocardial Infarction. Eur. Heart J. 2014, 35, 920–931. [Google Scholar] [CrossRef] [Green Version]
- Engineer, A.; Saiyin, T.; Lu, X.; Kucey, A.S.; Urquhart, B.L.; Drysdale, T.A.; Norozi, K.; Feng, Q. Sapropterin Treatment Prevents Congenital Heart Defects Induced by Pregestational Diabetes Mellitus in Mice. J. Am. Heart Assoc. 2018, 7, e009624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boer, B.A.; van den Berg, G.; de Boer, P.A.; Moorman, A.F.; Ruijter, J.M. Growth of the Developing Mouse Heart: An Interactive Qualitative and Quantitative 3D Atlas. Dev. Biol. 2012, 368, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Phillips, H.M.; Hildreth, V.; Peat, J.D.; Murdoch, J.N.; Kobayashi, K.; Chaudhry, B.; Henderson, D.J. Non-Cell-Autonomous Roles for the Planar Cell Polarity Gene Vangl2 in Development of the Coronary Circulation. Circ. Res. 2008, 102, 615–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, H.M.; Rhee, H.J.; Murdoch, J.N.; Hildreth, V.; Peat, J.D.; Anderson, R.H.; Copp, A.J.; Chaudhry, B.; Henderson, D.J. Disruption of Planar Cell Polarity Signaling Results in Congenital Heart Defects and Cardiomyopathy Attributable to Early Cardiomyocyte Disorganization. Circ. Res. 2007, 101, 137–145. [Google Scholar] [CrossRef]
- Sinha, T.; Wang, B.; Evans, S.; Wynshaw-Boris, A.; Wang, J. Disheveled Mediated Planar Cell Polarity Signaling Is Required in the Second Heart Field Lineage for Outflow Tract Morphogenesis. Dev. Biol. 2012, 370, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Simons, M.; Mlodzik, M. Planar Cell Polarity Signaling: From Fly Development to Human Disease. Annu. Rev. Genet. 2008, 42, 517–540. [Google Scholar] [CrossRef] [Green Version]
- Sedmera, D.; Hu, N.; Weiss, K.M.; Keller, B.B.; Denslow, S.; Thompson, R.P. Cellular Changes in Experimental Left Heart Hypoplasia. Anat. Rec. 2002, 267, 137–145. [Google Scholar] [CrossRef]
- Moses, K.A.; DeMayo, F.; Braun, R.M.; Reecy, J.L.; Schwartz, R.J. Embryonic Expression of an Nkx2-5/Cre Gene Using ROSA26 Reporter Mice. Genesis 2001, 31, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Hamblet, N.S.; Lijam, N.; Ruiz-Lozano, P.; Wang, J.; Yang, Y.; Luo, Z.; Mei, L.; Chien, K.R.; Sussman, D.J.; Wynshaw-Boris, A. Dishevelled 2 Is Essential for Cardiac Outflow Tract Development, Somite Segmentation and Neural Tube Closure. Development 2002, 129, 5827–5838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, C.; Liu, Y.; Lu, X.; Kim, M.; Drysdale, T.A.; Feng, Q. Rac1 Signaling Is Required for Anterior Second Heart Field Cellular Organization and Cardiac Outflow Tract Development. J. Am. Heart Assoc. 2016, 5, e002508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlessinger, K.; Hall, A.; Tolwinski, N. Wnt Signaling Pathways Meet Rho GTPases. Genes Dev. 2009, 23, 265–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niehrs, C. The Complex World of WNT Receptor Signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef]
- Engineer, A.; Saiyin, T.; Greco, E.R.; Feng, Q. Say NO to ROS: Their Roles in Embryonic Heart Development and Pathogenesis of Congenital Heart Defects in Maternal Diabetes. Antioxidants 2019, 8, 436. [Google Scholar] [CrossRef] [Green Version]
