
The Role of Cell Tracing and Fate Mapping Experiments in Cardiac Outflow Tract Development, New Opportunities through Emerging Technologies

 , ,
, ,  and
and
Abstract
:1. General Introduction
1.1. Basic Cardiac Development and Anatomy of the OFT
1.2. OFT Development
2. Cell Tracing Techniques Applied to OFT Development
2.1. Vital Dye and Viral Labelling Experiments
2.1.1. Basics of Vital Dye and Viral Labelling Experiments during Heart Development
2.1.2. Advantages of Vital Dye Labelling Experiments
2.1.3. Limitations of Vital Dye and Viral Labelling
2.1.4. Vital Dye and Viral Labelling Experiments Aimed at Understanding OFT Development
2.2. Lineage Tracing Using the Quail-Chick Chimeric System
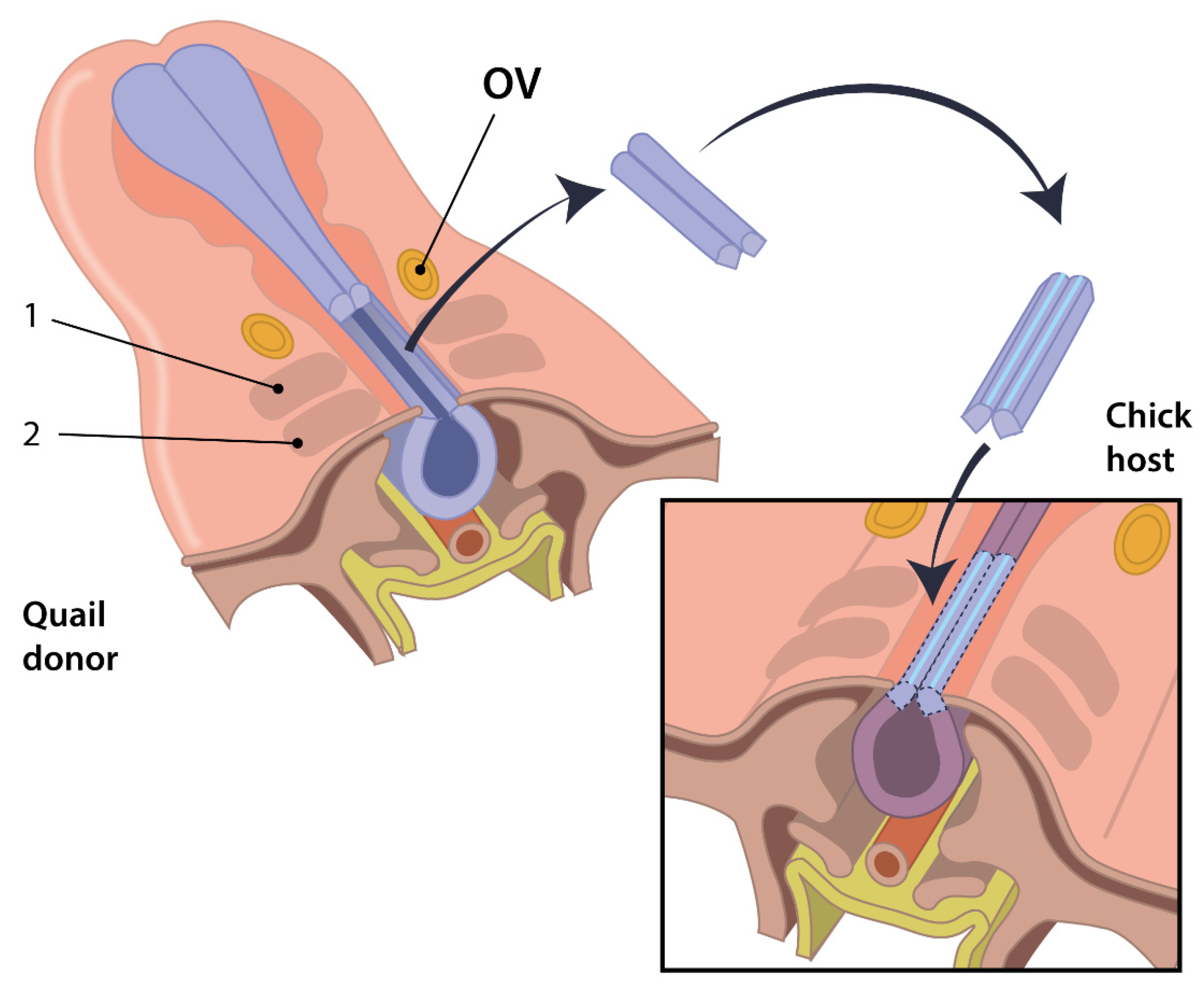
2.2.1. Basics of the Quail-Chick Chimeric System
2.2.2. Advantages of the Quail-Chick Chimeric System
2.2.3. Disadvantages of the Quail-Chick Chimeric System
2.2.4. The Quail-Chick Chimeric System and Insight into OFT Development
2.3. Genetic Lineage Tracing
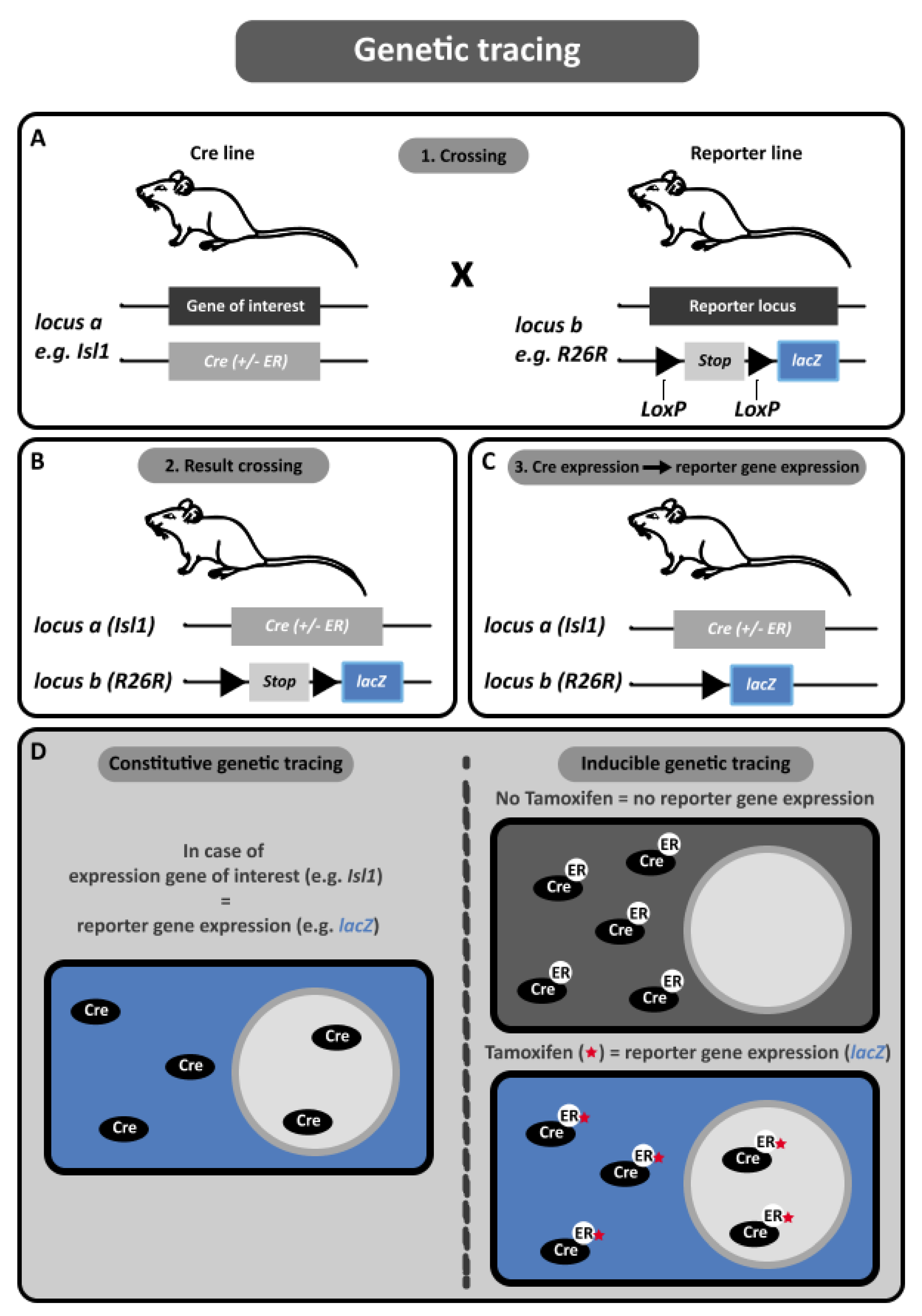
2.3.1. Basics of Genetic Lineage Tracing
2.3.2. Advantages of Genetic Tracing
2.3.3. Disadvantages of Genetic Tracing
2.3.4. Using Genetic Lineage Tracing to Study OFT Development

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transgenic Mouse Lines | Commonly Used as Cell Lineage Marker for: | Observed Tissue Expression | Possible Lineage Conflicts |
|---|---|---|---|
| Hcn4Cre-ert2 | First heart field [90] | Cardiac conduction system [90] Myocardium [90] | Second heart field |
| Hoxa1Cre | Cardiac precursors [91] | Aortopulmonary septum [91] Cardiac conduction system [91] Coronary arteries [91] Endothelial lining [91] Myocardium [91] Semilunar valves [91] | Endothelial Neural Crest Second heart field |
| Isl1Cre | Second heart field [81] | Atrioventricular valves [73] Cardiac conduction System [90] Cushion mesenchyme [73] Endocardium [73] Myocardium [92] Proepicardium [93] Semilunar Valves [73] | Endothelial Epicardial First heart field Neural Crest |
| Krox20Cre | Neural Crest [94] | Endocardium [94] Semilunar valves [94] | Endothelial |
| Mef2cCre | Second heart field [87] | Ascending aorta [95] Coronary arteries [96] Cushion mesenchyme [95] Endocardium [95] Myocardium [87,95] Semilunar valves [95] | Endothelial Epicardial |
| Nkx2-5Cre | First and Second heart field [97] | Ascending aorta [98] Coronary artery [98] Endocardium [73] Epicardium [93] Myocardium [97], Semilunar valves [98] | Endothelial Epicardial |
| Pax3Cre | Neural Crest [99] | Aortopulmonary septum [100] Ascending aorta [99] Cushion mesenchyme [100] Semilunar valves [100] | |
| Tbx18Cre | Proepicardium/epicardium [77] | Cardiac conduction system [90] Epicardium [77] Myocardium [77] | First heart field Second heart field |
| Tbx2Cre | Proepicardium/epicardium [101] | Cardiac conduction system [102] Coronary arteries [101] Epicardium [101] Myocardium [101,103] | First heart field Second heart field |
| Tie2Cre | Endothelium [22] | Atrioventricular valves [22,104] Coronary arteries [105] Cushion mesenchyme [22,104] Endocardium [22,104] Hematopoietic cells [106] Semilunar valves [104] | Hematopoietic |
| Tnnt2Cre | Myocardium [107] | Ascending aorta [25] Myocardium [25,107] Semilunar valves [25] | |
| Wnt11CreER | Cardiac precursors [108] | Endocardium [108] Epicardium [108] Myocardium [108] Semilunar valves [108] | First heart field Endothelial Epicardial Second heart field |
| Wnt1Cre | Neural Crest [20] | Aortopulmonary septum [20] Ascending aorta [20] Cardiac conduction system [109] Coronary arteries [71] Cushion mesenchyme [20] Epicardium [110] Myocardium [110,111] Semilunar valves [20] | Epicardial Second heart field |
| WT1Cre | Proepicardium/epicardium [112] | Coronary arteries [112] Epicardium [93] Myocardium [93] | Endothelial lineage First heart field Second heart field |
2.4. Retrospective Clonal Analysis
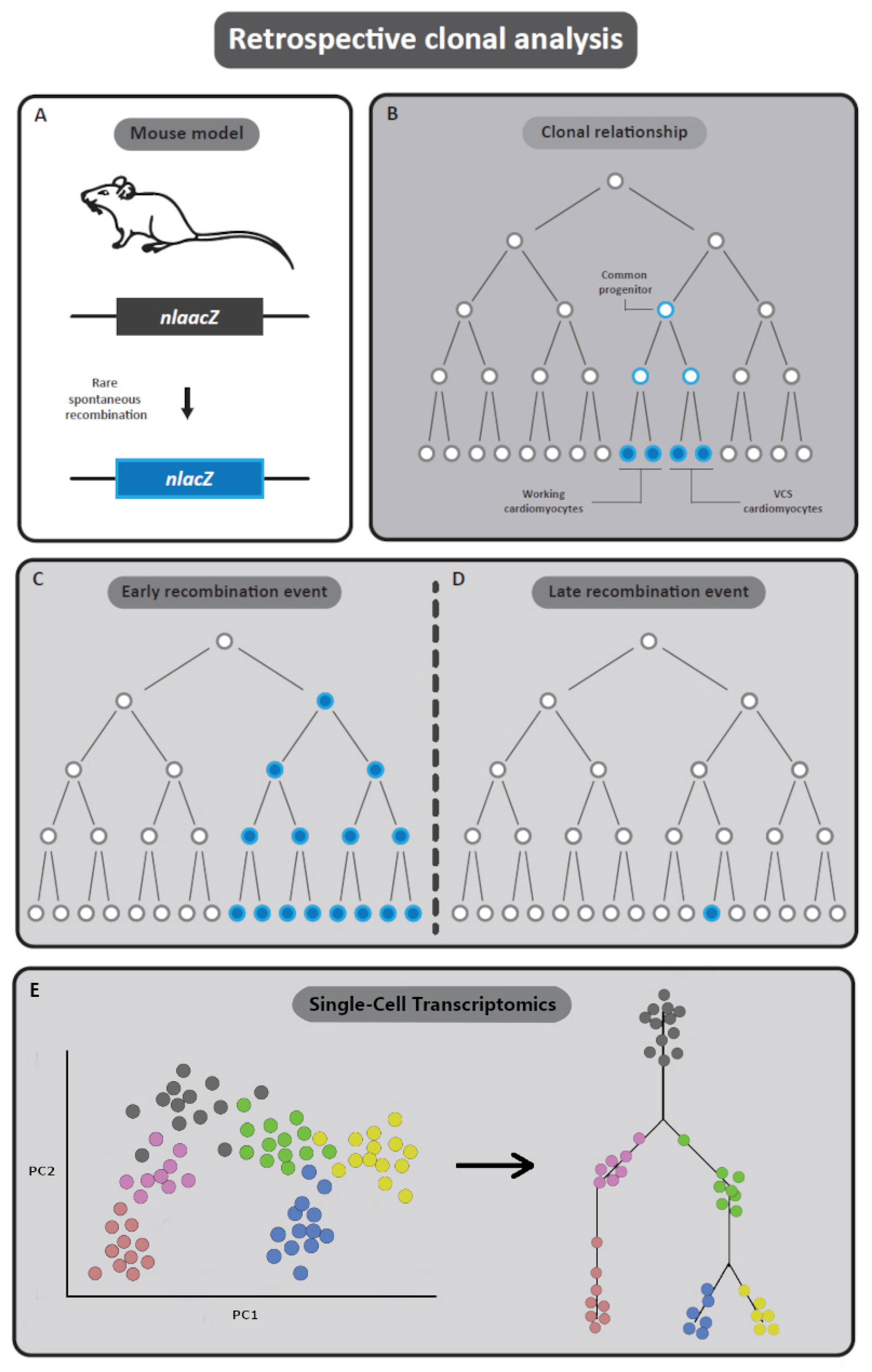
2.4.1. Basics of Retrospective Clonal Analysis
2.4.2. Advantages of Retrospective Clonal Analysis
2.4.3. Limitations of Retrospective Clonal Analysis
2.4.4. Retrospective Clonal Analysis Aimed at the Development of the OFT
3. New Technologies for Lineage Tracing in OFT Development
3.1. Spatially Resolved Transcriptomics
3.2. Lineage Tracing through DNA Barcoding
3.3. Multi-Omics Lineage Tracing
3.4. Reference Maps for Lineage Tracing
4. Future Perspectives, Combining Old Strengths with New Technologies
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMC | arterial mesothelial cells |
| SHF | second heart field |
| AVC | atrioventricular canal |
| SNV | single nucleotide variation |
| CHD | congenital heart disease |
| TIVA | transcriptome in vivo analysis |
| CNV | copy number variations |
| VIC | valvular interstitial cell |
| E | embryonic day |
| ER | estrogen receptor |
| FHF | first heart field |
