Outflow Tract Formation—Embryonic Origins of Conotruncal Congenital Heart Disease




{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Embryonic Origins of the OFT
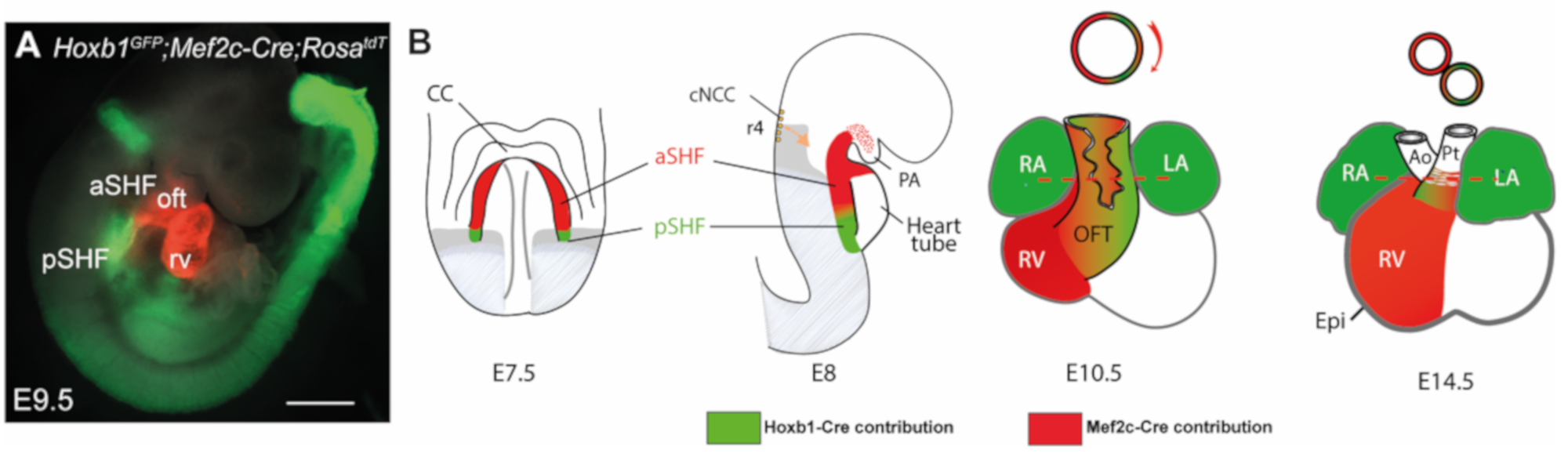
2.1. Myocardial Progenitors
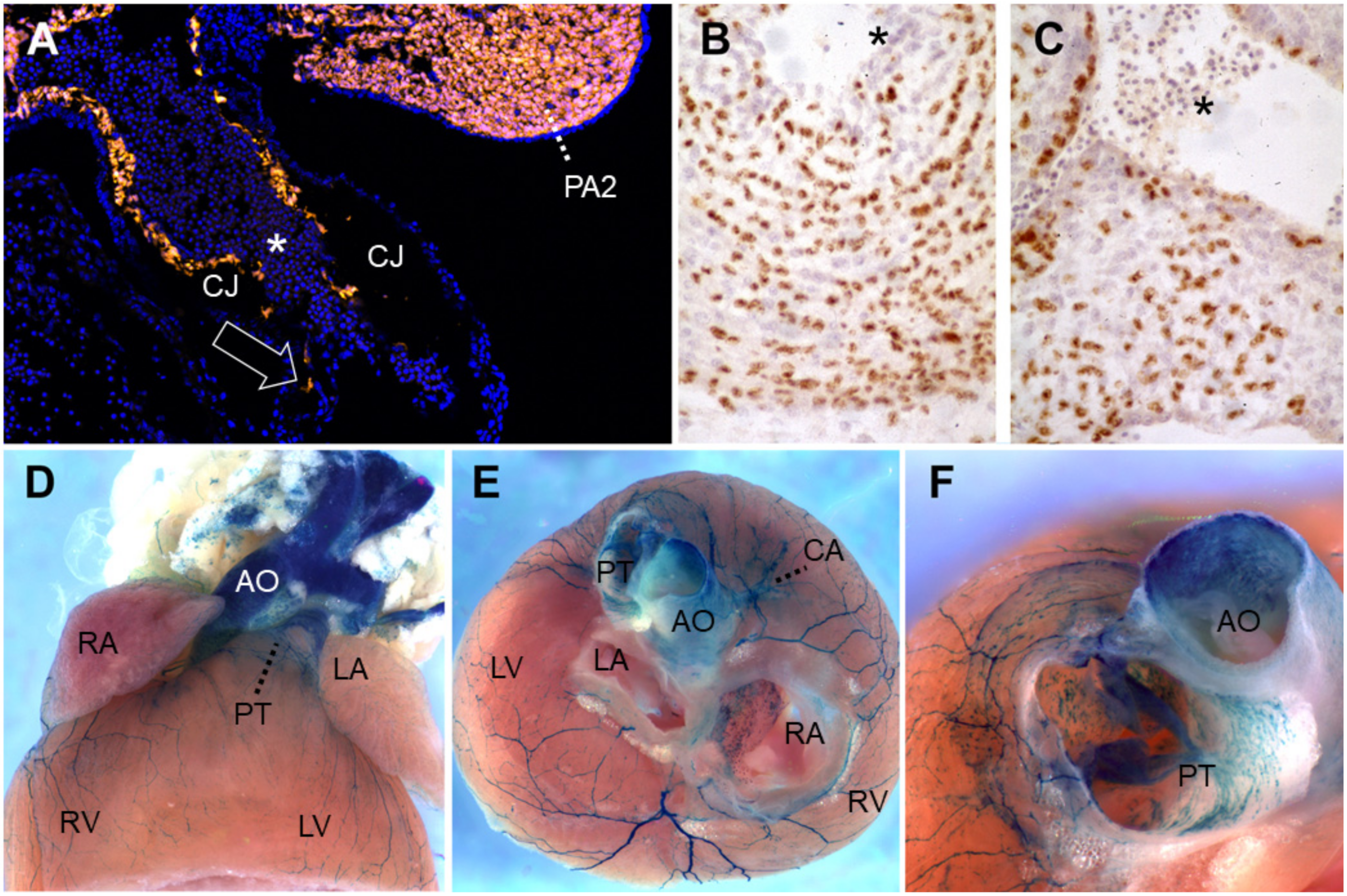
2.2. Endocardial Cushions
2.3. Cardiac Neural Crest
3. Retinoic Acid Signaling
3.1. RA Is Required for the Differentiation of Heart Progenitors into the Myocardium of the OFT
3.2. Cell-Autonomous RA Signaling Is Dispensable for the Migration and Fate Differentiation of CNCC
3.3. RA Signaling Defects Are Associated with DiGeorge Syndrome
4. Hox Transcription Factors
5. Other Signaling Pathways
6. Future Direction and Clinical Implications

Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hoffman, J. The global burden of congenital heart disease. Cardiovasc. J. Afr. 2013, 24, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, J.I.; Kaplan, S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 2002, 39, 1890–1900. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.H.; Tretter, J.T.; Spicer, D.E.; Mori, S. The Fate of the Outflow Tract Septal Complex in Relation to the Classification of Ventricular Septal Defects. J. Cardiovasc. Dev. Dis. 2019, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Bajolle, F.; Zaffran, S.; Kelly, R.G.; Hadchouel, J.; Bonnet, D.; Brown, N.A.; Buckingham, M.E. Rotation of the myocardial wall of the outflow tract is implicated in the normal positioning of the great arteries. Circ. Res. 2006, 98, 421–428. [Google Scholar] [CrossRef]
- Saga, Y.; Miyagawa-Tomita, S.; Takagi, A.; Kitajima, S.; Miyazaki, J.; Inoue, T. MesP1 is expressed in the heart precursor cells and required for the formation of a single heart tube. Development 1999, 126, 3437–3447. [Google Scholar] [PubMed]
- Hatada, Y.; Stern, C.D. A fate map of the epiblast of the early chick embryo. Development 1994, 120, 2879–2889. [Google Scholar] [PubMed]
- Stalsberg, H.; DeHaan, R.L. The precardiac areas and formation of the tubular heart in the chick embryo. Dev. Biol. 1969, 19, 128–159. [Google Scholar] [CrossRef]
- Christoffels, V.; Jensen, B. Cardiac Morphogenesis: Specification of the Four-Chambered Heart. Cold Spring Harb. Perspect. Biol. 2020, 12, a037143. [Google Scholar] [CrossRef]
- Kelly, G.R.; Buckingham, M.E.; Moorman, A.F. Heart fields and cardiac morphogenesis. Cold Spring Harb. Perspect. Med. 2014, 4, a015750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaffran, S.; Kelly, R.G. New developments in the second heart field. Differentiation 2012, 84, 17–24. [Google Scholar] [CrossRef]
- Prall, O.W.; Menon, M.K.; Solloway, M.J.; Watanabe, Y.; Zaffran, S.; Bajolle, F.; Biben, C.; McBride, J.J.; Robertson, B.R.; Chaulet, H.; et al. An Nkx2-5/Bmp2/Smad1 negative feedback loop controls heart progenitor specification and proliferation. Cell 2007, 128, 947–959. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Nomura-Kitabayashi, A.; Sultana, N.; Cai, W.; Cai, X.; Moon, A.M.; Cai, C.L. Mesodermal Nkx2.5 is necessary and sufficient for early second heart field development. Dev. Biol. 2014, 390, 68–79. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.L.; Liang, X.; Shi, Y.; Chu, P.H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, N.; Roux, M.; Ryckebüsch, L.; Niederreither, K.; Dollé, P.; Moon, A.; Capecchi, M.; Zaffran, S. Hox genes define distinct progenitor sub-domains within the second heart field. Dev. Biol. 2011, 353, 266–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanovic, S.; Laforest, B.; Desvignes, J.P.; Lescroart, F.; Argiro, L.; Maurel-Zaffran, C.; Salgado, D.; Plaindoux, E.; De Bono, C.; Pazur, K.; et al. Hox-dependent coordination of mouse cardiac progenitor cell patterning and differentiation. Elife 2020, 9, e55124. [Google Scholar] [CrossRef] [PubMed]
- Briggs, L.E.; Kakarla, J.; Wessels, A. The pathogenesis of atrial and atrioventricular septal defects with special emphasis on the role of the dorsal mesenchymal protrusion. Differentiation 2012, 84, 117–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bono, C.; Thellier, C.; Bertrand, N.; Sturny, R.; Jullian, E.; Cortes, C.; Stefanovic, S.; Zaffran, S.; Théveniau-Ruissy, M.; Kelly, R.G. T-box genes and retinoic acid signaling regulate the segregation of arterial and venous pole progenitor cells in the murine second heart field. Hum. Mol. Genet. 2018, 27, 3747–3760. [Google Scholar] [CrossRef] [PubMed]