- Covarrubias, L.; Hernandez-Garcia, D.; Schnabel, D.; Salas-Vidal, E.; Castro-Obregon, S. Function of Reactive Oxygen Species during Animal Development: Passive or Active? Dev. Biol. 2008, 320, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, A.R.; Ghosh, I.; Datta, K. Excessive Reactive Oxygen Species Induces Apoptosis in Fibroblasts: Role of Mitochondrially Accumulated Hyaluronic Acid Binding Protein 1 (HABP1/P32/GC1qR). Exp. Cell Res. 2008, 314, 651–667. [Google Scholar] [CrossRef]
- Moazzen, H.; Lu, X.; Liu, M.; Feng, Q. Pregestational Diabetes Induces Fetal Coronary Artery Malformation via Reactive Oxygen Species Signaling. Diabetes 2015, 64, 1431–1443. [Google Scholar] [CrossRef] [Green Version]
- Moazzen, H.; Lu, X.; Ma, N.L.; Velenosi, T.J.; Urquhart, B.L.; Wisse, L.J.; Groot, A.C.; Feng, Q. N-Acetylcysteine Prevents Congenital Heart Defects Induced by Pregestational Diabetes. Cardiovasc. Diabetol. 2014, 13, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moazzen, H.; Wu, Y.; Engineer, A.; Lu, X.; Aulakh, S.; Feng, Q. NOX2 Is Critical to Endocardial to Mesenchymal Transition and Heart Development. Oxid. Med. Cell Longev. 2020, 2020, 1679045. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mice | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| Nkx2.5-Cre | TGCCACGACCAAGTGACAGC | CCAGGTTACGGATATAGTTCATG |
| Rac1f/f | TCCAATCTGTGCTGCCCATC | GATGCTTCTAGGGGTGAGCC |
| mTmG | CTCTGCTGCCTCCTGGCTTCT | CGAGGCGGATCACAAGCAATA Mutant reverse: TCAATGGGCGGGGGTCGTT |
| Gene | Forward (5′-3′) | Primer Sequence (5′-3′) |
|---|---|---|
| Rac1 | TTGTCCAGCTGTGTCCCATA | AACCTGCCTGCTCATCAGTT |
| Gata4 | GCCTGCGATGTCTGAGTGAC | CACTATGGGCACAGCAGCTC |
| Nkx2.5 | GACAGCGGCAGGACCAGACT | CGTTGTAGCCATAGGCATTG |
| Tbx5 | AGGAGCACAGTGAGGCACAA | GGGCCAGAGACACCATTCTC |
| Tbx20 | CACCTATGGGGAAGAGGATGTTC | GTCGCTATGGATGCTGTACTGGT |
| Mef2c | TACCCCGGTGGTTTCCGTAG | CCCAACTGACTGAGGGCAGA |
| Scrib | AGGAGGAGAACAGGGATGAGGAG | CCTTTGTAGGGGGTAGAGCCTTT |
| Bmp10 | CCACTCGGATCAGGAGGAAC | CACACAGCAGGCTTTGGAAG |
| Hand1 | TGGCTACCAGTTACATCGCCTAC | GTGCGCCCTTTAATCCTCTTCT |
| Hand2 | GCTACATCGCCTACCTCATGGAT | TCTTGTCGTTGCTGCTCACTGT |
| 28S | ACATTGTTCCAACATGCCAG | TTGAAAATCCGGGGGAGAG |
| Bifid Apex | VSD | DORV | Overriding Aorta | Thin Compact Myocardium | |
|---|---|---|---|---|---|
| N = 17 | 17 | 17 | 11 | 6 | 17 |
| % | 100 | 100 | 64.7 | 35.3 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leung, C.; Engineer, A.; Kim, M.Y.; Lu, X.; Feng, Q. Myocardium-Specific Deletion of Rac1 Causes Ventricular Noncompaction and Outflow Tract Defects. J. Cardiovasc. Dev. Dis. 2021, 8, 29. https://doi.org/10.3390/jcdd8030029
Leung C, Engineer A, Kim MY, Lu X, Feng Q. Myocardium-Specific Deletion of Rac1 Causes Ventricular Noncompaction and Outflow Tract Defects. Journal of Cardiovascular Development and Disease. 2021; 8(3):29. https://doi.org/10.3390/jcdd8030029
Chicago/Turabian StyleLeung, Carmen, Anish Engineer, Mella Y. Kim, Xiangru Lu, and Qingping Feng. 2021. "Myocardium-Specific Deletion of Rac1 Causes Ventricular Noncompaction and Outflow Tract Defects" Journal of Cardiovascular Development and Disease 8, no. 3: 29. https://doi.org/10.3390/jcdd8030029
APA StyleLeung, C., Engineer, A., Kim, M. Y., Lu, X., & Feng, Q. (2021). Myocardium-Specific Deletion of Rac1 Causes Ventricular Noncompaction and Outflow Tract Defects. Journal of Cardiovascular Development and Disease, 8(3), 29. https://doi.org/10.3390/jcdd8030029