| GFP | green fluorescent protein |
| HH | Hamburger and Hamilton |
| HCA | Human Cell Atlas |
| LBD | ligand-binding domain |
| mT/mG | membrane-bound Tomato/membrane-bound GFP |
| NCCs | neural crest cells |
| OFT | outflow tract |
| PHT | primary heart tube |
| R26R | Rosa26 reporter |
| scRNA-seq | single cell RNA sequencing |
References
- Thompson, P. Description of a Human Embryo of Twenty-Three Paired Somites. J. Anat. Physiol. 1907, 41, 159–171. [Google Scholar] [PubMed]
- Garg, V.; Muth, A.N.; Ransom, J.F.; Schluterman, M.K.; Barnes, R.; King, I.N.; Grossfeld, P.D.; Srivastava, D. Mutations in NOTCH1 cause aortic valve disease. Nature 2005, 437, 270–274. [Google Scholar] [CrossRef]
- Laforest, B.; Andelfinger, G.; Nemer, M. Loss of Gata5 in mice leads to bicuspid aortic valve. J. Clin. Investig. 2011, 121, 2876–2887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, J.C.; Chughtai, M.; Wisse, L.J.; Gittenberger-de Groot, A.C.; Feng, Q.; Goumans, M.-J.T.H.; VanMunsteren, J.C.; Jongbloed, M.R.M.; DeRuiter, M.C. Bicuspid aortic valve formation: Nos3 mutation leads to abnormal lineage patterning of neural crest cells and the second heart field. Dis. Model. Mech. 2018, 11, 655–658. [Google Scholar] [CrossRef] [Green Version]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Perino, M.; Veenstra, G.J.C. Chromatin Control of Developmental Dynamics and Plasticity. Dev. Cell 2016, 38, 610–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, A.G. Cellular identity and lineage choice. Nat. Rev. Immunol. 2002, 2, 977–982. [Google Scholar] [CrossRef]
- Peaston, A.E.; Whitelaw, E. Epigenetics and phenotypic variation in mammals. Mamm. Genome 2006, 17, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Merrell, A.J.; Stanger, B.Z. Adult cell plasticity in vivo: De-differentiation and transdifferentiation are back in style. Nat. Rev. Mol. Cell Biol. 2016, 17, 413–425. [Google Scholar] [CrossRef]
- Olson, E.N.; Srivastava, D. Molecular pathways controlling heart development. Science 1996, 272, 671–676. [Google Scholar] [CrossRef]
- Laverriere, A.C.; MacNeill, C.; Mueller, C.; Poelmann, R.E.; Burch, J.B.E.; Evans, T. GATA-4/5/6, a subfamily of three transcription factors transcribed in developing heart and gut. J. Biol. Chem. 1994, 269, 23177–23184. [Google Scholar] [CrossRef]
- Buckingham, M.; Meilhac, S.; Zaffran, S. Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 2005, 6, 826–835. [Google Scholar] [CrossRef]
- Abu-Issa, R.; Kirby, M.L. Heart field: From mesoderm to heart tube. Annu. Rev. Cell Dev. Biol. 2007, 23, 45–68. [Google Scholar] [CrossRef] [PubMed]
- DeRuiter, M.C.; Poelmann, R.E.; VanderPlas-de Vries, I.; Mentink, M.M.T.; Gittenberger-de Groot, A.C. The development of the myocardium and endocardium in mouse embryos—Fusion of two heart tubes? Anat. Embryol. 1992, 185, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Tyser, R.C.V.; Srinivas, S. The first heartbeat—Origin of cardiac contractile activity. Cold Spring Harb. Perspect. Biol. 2020, 12, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moorman, A.F.M.; Christoffels, V.M. Cardiac chamber formation: Development, genes, and evolution. Physiol. Rev. 2003. [Google Scholar] [CrossRef]
- Combs, M.D.; Yutzey, K.E. Heart valve development: Regulatory networks in development and disease. Circ. Res. 2009, 105, 408–421. [Google Scholar] [CrossRef] [Green Version]
- Kirby, M.L. Cardiac Development; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Waldo, K.; Miyagawa-Tomita, S.; Kumiski, D.; Kirby, M.L. Cardiac neural crest cells provide new insight into septation of the cardiac outflow tract: Aortic sac to ventricular septal closure. Dev. Biol. 1998, 196, 129–144. [Google Scholar] [CrossRef]
- Jiang, X.; Rowitch, D.H.; Soriano, P.; McMahon, A.P.; Sucov, H.M. Fate of the mammalian cardiac neural crest. Development 2000, 127, 1607–1616. [Google Scholar] [CrossRef]
- Eisenberg, L.M.; Markwald, R.R. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ. Res. 1995, 77, 1–6. [Google Scholar] [CrossRef]
- Kisanuki, Y.Y.; Hammer, R.E.; Miyazaki, J.; Williams, S.C.; Richardson, J.A.; Yanagisawa, M. Tie2-Cre transgenic mice: A new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 2001, 230, 230–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaffran, S.; Kelly, R.G. New developments in the second heart field. Differentiation 2012, 84, 17–24. [Google Scholar] [CrossRef]
- Eley, L.; Alqahtani, A.M.S.; Macgrogan, D.; Richardson, R.V.; Murphy, L.; Salguero-jimenez, A.; Sintes, M.; San, R.; Tiurma, S.; Mccutcheon, L.; et al. A novel source of arterial valve cells linked to bicuspid aortic valve without raphe in mice. eLife 2018, 7, 1–27. [Google Scholar] [CrossRef]