- Verzi, M.P.; McCulley, D.J.; De Val, S.; Dodou, E.; Black, B.L. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev. Biol. 2005, 287, 134–145. [Google Scholar] [CrossRef] [Green Version]
- Dodou, E.; Verzi, M.P.; Anderson, J.P.; Xu, S.M.; Black, B.L. Mef2c is a direct transcriptional target of ISL1 and GATA factors in the anterior heart field during mouse embryonic development. Development 2004, 131, 3931–3942. [Google Scholar] [CrossRef] [Green Version]
- Goddeeris, M.M.; Schwartz, R.; Klingensmith, J.; Meyers, E.N. Independent requirements for Hedgehog signaling by both the anterior heart field and neural crest cells for outflow tract development. Development 2007, 134, 1593–1604. [Google Scholar] [CrossRef] [Green Version]
- Kelly, G.R.; Brown, N.A.; Buckingham, M.E. The arterial pole of the mouse heart forms from Fgf10-expressing cells in pharyngeal mesoderm. Dev. Cell 2001, 1, 435–440. [Google Scholar] [CrossRef] [Green Version]
- Golzio, C.; Havis, E.; Daubas, P.; Nuel, G.; Babarit, C.; Munnich, A.; Vekemans, M.; Zaffran, S.; Lyonnet, S.; Etchevers, H.C. ISL1 directly regulates FGF10 transcription during human cardiac outflow formation. PLoS ONE 2012, 7, e30677. [Google Scholar] [CrossRef] [Green Version]
- Stefanovic, S.; Zaffran, S. Mechanisms of retinoic acid signaling during cardiogenesis. Mech. Dev. 2017, 143, 9–19. [Google Scholar] [CrossRef]
- Hochgreb, T.; Linhares, V.L.; Menezes, D.C.; Sampaio, A.C.; Yan, C.Y.; Cardoso, W.V.; Rosenthal, N.; Xavier-Neto, J. A caudorostral wave of RALDH2 conveys anteroposterior information to the cardiac field. Development 2003, 130, 5363–5374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diman, N.Y.; Remacle, S.; Bertrand, N.; Picard, J.J.; Zaffran, S.; Rezsohazy, R. A retinoic acid responsive Hoxa3 transgene expressed in embryonic pharyngeal endoderm, cardiac neural crest and a subdomain of the second heart field. PLoS ONE 2011, 6, e27624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buenrostro, J.D.; Wu, B.; Litzenburger, U.M.; Ruff, D.; Gonzales, M.L.; Snyder, M.P.; Chang, H.Y.; Greenleaf, W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 2015, 523, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Moorman, F.A.; Christoffels, V.M. Cardiac chamber formation: Development, genes, and evolution. Physiol. Rev. 2003, 83, 1223–1267. [Google Scholar] [CrossRef]
- Eisenberg, M.L.; Markwald, R.R. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ. Res. 1995, 77, 1–6. [Google Scholar] [CrossRef]
- MacGrogan, D.; Luxán, G.; Driessen-Mol, A.; Bouten, C.; Baaijens, F.; de la Pompa, J.L. How to make a heart valve: From embryonic development to bioengineering of living valve substitutes. Cold Spring Harb. Perspect Med. 2014, 4, a013912. [Google Scholar] [CrossRef]
- Poelmann, R.E.; Gittenberger-de Groot, A.C. Hemodynamics in Cardiac Development. J. Cardiovasc. Dev. Dis. 2018, 5, 54. [Google Scholar] [CrossRef] [Green Version]
- McCulley, D.J.; Kang, J.O.; Martin, J.F.; Black, B.L. BMP4 is required in the anterior heart field and its derivatives for endocardial cushion remodeling, outflow tract septation, and semilunar valve development. Dev. Dyn. 2008, 237, 3200–3209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Lu, M.F.; Schwartz, R.J.; Martin, J.F. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development 2005, 132, 5601–5611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habets, P.E.; Moorman, A.F.; Clout, D.E.; van Roon, M.A.; Lingbeek, M.; van Lohuizen, M.; Campione, M.; Christoffels, V.M. Cooperative action of Tbx2 and Nkx2.5 inhibits ANF expression in the atrioventricular canal: Implications for cardiac chamber formation. Genes Dev. 2002, 16, 1234–1246. [Google Scholar] [CrossRef] [Green Version]
- Hoogaars, W.M.; Tessari, A.; Moorman, A.F.; de Boer, P.A.; Hagoort, J.; Soufan, A.T.; Campione, M.; Christof-fels, V.M. The transcriptional repressor Tbx3 delineates the developing central conduction system of the heart. Cardiovasc. Res. 2004, 62, 489–499. [Google Scholar] [CrossRef]
- Yamada, M.; Revelli, J.P.; Eichele, G.; Barron, M.; Schwartz, R.J. Expression of chick Tbx-2, Tbx-3, and Tbx-5 genes during early heart development: Evidence for BMP2 induction of Tbx2. Dev. Biol. 2000, 228, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Rutenberg, J.B.; Fischer, A.; Jia, H.; Gessler, M.; Zhong, T.P.; Mercola, M. Developmental patterning of the cardiac atrioventricular canal by Notch and Hairy-related transcription factors. Development 2006, 133, 4381–4390. [Google Scholar] [CrossRef] [Green Version]
- Papoutsi, T.; Luna-Zurita, L.; Prados, B.; Zaffran, S.; de la Pompa, J.L. Bmp2 and Notch cooperate to pattern the embryonic endocardium. Development 2018, 145. [Google Scholar] [CrossRef] [Green Version]
- De Lange, F.J.; Moorman, A.F.; Anderson, R.H.; Männer, J.; Soufan, A.T.; de Gier-de Vries, C.; Schneider, M.D.; Webb, S.; van den Hoff, M.J.; Christoffels, V.M. Lineage and morphogenetic analysis of the cardiac valves. Circ. Res. 2004, 95, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Rowitch, D.H.; Soriano, P.; McMahon, A.P.; Sucov, H.M. Fate of the mammalian cardiac neural crest. Development 2000, 127, 1607–1616. [Google Scholar]
- Odelin, G.; Faure, E.; Coulpier, F.; Di Bonito, M.; Bajolle, F.; Studer, M.; Avierinos, J.F.; Charnay, P.; Topilko, P.; Zaf-fran, S. Krox20 defines a subpopulation of cardiac neural crest cells contributing to arterial valves and bicuspid aortic valve. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura-Kitabayashi, A.; Phoon, C.K.; Kishigami, S.; Rosenthal, J.; Yamauchi, Y.; Abe, K.; Yamamura, K.; Samtani, R.; Lo, C.W.; Mishina, Y. Outflow tract cushions perform a critical valve-like function in the early embryonic heart requiring BMPRIA-mediated signaling in cardiac neural crest. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1617–H1628. [Google Scholar] [CrossRef] [Green Version]
- Eley, L.; Alqahtani, A.M.; MacGrogan, D.; Richardson, R.V.; Murphy, L.; Salguero-Jimenez, A.; Sintes Rodriguez San Pedro, M.; Tiurma, S.; McCutcheon, L.; Gilmore, A.; et al. A novel source of arterial valve cells linked to bicuspid aortic valve without raphe in mice. Elife 2018, 7, e34110. [Google Scholar] [CrossRef] [PubMed]
- Mifflin, J.J.; Dupuis, L.; Alcala, N.E.; Russell, L.G.; Kern, C.B. Intercalated cushion cells within the cardiac outflow tract are derived from the myocardial troponin T type 2 (Tnnt2) Cre lineage. Dev. Dyn. 2018, 247, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, W.; Li, W.; Li, Y.; Priest, J.R.; Zhou, B.; Wang, J.; Zhou, Z. Single-Cell RNA-Seq of the Developing Cardiac Outflow Tract Reveals Convergent Development of the Vascular Smooth Muscle Cells. Cell Rep. 2019, 28, 1346–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldo, K.L.; Hutson, M.R.; Ward, C.C.; Zdanowicz, M.; Stadt, H.A.; Kumiski, D.; Abu-Issa, R.; Kirby, M.L. Secondary heart field contributes myocardium and smooth muscle to the arterial pole of the developing heart. Dev. Biol. 2005, 281, 78–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etchevers, C.H.; Dupin, E.; le Douarin, N.M. The diverse neural crest: From embryology to human pathology. Development 2019, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Lièvre, C.; Le Douarin, N. Role of mesectoderm in the genesis of aortic arches of the bird embryo. Comptes Rendus Acad. Hebd. Seances Acad. Sci. D 1973, 276, 383–386. [Google Scholar]
- Hutchins, E.J.; Kunttas, E.; Piacentino, M.L.; Howard, A.G.A.t.; Bronner, M.E.; Uribe, R.A. Migration and diversification of the vagal neural crest. Dev. Biol. 2018, 444 (Suppl. S1), S98–S109. [Google Scholar] [CrossRef] [PubMed]
- Kuratani, C.S.; Kirby, M.L. Initial migration and distribution of the cardiac neural crest in the avian embryo: An introduction to the concept of the circumpharyngeal crest. Am. J. Anat. 1991, 191, 215–227. [Google Scholar] [CrossRef]
- Boot, M.J.; Gittenberger-De Groot, A.C.; Van Iperen, L.; Hierck, B.P.; Poelmann, R.E. Spatiotemporally separated cardiac neural crest subpopulations that target the outflow tract septum and pharyngeal arch arteries. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 2003, 275, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Bergwerff, M.; Verberne, M.E.; DeRuiter, M.C.; Poelmann, R.E.; Gittenberger-de Groot, A.C. Neural crest cell contribution to the developing circulatory system: Implications for vascular morphology? Circ. Res. 1998, 82, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Etchevers, H.C.; Vincent, C.; Le Douarin, N.M.; Couly, G.F. The cephalic neural crest provides pericytes and smooth muscle cells to all blood vessels of the face and forebrain. Development 2001, 128, 1059–1068. [Google Scholar]