- Mifflin, J.J.; Dupuis, L.E.; Alcala, N.E.; Russell, L.G.; Kern, C.B. Intercalated cushion cells within the cardiac outflow tract are derived from the myocardial troponin T type 2 (Tnnt2) Cre lineage. Dev. Dyn. 2018, 247, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Miquerol, L.; Kelly, R.G. Organogenesis of the vertebrate heart. Wiley Interdiscip. Rev. Dev. Biol. 2013. [Google Scholar] [CrossRef]
- Scherptong, R.W.; Jongbloed, M.R.; Wisse, L.J.; Vicente-Steijn, R.; Bartelings, M.M.; Poelmann, R.E.; Schalij, M.J.; Gittenberger-De Groot, A.C. Morphogenesis of outflow tract rotation during cardiac development: The pulmonary push concept. Dev. Dyn 2012, 241, 1413–1422. [Google Scholar] [CrossRef]
- Gittenberger-De Groot, A.C.; Bartelings, M.M.; Deruiter, M.C.; Poelmann, R.E. Basics of cardiac development for the understanding of congenital heart malformations. Pediatric Res. 2005, 57, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Gittenberger-de Groot, A.C.; Bartelings, M.M.; Poelmann, R.E.; Haak, M.C.; Jongbloed, M.R.M. Embryology of the heart and its impact on understanding fetal and neonatal heart disease. Semin. Fetal Neonatal Med. 2013, 18, 237–244. [Google Scholar] [CrossRef]
- Gittenberger-de Groot, A.C.; Calkoen, E.E.; Poelmann, R.E.; Bartelings, M.M.; Jongbloed, M.R.M. Morphogenesis and molecular considerations on congenital cardiac septal defects. Ann. Med. 2014, 46, 640–652. [Google Scholar] [CrossRef] [PubMed]
- Bartelings, M.M.; Gittenberger-de Groot, A.C. Morphogenetic considerations on congenital malformations of the outflow tract. Part 1: Common arterial trunk and tetralogy of Fallot. Int. J. Cardiol. 1991, 32, 213–230. [Google Scholar] [CrossRef]
- Poelmann, R.E.; Gittenberger-de Groot, A.C.; Biermans, M.W.M.; Dolfing, A.I.; Jagessar, A.; van Hattum, S.; Hoogenboom, A.; Wisse, L.J.; Vicente-Steijn, R.; de Bakker, M.A.G.; et al. Outflow tract septation and the aortic arch system in reptiles: Lessons for understanding the mammalian heart. Evodevo 2017, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Poelmann, R.E.; Mikawa, T.; Gittenberger-De Groot, A.C. Neural crest cells in outflow tract septation of the embryonic chicken heart: Differentiation and apoptosis. Dev. Dyn. 1998, 212, 373–384. [Google Scholar] [CrossRef]
- Kirby, M.L.; Gale, T.F.; Stewart, D.E. Neural crest cells contribute to normal aorticopulmonary septation. Science 1983, 220, 1059–1061. [Google Scholar] [CrossRef]
- Anderson, R.; Mohun, T.; Spicer, D.; Bamforth, S.; Brown, N.; Chaudhry, B.; Henderson, D. Myths and Realities Relating to Development of the Arterial Valves. J. Cardiovasc. Dev. Dis. 2014, 1, 177–200. [Google Scholar] [CrossRef] [Green Version]
- Gittenberger-de Groot, A.C.; Bartelings, M.M.; Bogers, A.J.J.C.; Boot, M.J.; Poelmann, R.E. The embryology of the common arterial trunk. Prog. Pediatric Cardiol. 2002, 15, 1–8. [Google Scholar] [CrossRef]
- DeHaan, R.L. Migration patterns of the precardiac mesoderm in the early chick embryo. Exp. Cell Res. 1963, 29, 544–560. [Google Scholar] [CrossRef]
- Davis, C. Development of the human heart from its first appearance to the stage found in embryos of twenty paired somites. Carnegie Inst. Contr. Embryol. 1927, 19, 245–284. [Google Scholar]
- Axelrod, D. Carbocyanine dye orientation in red cell membrane studied by microscopic fluorescence polarization. Biophys. J. 1979, 26, 557–573. [Google Scholar] [CrossRef] [Green Version]
- Kelder, T.P.; Vicente-Steijn, R.; Harryvan, T.J.T.; Kosmidis, G.; Gittenberger-de Groot, A.C.; Poelmann, R.R.E.; Schalij, M.M.J.; DeRuiter, M.M.C.; Jongbloed, M.R.M. The sinus venosus myocardium contributes to the atrioventricular canal: Potential role during atrioventricular node development? J. Cell. Mol. Med. 2015, 19, 1375–1389. [Google Scholar] [CrossRef] [PubMed]
- Sanes, J.R. Analysing cell lineage with a recombinant retrovirus. Trends Neurosci. 1989, 12, 21–28. [Google Scholar] [CrossRef]
- Sanes, J.R.; Rubenstein, J.L.; Nicolas, J.F. Use of a recombinant retrovirus to study post-implantation cell lineage in mouse embryos. EMBO J. 1986, 5, 3133–3142. [Google Scholar] [CrossRef]
- Price, J.; Turner, D.; Cepko, C. Lineage analysis in the vertebrate nervous system by retrovirus-mediated gene transfer. Proc. Natl. Acad. Sci. USA 1987, 84, 156–160. [Google Scholar] [CrossRef] [Green Version]
- Pacak, C.A.; Byrne, B.J. AAV vectors for cardiac gene transfer: Experimental tools and clinical opportunities. Mol. Ther. 2011, 19, 1582–1590. [Google Scholar] [CrossRef] [Green Version]
- Stainier, D.Y.R.; Lee, R.K.; Fishman, M.C. Cardiovascular development in the zebrafish: I. Myocardial fate map and heart tube formation. Development 1993, 119, 31–40. [Google Scholar] [CrossRef]
- Krotoski, D.M.; Fraser, S.E.; Bronner-Fraser, M. Mapping of neural crest pathways in Xenopus laevis using inter- and intra-specific cell markers. Dev. Biol. 1988, 127, 119–132. [Google Scholar] [CrossRef]