- Teillet, A.M.; Kalcheim, C.; le Douarin, N.M. Formation of the dorsal root ganglia in the avian embryo: Segmental origin and migratory behavior of neural crest progenitor cells. Dev. Biol. 1987, 120, 329–347. [Google Scholar] [CrossRef]
- Le Lièvre, C.S.; Le Douarin, N.M. Mesenchymal derivatives of the neural crest: Analysis of chimaeric quail and chick embryos. J. Embryol. Exp. Morphol. 1975, 34, 125–154. [Google Scholar] [PubMed]
- Poelmann, R.E.; Gittenberger-de Groot, A.C. A subpopulation of apoptosis-prone cardiac neural crest cells targets to the venous pole: Multiple functions in heart development? Dev. Biol. 1999, 207, 271–286. [Google Scholar] [CrossRef]
- Kirby, M.L.; Stewart, D.E. Neural crest origin of cardiac ganglion cells in the chick embryo: Identification and extirpation. Dev. Biol. 1983, 97, 433–443. [Google Scholar] [CrossRef]
- Verberne, M.E.; Gittenberger-de Groot, A.C.; Poelmann, R.E. Lineage and development of the parasympathetic nervous system of the embryonic chick heart. Anat. Embryol. 1998, 198, 171–184. [Google Scholar] [CrossRef]
- Arima, Y.; Miyagawa-Tomita, S.; Maeda, K.; Asai, R.; Seya, D.; Minoux, M.; Rijli, F.M.; Nishiyama, K.; Kim, K.S.; Uchijima, Y.; et al. Preotic neural crest cells contribute to coronary artery smooth muscle involving endothelin signalling. Nat. Commun. 2012, 3, 1267. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.; Engleka, K.A.; Rentschler, S.L.; Manderfield, L.J.; Li, L.; Yuan, L.; Epstein, J.A. Cardiac neural crest orchestrates remodeling and functional maturation of mouse semilunar valves. J. Clin. Invest. 2011, 121, 422–430. [Google Scholar] [CrossRef] [Green Version]
- Phillips, H.M.; Mahendran, P.; Singh, E.; Anderson, R.H.; Chaudhry, B.; Henderson, D.J. Neural crest cells are required for correct positioning of the developing outflow cushions and pattern the arterial valve leaflets. Cardiovasc. Res. 2013, 99, 452–460. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Colbert, M.C.; Robbins, J. Neural crest cells retain multipotential characteristics in the developing valves and label the cardiac conduction system. Circ. Res. 2006, 98, 1547–1554. [Google Scholar] [CrossRef] [Green Version]
- El Robrini, N.; Etchevers, H.C.; Ryckebüsch, L.; Faure, E.; Eudes, N.; Niederreither, K.; Zaffran, S.; Bertrand, N. Cardiac outflow morphogenesis depends on effects of retinoic acid signaling on multiple cell lineages. Dev. Dyn. 2016, 245, 388–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatzistergos, K.E.; Takeuchi, L.M.; Saur, D.; Seidler, B.; Dymecki, S.M.; Mai, J.J.; White, I.A.; Balkan, W.; Kanashiro-Takeuchi, R.M.; Schally, A.V.; et al. cKit+ cardiac progenitors of neural crest origin. Proc. Natl. Acad. Sci. USA 2015, 112, 13051–13056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soldatov, R.; Kaucka, M.; Kastriti, M.E.; Petersen, J.; Chontorotzea, T.; Englmaier, L.; Akkuratova, N.; Yang, Y.; Häring, M.; Dyachuk, V.; et al. Spatiotemporal structure of cell fate decisions in murine neural crest. Science 2019, 364. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Martik, M.L.; Li, Y.; Bronner, M.E. Cardiac neural crest contributes to cardiomyocytes in amniotes and heart regeneration in zebrafish. Elife 2019, 8, e47929. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Yost, H.J. Cardiac neural crest contributes to cardiomyogenesis in zebrafish. Dev. Biol. 2003, 257, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.X.; Zdanowicz, M.; Young, L.; Kumiski, D.; Leatherbury, L.; Kirby, M.L. Cardiac neural crest in zebrafish embryos contributes to myocardial cell lineage and early heart function. Dev. Dyn. 2003, 226, 540–550. [Google Scholar] [CrossRef]
- Lee, Y.H.; Saint-Jeannet, J.P. Cardiac neural crest is dispensable for outflow tract septation in Xenopus. Development 2011, 138, 2025–2034. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, S.; Ezin, M.; Bronner, M.E. Reprogramming Axial Level Identity to Rescue Neural-Crest-Related Congenital Heart Defects. Dev. Cell. 2020, 53, 300–315. [Google Scholar] [CrossRef]
- Poelmann, R.E.; Gittenberger-de Groot, A.C.; Biermans, M.W.M.; Dolfing, A.I.; Jagessar, A.; van Hattum, S.; Hoogenboom, A.; Wisse, L.J.; Vicente-Steijn, R.; de Bakker, M.A.G.; et al. Outflow tract septation and the aortic arch system in reptiles: Lessons for understanding the mammalian heart. Evodevo 2017, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Bockman, D.E.; Kirby, M.L. Dependence of thymus development on derivatives of the neural crest. Science 1984, 223, 498–500. [Google Scholar] [CrossRef]