- De la Cruz, M.V.; Sánchez Gómez, C.; Arteaga, M.M.; Argüello, C. Experimental study of the development of the truncus and the conus in the chick embryo. J. Anat. 1977, 123, 661–686. [Google Scholar]
- Kelder, T.P.; Vicente-Steijn, R.; Poelmann, R.E.; Mummery, C.L.; DeRuiter, M.C.; Jongbloed, M.R.M. The avian embryo to study development of the cardiac conduction system. Differentiation 2016, 91, 90–103. [Google Scholar] [CrossRef]
- Domínguez, J.N.; Meilhac, S.M.; Bland, Y.S.; Buckingham, M.E.; Brown, N.A. Asymmetric fate of the posterior part of the second heart field results in unexpected left/right contributions to both poles of the heart. Circ. Res. 2012, 111, 1323–1335. [Google Scholar] [CrossRef] [Green Version]
- Honig, M.G.; Hume, R.I. Fluorescent carbocyanine dyes allow living neurons of identified origin to be studied in long-term cultures. J. Cell Biol. 1986, 103, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Progatzky, F.; Dallman, M.J.; Lo Celso, C. From seeing to believing: Labelling strategies for in vivo cell-tracking experiments. Interface Focus 2013, 3, 20130001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckingham, M.E.; Meilhac, S.M. Tracing cells for tracking cell lineage and clonal behavior. Dev. Cell 2011, 21, 394–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bressan, M.; Liu, G.; Mikawa, T. Early mesodermal cues assign avian cardiac pacemaker fate potential in a tertiary heart field. Science 2013, 340, 744–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Celso, C.; Fleming, H.E.; Wu, J.W.; Zhao, C.X.; Miake-Lye, S.; Fujisaki, J.; Côté, D.; Rowe, D.W.; Lin, C.P.; Scadden, D.T. Live-animal tracking of individual haematopoietic stem/progenitor cells in their niche. Nature 2009, 457, 92–96. [Google Scholar] [CrossRef] [Green Version]
- Sipkins, D.A.; Wei, X.; Wu, J.W.; Runnels, J.M.; Côté, D.; Means, T.K.; Luster, A.D.; Scadden, D.T.; Lin, C.P. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature 2005, 435, 969–973. [Google Scholar] [CrossRef]
- Honig, M.G.; Hume, R.I. Dil and diO: Versatile fluorescent dyes for neuronal labelling and pathway tracing. Trends Neurosci. 1989, 12, 333–335, 340–341. [Google Scholar] [CrossRef] [Green Version]
- Mikawa, T.; Hyer, J.; Itoh, N.; Wei, Y. Retroviral vectors to study cardiovascular development. Trends Cardiovasc. Med. 1996, 6, 79–86. [Google Scholar] [CrossRef]
- Barbosky, L.; Lawrence, D.K.; Karunamuni, G.; Wikenheiser, J.C.; Doughman, Y.-Q.; Visconti, R.P.; Burch, J.B.E.; Watanabe, M. Apoptosis in the developing mouse heart. Dev. Dyn. 2006, 235, 2592–2602. [Google Scholar] [CrossRef]
- Schaefer, K.S.; Doughman, Y.Q.; Fisher, S.A.; Watanabe, M. Dynamic patterns of apoptosis in the developing chicken heart. Dev. Dyn. 2004, 229, 489–499. [Google Scholar] [CrossRef]
- Le Douarin, N. Particularites du noyau interphasique chez la caille Japonaise (Coturnix coturnix japonica). Bull. Biol. Fr. Belg. 1969, 103, 435–452. [Google Scholar]
- Le Douarin, N. A biological cell labeling technique and its use in experimental embryology. Dev. Biol. 1973, 30, 217–222. [Google Scholar] [CrossRef]
- Le Douarin, N.M. The avian embryo as a model to study the development of the neural crest: A long and still ongoing story. Mech. Dev. 2004, 121, 1089–1102. [Google Scholar] [CrossRef]
- Selleck, M.A.; Bronner-Fraser, M. Origins of the avian neural crest: The role of neural plate-epidermal interactions. Development 1995, 121, 525–538. [Google Scholar] [CrossRef]
- Koga, M.; Kageura, H.; Yamana, K. Use of Hybrids between Xenopus laevis and Xenopus borealis in Chimera Formation: Dorsalization of Ventral Cells: (cell lineage/chimera/hybrid/Xenopus/dorsalization). Dev. Growth Differ. 1986, 28, 177–183. [Google Scholar] [CrossRef]
- Tam, P.P.L.; Rossant, J. Mouse embryonic chimeras: Tools for studying mammalian development. Development 2003, 130, 6155–6163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sereti, K.I.; Nguyen, N.B.; Kamran, P.; Zhao, P.; Ranjbarvaziri, S.; Park, S.; Sabri, S.; Engel, J.L.; Sung, K.; Kulkarni, R.P.; et al. Analysis of cardiomyocyte clonal expansion during mouse heart development and injury. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Le Lievre, C.S.; Le Douarin, N.M. Mesenchymal derivatives of the neural crest: Analysis of chimaeric quail and chick embryos. J. Embryol. Exp. Morphol. 1975, 34, 125–154. [Google Scholar] [PubMed]
- Baroffio, A.; Dupin, E.; Le Douarin, N.M. Common precursors for neural and mesectodermal derivatives in the cephalic neural crest. Development 1991, 112, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Takamura, K.; Okishima, T.; Ohdo, S.; Hayakawa, K. Association of cephalic neural crest cells with cardiovascular development, particularly that of the semilunar valves. Anat. Embryol. 1990, 182, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Rosenquist, T.H.; McCoy, J.R.; Waldo, K.L.; Kirby, M.L. Origin and propagation of elastogenesis in the developing cardiovascular system. Anat. Rec. 1988, 221, 860–871. [Google Scholar] [CrossRef] [PubMed]
- Arima, Y.; Miyagawa-Tomita, S.; Maeda, K.; Asai, R.; Seya, D.; Minoux, M.; Rijli, F.M.; Nishiyama, K.; Kim, K.S.; Uchijima, Y.; et al. Preotic neural crest cells contribute to coronary artery smooth muscle involving endothelin signalling. Nat. Commun. 2012, 3, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sauer, B. Inducible gene targeting in mice using the Cre/lox system. Methods 1998, 14, 381–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Q.; Zhou, B.; Pu, W.T. Reassessment of Isl1 and Nkx2-5 cardiac fate maps using a Gata4-based reporter of Cre activity. Dev. Biol. 2008, 323, 98–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feil, R.; Brocard, J.; Mascrez, B.; LeMeur, M.; Metzger, D.; Chambon, P. Ligand-activated site-specific recombination in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 10887–10890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indra, A.K.; Warot, X.; Brocard, J.; Bornert, J.M.; Xiao, J.H.; Chambon, P.; Metzger, D. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: Comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic Acids Res. 1999, 27, 4324–4327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livet, J.; Weissman, T.A.; Kang, H.; Draft, R.W.; Lu, J.; Bennis, R.A.; Sanes, J.R.; Lichtman, J.W. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature 2007, 450, 56–62. [Google Scholar] [CrossRef]
- Cai, C.-L.; Martin, J.C.; Sun, Y.; Cui, L.; Wang, L.; Ouyang, K.; Yang, L.; Bu, L.; Liang, X.; Zhang, X.; et al. A myocardial lineage derives from Tbx18 epicardial cells. Nature 2008, 454, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Christoffels, V.M.; Grieskamp, T.; Norden, J.; Mommersteeg, M.T.M.; Rudat, C.; Kispert, A. Tbx18 and the fate of epicardial progenitors. Nature 2009, 458, E8–E9. [Google Scholar] [CrossRef]
- Hayashi, S.; McMahon, A.P. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: A tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol. 2002, 244, 305–318. [Google Scholar] [CrossRef] [Green Version]
- Álvarez-Aznar, A.; Martínez-Corral, I.; Daubel, N.; Betsholtz, C.; Mäkinen, T.; Gaengel, K. Tamoxifen-independent recombination of reporter genes limits lineage tracing and mosaic analysis using CreERT2 lines. Transgenic Res. 2020, 29, 53–68. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.-L.; Liang, X.; Shi, Y.; Chu, P.-H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Liang, X.; Najafi, N.; Cass, M.; Lin, L.; Cai, C.-L.; Chen, J.; Evans, S.M. Islet 1 is expressed in distinct cardiovascular lineages, including pacemaker and coronary vascular cells. Dev. Biol. 2007, 304, 286–296. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Schoenwolf, G.C. Islet-1 marks the early heart rudiments and is asymmetrically expressed during early rotation of the foregut in the chick embryo. Anat. Rec. 2000, 260, 204–207. [Google Scholar] [CrossRef]
- Baardman, M.E.; Zwier, M.V.; Wisse, L.J.; Gittenberger-De Groot, A.C.; Kerstjens-Frederikse, W.S.; Hofstra, R.M.W.; Jurdzinski, A.; Hierck, B.P.; Jongbloed, M.R.M.; Berger, R.M.F.; et al. Common arterial trunk and ventricular non-compaction in Lrp2 knockout mice indicate a crucial role of LRP2 in cardiac development. DMM Dis. Model. Mech. 2016, 9, 413–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Colbert, M.C.; Robbins, J. Neural crest cells retain multipotential characteristics in the developing valves and label the cardiac conduction system. Circ. Res. 2006, 98, 1547–1554. [Google Scholar] [CrossRef] [Green Version]
- Bolender, D.L.; Markwald, R.R. Epithelial-mesenchymal transformation in chick atrioventricular cushion morphogenesis. Scan. Electron. Microsc. 1979, 3, 313–321. [Google Scholar]
- Verzi, M.P.; McCulley, D.J.; De Val, S.; Dodou, E.; Black, B.L. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev. Biol. 2005, 287, 134–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Yu, W.; Tang, M.; Tang, J.; Liu, X.; Liu, Q.; Li, Y.; He, L.; Zhang, L.; Evans, S.M.; et al. A dual genetic tracing system identifies diverse and dynamic origins of cardiac valve mesenchyme. Development 2018, 145. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Jin, H.; Zhou, B. Genetic lineage tracing with multiple DNA recombinases: A user’s guide for conducting more precise cell fate mapping studies. J. Biol. Chem. 2020, 295, 6413–6424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.; Wang, G.; Lin, L.; Lowe, J.; Zhang, Q.; Bu, L.; Chen, Y.; Chen, J.; Sun, Y.; Evans, S.M. HCN4 dynamically marks the first heart field and conduction system precursors. Circ. Res. 2013, 113, 399–407. [Google Scholar] [CrossRef]