- Zaffran, S.; Odelin, G.; Stefanovic, S.; Lescroart, F.; Etchevers, H.C. Ectopic expression of Hoxb1 induces cardiac and craniofacial malformations. Genesis 2018, 56, e23221. [Google Scholar] [CrossRef] [Green Version]
- Roux, M.; Laforest, B.; Eudes, N.; Bertrand, N.; Stefanovic, S.; Zaffran, S. Hoxa1 and Hoxb1 are required for pharyngeal arch artery development. Mech. Dev. 2017, 143, 1–8. [Google Scholar] [CrossRef]
- Frank, D.U.; Fotheringham, L.K.; Brewer, J.A.; Muglia, L.J.; Tristani-Firouzi, M.; Capecchi, M.R.; Moon, A.M. An Fgf8 mouse mutant phenocopies human 22q11 deletion syndrome. Development 2002, 129, 4591–4603. [Google Scholar] [PubMed]
- Bajolle, F.; Zaffran, S.; Meilhac, S.M.; Dandonneau, M.; Chang, T.; Kelly, R.G.; Buckingham, M.E. Myocardium at the base of the aorta and pulmonary trunk is prefigured in the outflow tract of the heart and in subdomains of the second heart field. Dev. Biol. 2008, 313, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Scherptong, R.W.; Jongbloed, M.R.; Wisse, L.J.; Vicente-Steijn, R.; Bartelings, M.M.; Poelmann, R.E.; Schalij, M.J.; Gittenberger-De Groot, A.C. Morphogenesis of outflow tract rotation during cardiac development: The pulmonary push concept. Dev. Dyn. 2012, 241, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Baardman, M.E.; Zwier, M.V.; Wisse, L.J.; Gittenberger-de Groot, A.C.; Kerstjens-Frederikse, W.S.; Hofstra, R.M.; Jurdzinski, A.; Hierck, B.P.; Jongbloed, M.R.; Berger, R.M.; et al. Common arterial trunk and ventricular non-compaction in Lrp2 knockout mice indicate a crucial role of LRP2 in cardiac development. Dis. Models Mech. 2016, 9, 413–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christ, A.; Marczenke, M.; Willnow, T.E. LRP2 controls sonic hedgehog-dependent differentiation of cardiac progenitor cells during outflow tract formation. Hum. Mol. Genet. 2020, 29, 3183–3196. [Google Scholar] [CrossRef] [PubMed]
- Theis, J.L.; Vogler, G.; Missinato, M.A.; Li, X.; Nielsen, T.; Zeng, X.I.; Martinez-Fernandez, A.; Walls, S.M.; Kervadec, A.; Kezos, J.N.; et al. Patient-specific genomics and cross-species functional analysis implicate LRP2 in hypoplastic left heart syndrome. Elife 2020, 9, e59554. [Google Scholar] [CrossRef]
- Sakabe, M.; Kokubo, H.; Nakajima, Y.; Saga, Y. Ectopic retinoic acid signaling affects outflow tract cushion development through suppression of the myocardial Tbx2-Tgfβ2 pathway. Development 2012, 139, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Yasui, H.; Nakazawa, M.; Morishima, M.; Miyagawa-Tomita, S.; Momma, K. Morphological observations on the pathogenetic process of transposition of the great arteries induced by retinoic acid in mice. Circulation 1995, 91, 2478–2486. [Google Scholar] [CrossRef]
- Golzio, C.; Martinovic-Bouriel, J.; Thomas, S.; Mougou-Zrelli, S.; Grattagliano-Bessieres, B.; Bonniere, M.; Delahaye, S.; Munnich, A.; Encha-Razavi, F.; Lyonnet, S.; et al. Matthew-Wood syndrome is caused by truncating mutations in the retinol-binding protein receptor gene STRA6. Am. J. Hum. Genet. 2007, 80, 1179–1187. [Google Scholar] [CrossRef] [Green Version]
- Pasutto, F.; Sticht, H.; Hammersen, G.; Gillessen-Kaesbach, G.; Fitzpatrick, D.R.; Nürnberg, G.; Brasch, F.; Schirmer-Zimmermann, H.; Tolmie, J.L.; Chitayat, D.; et al. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am. J. Hum. Genet. 2007, 80, 550–560. [Google Scholar] [CrossRef] [Green Version]
- Chassaing, N.; Golzio, C.; Odent, S.; Lequeux, L.; Vigouroux, A.; Martinovic-Bouriel, J.; Tiziano, F.D.; Masini, L.; Piro, F.; Maragliano, G.; et al. Phenotypic spectrum of STRA6 mutations: From Matthew-Wood syndrome to non-lethal anophthalmia. Hum. Mutat. 2009, 30, E673–E681. [Google Scholar] [CrossRef]
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 2007, 315, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Lammer, E.J.; Chen, D.T.; Hoar, R.M.; Agnish, N.D.; Benke, P.J.; Braun, J.T.; Curry, C.J.; Fernhoff, P.M.; Grix, A.W., Jr.; Lott, I.T.; et al. Retinoic acid embryopathy. N. Engl. J. Med. 1985, 313, 837–841. [Google Scholar] [CrossRef] [PubMed]
- Kastner, P.; Grondona, J.M.; Mark, M.; Gansmuller, A.; LeMeur, M.; Decimo, D.; Vonesch, J.L.; Dollé, P.; Chambon, P. Genetic analysis of RXR alpha developmental function: Convergence of RXR and RAR signaling pathways in heart and eye morphogenesis. Cell 1994, 78, 987–1003. [Google Scholar] [CrossRef]
- Kastner, P.; Mark, M.; Ghyselinck, N.; Krezel, W.; Dupé, V.; Grondona, J.M.; Chambon, P. Genetic evidence that the retinoid signal is transduced by heterodimeric RXR/RAR functional units during mouse development. Development 1997, 124, 313–326. [Google Scholar] [PubMed]
- Lee, R.Y.; Luo, J.; Evans, R.M.; Giguere, V.; Sucov, H.M. Compartment-selective sensitivity of cardiovascular morphogenesis to combinations of retinoic acid receptor gene mutations. Circ. Res. 1997, 80, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, C.; Lohnes, D.; Décimo, D.; Lufkin, T.; LeMeur, M.; Chambon, P.; Mark, M. Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development 1994, 120, 2749–2771. [Google Scholar] [PubMed]
- Li, P.; Pashmforoush, M.; Sucov, H.M. Retinoic acid regulates differentiation of the secondary heart field and TGFbeta-mediated outflow tract septation. Dev. Cell 2010, 18, 480–485. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Baker, K.M. Retinoic acid and the heart. Vitam. Horm. 2007, 75, 257–283. [Google Scholar]
- Rhinn, M.; Dollé, P. Retinoic acid signalling during development. Development 2012, 139, 843–858. [Google Scholar] [CrossRef] [Green Version]
- Ryckebusch, L.; Wang, Z.; Bertrand, N.; Lin, S.-C.; Chi, X.; Schwartz, R.; Zaffran, S.; Niederreither, K. Retinoic acid deficiency alters second heart field formation. Proc. Natl. Acad. Sci. USA 2008, 105, 2913–2918. [Google Scholar] [CrossRef] [Green Version]
- Sirbu, O.I.; Zhao, X.; Duester, G. Retinoic acid controls heart anteroposterior patterning by down-regulating Isl1 through the Fgf8 pathway. Dev. Dyn. 2008, 237, 1627–1635. [Google Scholar] [CrossRef] [Green Version]
- Duong, T.B.; Holowiecki, A.; Waxman, J.S. Retinoic acid signaling restricts the size of the first heart field within the anterior lateral plate mesoderm. Dev. Biol. 2021, 473, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.; Ivins, S.M.; James, C.T.; Scambler, P.J. Retinoic acid down-regulates Tbx1 expression in vivo and in vitro. Dev. Dyn. 2005, 232, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Blentic, A.; Gale, E.; Maden, M. Retinoic acid signalling centres in the avian embryo identified by sites of expression of synthesising and catabolising enzymes. Dev. Dyn. 2003, 227, 114–127. [Google Scholar] [CrossRef]
- Guris, D.L.; Duester, G.; Papaioannou, V.E.; Imamoto, A. Dose-dependent interaction of Tbx1 and Crkl and locally aberrant RA signaling in a model of del22q11 syndrome. Dev. Cell. 2006, 10, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Reijntjes, S.; Gale, E.; Maden, M. Generating gradients of retinoic acid in the chick embryo: Cyp26C1 expression and a comparative analysis of the Cyp26 enzymes. Dev. Dyn. 2004, 230, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Choudhary, B.; Merki, E.; Chien, K.R.; Maxson, R.E.; Sucov, H.M. Normal fate and altered function of the cardiac neural crest cell lineage in retinoic acid receptor mutant embryos. Mech. Dev. 2002, 117, 115–122. [Google Scholar] [CrossRef]
- Li, J.; Molkentin, J.D.; Colbert, M.C. Retinoic acid inhibits cardiac neural crest migration by blocking c-Jun N-terminal kinase activation. Dev. Biol. 2001, 232, 351–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eferl, R.; Sibilia, M.; Hilberg, F.; Fuchsbichler, A.; Kufferath, I.; Guertl, B.; Zenz, R.; Wagner, E.F.; Zatloukal, K. Functions of c-Jun in liver and heart development. J. Cell Biol. 1999, 145, 1049–1061. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Liu, J.; Zhang, J.; Thekkethottiyil, E.B.; Macatee, T.L.; Ismat, F.A.; Wang, F.; Stoller, J.Z. Jun is required in Isl1-expressing progenitor cells for cardiovascular development. PLoS ONE 2013, 8, e57032. [Google Scholar] [CrossRef] [Green Version]
- Jerome, L.A.; Papaioannou, V.E. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat. Genet. 2001, 27, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Ilagan, R.; Abu-Issa, R.; Brown, D.; Yang, Y.P.; Jiao, K.; Schwartz, R.J.; Klingensmith, J.; Meyers, E.N. Fgf8 is required for anterior heart field development. Development 2006, 133, 2435–2445. [Google Scholar] [CrossRef] [Green Version]
- Vermot, J.; Niederreither, K.; Garnier, J.M.; Chambon, P.; Dollé, P. Decreased embryonic retinoic acid synthesis results in a DiGeorge syndrome phenotype in newborn mice. Proc. Natl. Acad. Sci. USA 2003, 100, 1763–1768. [Google Scholar] [CrossRef] [Green Version]