- Makki, N.; Capecchi, M.R. Hoxa1 lineage tracing indicates a direct role for Hoxa1 in the development of the inner ear, the heart, and the third rhombomere. Dev. Biol. 2010, 341, 499–509. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Cai, C.L.; Lin, L.; Qyang, Y.; Chung, C.; Monteiro, R.M.; Mummery, C.L.; Fishman, G.I.; Cogen, A.; Evans, S. Isl1 Cre reveals a common Bmp pathway in heart and limb development. Development 2006, 133, 1575–1585. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Ma, Q.; Rajagopal, S.; Wu, S.M.; Domian, I.; Rivera-Feliciano, J.; Jiang, D.; von Gise, A.; Ikeda, S.; Chien, K.R.; et al. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 2008, 454, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Odelin, G.; Faure, E.; Coulpier, F.; Di Bonito, M.; Bajolle, F.; Studer, M.; Avierinos, J.-F.; Charnay, P.; Topilko, P.; Zaffran, S. Krox20 defines a subpopulation of cardiac neural crest cells contributing to arterial valves and bicuspid aortic valve. Development 2017, 145, dev151944. [Google Scholar] [CrossRef] [Green Version]
- Crucean, A.; Alqahtani, A.; Barron, D.J.; Brawn, W.J.; Richardson, R.V.; O’Sullivan, J.; Anderson, R.H.; Henderson, D.J.; Chaudhry, B. Re-evaluation of hypoplastic left heart syndrome from a developmental and morphological perspective. Orphanet J. Rare Dis. 2017, 12, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Hu, T.; Zhang, H.; He, L.; Huang, X.; Liu, Q.; Yu, W.; He, L.; Yang, Z.; Zhang, Z.; et al. Subepicardial endothelial cells invade the embryonic ventricle wall to form coronary arteries. Cell Res. 2013, 23, 1075–1090. [Google Scholar] [CrossRef] [Green Version]
- Moses, K.A.; DeMayo, F.; Braun, R.M.; Reecy, J.L.; Schwartz, R.J. Embryonic expression of an Nkx2-5/Cre gene using ROSA26 reporter mice. Genesis 2001, 31, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Harmon, A.W.; Nakano, A. Nkx2-5 lineage tracing visualizes the distribution of second heart field-derived aortic smooth muscle. Genesis 2013, 51, 862–869. [Google Scholar] [CrossRef] [Green Version]
- Epstein, J.A.; Li, J.; Lang, D.; Chen, F.; Brown, C.B.; Jin, F.; Lu, M.M.; Thomas, M.; Liu, E.; Wessels, A.; et al. Migration of cardiac neural crest cells in Splotch embryos. Development 2000, 127, 1869–1878. [Google Scholar] [CrossRef]
- Epstein, J.; Buck, C.A. Transcriptional regulation of cardiac development: Implications for congenital heart disease and DiGeorge syndrome. Pediatric Res. 2000, 48, 717–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greulich, F.; Rudat, C.; Farin, H.F.; Christoffels, V.M.; Kispert, A. Lack of Genetic Interaction between Tbx18 and Tbx2/Tbx20 in Mouse Epicardial Development. PLoS ONE 2016, 11, e0156787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aanhaanen, W.T.J.; Brons, J.F.; Domínguez, J.N.; Rana, M.S.; Norden, J.; Airik, R.; Wakker, V.; De Gier-De Vries, C.; Brown, N.A.; Kispert, A.; et al. The Tbx2+ primary myocardium of the atrioventricular canal forms the atrioventricular node and the base of the left ventricle. Circ. Res. 2009, 104, 1267–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Hoogaars, W.M.; Barnett, P.; Grieskamp, T.; Rana, M.S.; Buermans, H.; Farin, H.F.; Petry, M.; Heallen, T.; Martin, J.F.; et al. Tbx2 and Tbx3 induce atrioventricular myocardial development and endocardial cushion formation. Cell. Mol. Life Sci. 2012, 69, 1377–1389. [Google Scholar] [CrossRef] [Green Version]
- De Lange, F.J.; Moorman, A.F.M.; Anderson, R.H.; Männer, J.; Soufan, A.T.; De Gier-De Vries, C.; Schneider, M.D.; Webb, S.; van den Hoff, M.J.B.; Christoffels, V.M.; et al. Lineage and morphogenetic analysis of the cardiac valves. Circ. Res. 2004, 95, 645–654. [Google Scholar] [CrossRef]
- Payne, S.; De Val, S.; Neal, A. Endothelial-specific cre mouse models is your cre CREdibile? Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2550–2561. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Harrington, A.; Yang, X.; Friesel, R.E.; Liaw, L. The contribution of the Tie2+ lineage to primitive and definitive hematopoietic cells. Genesis 2010, 48, 563–567. [Google Scholar] [CrossRef] [Green Version]
- Jiao, K.; Kulessa, H.; Tompkins, K.; Zhou, Y.; Batts, L.; Baldwin, H.S.; Hogan, B.L.M. An essential role of Bmp4 in the atrioventricular septation of the mouse heart. Genes Dev. 2003, 17, 2362–2367. [Google Scholar] [CrossRef] [Green Version]
- Sinha, T.; Lin, L.; Li, D.; Davis, J.; Evans, S.; Wynshaw-Boris, A.; Wang, J. Mapping the dynamic expression of Wnt11 and the lineage contribution of Wnt11-expressing cells during early mouse development. Dev. Biol. 2015, 398, 177–192. [Google Scholar] [CrossRef] [Green Version]
- Poelmann, R.E.; Jongbloed, M.R.M.; Molin, D.G.M.; Fekkes, M.L.; Wang, Z.; Fishman, G.I.; Doetschman, T.; Azhar, M.; Gittenberger-De Groot, A.C. The neural crest is contiguous with the cardiac conduction system in the mouse embryo: A role in induction? Anat. Embryol. 2004, 208, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Stottmann, R.W.; Choi, M.; Mishina, Y.; Meyers, E.N.; Klingensmith, J. BMP receptor IA is required in mammalian neural crest cells for development of the cardiac outflow tract and ventricular myocardium. Development 2004, 131, 2205–2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatzistergos, K.E.; Durante, M.A.; Valasaki, K.; Wanschel, A.C.B.A.; William Harbour, J.; Hare, J.M. A novel cardiomyogenic role for Isl1+ neural crest cells in the inflow tract. Sci. Adv. 2020, 6, eaba9950. [Google Scholar] [CrossRef]
- Zhou, B.; Pu, W.T. Genetic Cre-loxP assessment of epicardial cell fate using Wt1-Driven cre alleles. Circ. Res. 2012, 111, e276. [Google Scholar] [CrossRef] [Green Version]
- Bonnerot, C.; Nicolas, J.F. Clonal analysis in the intact mouse embryo by intragenic homologous recombination. Comptes Rendus Acad. Sci. III 1993, 316, 1207–1217. [Google Scholar]
- Meilhac, S.M.; Kelly, R.G.; Rocancourt, D.; Eloy-Trinquet, S.; Nicolas, J.-F.; Buckingham, M.E. A retrospective clonal analysis of the myocardium reveals two phases of clonal growth in the developing mouse heart. Development 2003, 130, 3877–3889. [Google Scholar] [CrossRef] [Green Version]
- Miquerol, L.; Moreno-Rascon, N.; Beyer, S.; Dupays, L.; Meilhac, S.M.; Buckingham, M.E.; Franco, D.; Kelly, R.G. Biphasic development of the mammalian ventricular conduction system. Circ. Res. 2010, 107, 153–161. [Google Scholar] [CrossRef]
- Miquerol, L.; Bellon, A.; Moreno, N.; Beyer, S.; Meilhac, S.M.; Buckingham, M.; Franco, D.; Kelly, R.G. Resolving cell lineage contributions to the ventricular conduction system with a Cx40-GFP allele: A dual contribution of the first and second heart fields. Dev. Dyn. 2013, 242, 665–677. [Google Scholar] [CrossRef]
- Luria, S.E.; Delbrück, M. Mutations of Bacteria from Virus Sensitivity to Virus Resistance. Genetics 1943, 28, 491–511. [Google Scholar] [CrossRef] [PubMed]
- Lescroart, F.; Mohun, T.; Meilhac, S.M.; Bennett, M.; Buckingham, M. Lineage tree for the venous pole of the heart: Clonal analysis clarifies controversial genealogy based on genetic tracing. Circ. Res. 2012, 111, 1313–1322. [Google Scholar] [CrossRef] [Green Version]
- Cannoodt, R.; Saelens, W.; Saeys, Y. Computational methods for trajectory inference from single-cell transcriptomics. Eur. J. Immunol. 2016, 46, 2496–2506. [Google Scholar] [CrossRef] [PubMed]
- Todorov, H.; Cannoodt, R.; Saelens, W.; Saeys, Y. Network Inference from Single-Cell Transcriptomic Data. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2019; Volume 1883, pp. 235–249. [Google Scholar]
- Lescroart, F.; Kelly, R.G.; Le Garrec, J.-F.; Nicolas, J.-F.; Meilhac, S.M.; Buckingham, M. Clonal analysis reveals common lineage relationships between head muscles and second heart field derivatives in the mouse embryo. Development 2010, 137, 3269–3279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bredman, J.J.; Weijs, W.A.; Korfage, H.A.; Brugman, P.; Moorman, A.F. Myosin heavy chain expression in rabbit masseter muscle during postnatal development. J. Anat. 1992, 180 Pt 2, 263–274. [Google Scholar]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Waters, J.; Leung, M.L.; Unruh, A.; Roh, W.; Shi, X.; Chen, K.; Scheet, P.; Vattathil, S.; Liang, H.; et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 2014, 512, 155–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Nuno, K.; Litzenburger, U.M.; Qi, Y.; Corces, M.R.; Majeti, R.; Chang, H.Y. Single-cell lineage tracing by endogenous mutations enriched in transposase accessible mitochondrial DNA. eLife 2019, 8, e45105. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Lin, C.; Bar-Joseph, Z. Cell lineage inference from SNP and scRNA-Seq data. Nucleic Acids Res. 2019, 47, e56. [Google Scholar] [CrossRef] [Green Version]
- Lioux, G.; Liu, X.; Temiño, S.; Oxendine, M.; Ayala, E.; Ortega, S.; Kelly, R.G.; Oliver, G.; Torres, M. A Second Heart Field-Derived Vasculogenic Niche Contributes to Cardiac Lymphatics. Dev. Cell 2020, 52, 350–363. [Google Scholar] [CrossRef] [PubMed]
- Gittenberger-De Groot, A.C.; Vrancken Peeters, M.P.F.M.; Bergwerff, M.; Mentink, M.M.T.; Poelmann, R.E. Epicardial outgrowth inhibition leads to compensatory mesothelial outflow tract collar and abnormal cardiac septation and coronary formation. Circ. Res. 2000, 87, 969–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arques, C.G.; Doohan, R.; Sharpe, J.; Torres, M. Cell tracing reveals a dorsoventral lineage restriction plane in the mouse limb bud mesenchyme. Development 2007, 134, 3713–3722. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Chen, W.; Li, W.; Li, Y.; Priest, J.R.; Zhou, B.; Wang, J.; Zhou, Z. Single-Cell RNA-Seq of the Developing Cardiac Outflow Tract Reveals Convergent Development of the Vascular Smooth Muscle Cells. Cell Rep. 2019, 28, 1346–1361. [Google Scholar] [CrossRef] [Green Version]