- Roberts, C.; Ivins, S.; Cook, A.C.; Baldini, A.; Scambler, P.J. Cyp26 genes a1, b1 and c1 are down-regulated in Tbx1 null mice and inhibition of Cyp26 enzyme function produces a phenocopy of DiGeorge Syndrome in the chick. Hum. Mol. Genet. 2006, 15, 3394–3410. [Google Scholar] [CrossRef] [Green Version]
- Ryckebüsch, L.; Bertrand, N.; Mesbah, K.; Bajolle, F.; Niederreither, K.; Kelly, R.G.; Zaffran, S. Decreased levels of embryonic retinoic acid synthesis accelerate recovery from arterial growth delay in a mouse model of DiGeorge syndrome. Circ. Res. 2010, 106, 686–694. [Google Scholar] [CrossRef] [Green Version]
- Deschamps, J.; van Nes, J. Developmental regulation of the Hox genes during axial morphogenesis in the mouse. Development 2005, 132, 2931–2942. [Google Scholar] [CrossRef] [Green Version]
- Domínguez, J.N.; Meilhac, S.M.; Bland, Y.S.; Buckingham, M.E.; Brown, N.A. Asymmetric fate of the posterior part of the second heart field results in unexpected left/right contributions to both poles of the heart. Circ. Res. 2012, 111, 1323–1335. [Google Scholar] [CrossRef] [Green Version]
- Lescroart, F.; Mohun, T.; Meilhac, S.M.; Bennett, M.; Buckingham, M. Lineage tree for the venous pole of the heart: Clonal analysis clarifies controversial genealogy based on genetic tracing. Circ. Res. 2012, 111, 1313–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lescroart, F.; Wang, X.; Lin, X.; Swedlund, B.; Gargouri, S.; Sànchez-Dànes, A.; Moignard, V.; Dubois, C.; Paulissen, C.; Kinston, S.; et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science 2018, 359, 1177–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, M.S.; Théveniau-Ruissy, M.; De Bono, C.; Mesbah, K.; Francou, A.; Rammah, M.; Domínguez, J.N.; Roux, M.; Laforest, B.; Anderson, R.H.; et al. Tbx1 coordinates addition of posterior second heart field progenitor cells to the arterial and venous poles of the heart. Circ. Res. 2014, 115, 790–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, M.; Laforest, B.; Capecchi, M.; Bertrand, N.; Zaffran, S. Hoxb1 regulates proliferation and differentiation of second heart field progenitors in pharyngeal mesoderm and genetically interacts with Hoxa1 during cardiac outflow tract development. Dev. Biol. 2015, 406, 247–258. [Google Scholar] [CrossRef] [Green Version]
- Wendling, O.; Dennefeld, C.; Chambon, P.; Mark, M. Retinoid signaling is essential for patterning the endoderm of the third and fourth pharyngeal arches. Development 2000, 127, 1553–1562. [Google Scholar]
- Couly, G.; Creuzet, S.; Bennaceur, S.; Vincent, C.; Le Douarin, N.M. Interactions between Hox-negative cephalic neural crest cells and the foregut endoderm in patterning the facial skeleton in the vertebrate head. Development 2002, 129, 1061–1073. [Google Scholar]
- Chisaka, O.; Capecchi, M.R. Regionally restricted developmental defects resulting from targeted disruption of the mouse homeobox gene hox-1.5. Nature 1991, 350, 473–479. [Google Scholar] [CrossRef]
- Kirby, M.L. Plasticity and predetermination of mesencephalic and trunk neural crest transplanted into the region of the cardiac neural crest. Dev. Biol. 1989, 134, 402–412. [Google Scholar] [CrossRef]
- Wamstad, J.A.; Alexander, J.M.; Truty, R.M.; Shrikumar, A.; Li, F.; Eilertson, K.E.; Ding, H.; Wylie, J.N.; Pico, A.R.; Capra, J.A.; et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell 2012, 151, 206–220. [Google Scholar] [CrossRef] [Green Version]
- Paige, S.L.; Thomas, S.; Stoick-Cooper, C.L.; Wang, H.; Maves, L.; Sandstrom, R.; Pabon, L.; Reinecke, H.; Pratt, G.; Keller, G.; et al. A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell 2012, 151, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Stankunas, K.; Shang, C.; Twu, K.Y.; Kao, S.C.; Jenkins, N.A.; Copeland, N.G.; Sanyal, M.; Selleri, L.; Cleary, M.L.; Chang, C.P. Pbx/Meis deficiencies demonstrate multigenetic origins of congenital heart disease. Circ. Res. 2008, 103, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Simán, C.M.; Gittenberger-De Groot, A.C.; Wisse, B.; Eriksson, U.J. Malformations in offspring of diabetic rats: Morphometric analysis of neural crest-derived organs and effects of maternal vitamin E treatment. Teratology 2000, 61, 355–367. [Google Scholar] [CrossRef]
- Roest, P.A.; van Iperen, L.; Vis, S.; Wisse, L.J.; Poelmann, R.E.; Steegers-Theunissen, R.P.; Molin, D.G.; Eriksson, U.J.; Gittenberger-De Groot, A.C. Exposure of neural crest cells to elevated glucose leads to congenital heart defects, an effect that can be prevented by N-acetylcysteine. Birth Defects Res. A Clin. Mol. Teratol. 2007, 79, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Roest, P.A.; Molin, D.G.; Schalkwijk, C.G.; van Iperen, L.; Wentzel, P.; Eriksson, U.J.; Gittenberger-de Groot, A.C. Specific local cardiovascular changes of Nepsilon-(carboxymethyl)lysine, vascular endothelial growth factor, and Smad2 in the developing embryos coincide with maternal diabetes-induced congenital heart defects. Diabetes 2009, 58, 1222–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Soysa, T.Y.; Ranade, S.S.; Okawa, S.; Ravichandran, S.; Huang, Y.; Salunga, H.T.; Schricker, A.; Del Sol, A.; Gifford, C.A.; Srivastava, D. Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nature 2019, 572, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Pijuan-Sala, B.; Griffiths, J.A.; Guibentif, C.; Hiscock, T.W.; Jawaid, W.; Calero-Nieto, F.J.; Mulas, C.; Ibarra-Soria, X.; Tyser, R.C.V.; Ho, D.L.L.; et al. A single-cell molecular map of mouse gastrulation and early organogenesis. Nature 2019, 566, 490–495. [Google Scholar] [CrossRef]
- DeLaughter, D.M.; Bick, A.G.; Wakimoto, H.; McKean, D.; Gorham, J.M.; Kathiriya, I.S.; Hinson, J.T.; Homsy, J.; Gray, J.; Pu, W.; et al. Single-Cell Resolution of Temporal Gene Expression during Heart Development. Dev. Cell 2016, 39, 480–490. [Google Scholar] [CrossRef] [Green Version]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Xu, A.; Sim, S.; Priest, J.R.; Tian, X.; Khan, T.; Quertermous, T.; Zhou, B.; Tsao, P.S.; Quake, S.R.; et al. Transcriptomic Profiling Maps Anatomically Patterned Subpopulations among Single Embryonic Cardiac Cells. Dev. Cell 2016, 39, 491–507. [Google Scholar] [CrossRef] [Green Version]
- Skelly, D.A.; Squiers, G.T.; McLellan, M.A.; Bolisetty, M.T.; Robson, P.; Rosenthal, N.A.; Pinto, A.R. Single-Cell Transcriptional Profiling Reveals Cellular Diversity and Intercommunication in the Mouse Heart. Cell Rep. 2018, 22, 600–610. [Google Scholar] [CrossRef] [Green Version]
- Poelmann, R.E.; Jongbloed, M.R.; Molin, D.G.; Fekkes, M.L.; Wang, Z.; Fishman, G.I.; Doetschman, T.; Azhar, M.; Gittenberger-de Groot, A.C. The neural crest is contiguous with the cardiac conduction system in the mouse embryo: A role in induction? Anat. Embryol. 2004, 208, 389–393. [Google Scholar] [CrossRef]
- Mjaatvedt, C.H.; Kern, C.B.; Norris, R.A.; Fairey, S.; Cave, C.L. Normal distribution of melanocytes in the mouse heart. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 2005, 285, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Balani, K.; Brito, F.C.; Kos, L.; Agarwal, A. Melanocyte pigmentation stiffens murine cardiac tricuspid valve leaflet. J. R. Soc. Interface 2009, 6, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Yajima, I.; Colombo, S.; Puig, I.; Champeval, D.; Kumasaka, M.; Belloir, E.; Bonaventure, J.; Mark, M.; Yamamoto, H.; Taketo, M.M.; et al. A subpopulation of smooth muscle cells, derived from melanocyte-competent precursors, prevents patent ductus arteriosus. PLoS ONE 2013, 8, e53183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulin, A.; Hortells, L.; Gomez-Stallons, M.V.; O’Donnell, A.; Chetal, K.; Adam, M.; Lancellotti, P.; Oury, C.; Potter, S.S.; Salomonis, N.; et al. Maturation of heart valve cell populations during postnatal remodeling. Development 2019, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stefanovic, S.; Etchevers, H.C.; Zaffran, S. Outflow Tract Formation—Embryonic Origins of Conotruncal Congenital Heart Disease. J. Cardiovasc. Dev. Dis. 2021, 8, 42. https://doi.org/10.3390/jcdd8040042
Stefanovic S, Etchevers HC, Zaffran S. Outflow Tract Formation—Embryonic Origins of Conotruncal Congenital Heart Disease. Journal of Cardiovascular Development and Disease. 2021; 8(4):42. https://doi.org/10.3390/jcdd8040042
Chicago/Turabian StyleStefanovic, Sonia, Heather C. Etchevers, and Stéphane Zaffran. 2021. "Outflow Tract Formation—Embryonic Origins of Conotruncal Congenital Heart Disease" Journal of Cardiovascular Development and Disease 8, no. 4: 42. https://doi.org/10.3390/jcdd8040042
APA StyleStefanovic, S., Etchevers, H. C., & Zaffran, S. (2021). Outflow Tract Formation—Embryonic Origins of Conotruncal Congenital Heart Disease. Journal of Cardiovascular Development and Disease, 8(4), 42. https://doi.org/10.3390/jcdd8040042