- Kruse, F.; Junker, J.P.; van Oudenaarden, A.; Bakkers, J. Tomo-seq: A method to obtain genome-wide expression data with spatial resolution. Methods Cell Biol. 2016, 135, 299–307. [Google Scholar] [CrossRef]
- Junker, J.P.; Noel, E.S.; Guryev, V.; Peterson, K.A.; Shah, G.; Huisken, J.; McMahon, A.P.; Berezikov, E.; Bakkers, J.; Van Oudenaarden, A.; et al. Genome-wide RNA Tomography in the Zebrafish Embryo. Cell 2014, 159, 662–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovatt, D.; Ruble, B.K.; Lee, J.; Dueck, H.; Kim, T.K.; Fisher, S.; Francis, C.; Spaethling, J.M.; Wolf, J.A.; Grady, M.S.; et al. Transcriptome in vivo analysis (TIVA) of spatially defined single cells in live tissue. Nat. Methods 2014, 11, 190–196. [Google Scholar] [CrossRef] [Green Version]
- Rutkovskiy, A.; Malashicheva, A.; Sullivan, G.; Bogdanova, M.; Kostareva, A.; Stensløkken, K.O.; Fiane, A.; Vaage, J. Valve interstitial cells: The key to understanding the pathophysiology of heart valve calcification. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Raj, B.; Wagner, D.E.; McKenna, A.; Pandey, S.; Klein, A.M.; Shendure, J.; Gagnon, J.A.; Schier, A.F. Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain. Nat. Biotechnol. 2018, 36, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Alemany, A.; Florescu, M.; Baron, C.S.; Peterson-Maduro, J.; Van Oudenaarden, A. Whole-organism clone tracing using single-cell sequencing. Nature 2018, 556, 108–112. [Google Scholar] [CrossRef]
- Spanjaard, B.; Hu, B.; Mitic, N.; Olivares-Chauvet, P.; Janjuha, S.; Ninov, N.; Junker, J.P. Simultaneous lineage tracing and cell-type identification using CrIsPr-Cas9-induced genetic scars. Nat. Biotechnol. 2018, 36, 469–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junker, J.P.; Spanjaard, B.; Peterson-Maduro, J.; Alemany, A.; Hu, B.; Florescu, M.; van Oudenaarden, A. Massively parallel clonal analysis using CRISPR/Cas9 induced genetic scars. bioRxiv 2016, 56499. [Google Scholar] [CrossRef] [Green Version]
- Weinreb, C.; Rodriguez-Fraticelli, A.; Camargo, F.D.; Klein, A.M. Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 2020, 367, eaaw3381. [Google Scholar] [CrossRef]
- Pei, W.; Feyerabend, T.B.; Rössler, J.; Wang, X.; Postrach, D.; Busch, K.; Rode, I.; Klapproth, K.; Dietlein, N.; Quedenau, C.; et al. Polylox barcoding reveals haematopoietic stem cell fates realized in vivo. Nature 2017, 548, 456–460. [Google Scholar] [CrossRef] [Green Version]
- Wagner, D.E.; Klein, A.M. Lineage tracing meets single-cell omics: Opportunities and challenges. Nat. Rev. Genet. 2020, 21, 410–427. [Google Scholar] [CrossRef]
- Wei, C.J.Y.; Zhang, K. Retrace: Simultaneous retrospective lineage tracing and methylation profiling of single cells. Genome Res. 2020, 30, 602–610. [Google Scholar] [CrossRef] [Green Version]
- Biezuner, T.; Spiro, A.; Raz, O.; Amir, S.; Milo, L.; Adar, R.; Chapal-Ilani, N.; Berman, V.; Fried, Y.; Ainbinder, E.; et al. A generic, cost-effective, and scalable cell lineage analysis platform. Genome Res. 2016, 26, 1588–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Siejka-Zielińska, P.; Velikova, G.; Bi, Y.; Yuan, F.; Tomkova, M.; Bai, C.; Chen, L.; Schuster-Böckler, B.; Song, C.X. Bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution. Nat. Biotechnol. 2019, 37, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Ku, W.L.; Nakamura, K.; Gao, W.; Cui, K.; Hu, G.; Tang, Q.; Ni, B.; Zhao, K. Single-cell chromatin immunocleavage sequencing (scChIC-seq) to profile histone modification. Nat. Methods 2019, 16, 323–325. [Google Scholar] [CrossRef] [PubMed]
- Regev, A.; Teichmann, S.A.; Lander, E.S.; Amit, I.; Benoist, C.; Birney, E.; Bodenmiller, B.; Campbell, P.; Carninci, P.; Clatworthy, M.; et al. Science forum: The human cell atlas. eLife 2017, 6, e27041. [Google Scholar] [CrossRef] [PubMed]
- Pullen, L.C. Human Cell Atlas Poised to Transform Our Understanding of Organs. Am. J. Transplant. 2018. [Google Scholar] [CrossRef] [Green Version]
- Regev, A.; Teichmann, S.; Rozenblatt-Rosen, O.; Stubbington, M.; Ardlie, K.; Amit, I.; Arlotta, P.; Bader, G.; Benoist, C.; Biton, M.; et al. The Human Cell Atlas White Paper. arXiv 2018, arXiv:1810.05192. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peterson, J.C.; Kelder, T.P.; Goumans, M.J.T.H.; Jongbloed, M.R.M.; DeRuiter, M.C. The Role of Cell Tracing and Fate Mapping Experiments in Cardiac Outflow Tract Development, New Opportunities through Emerging Technologies. J. Cardiovasc. Dev. Dis. 2021, 8, 47. https://doi.org/10.3390/jcdd8050047
Peterson JC, Kelder TP, Goumans MJTH, Jongbloed MRM, DeRuiter MC. The Role of Cell Tracing and Fate Mapping Experiments in Cardiac Outflow Tract Development, New Opportunities through Emerging Technologies. Journal of Cardiovascular Development and Disease. 2021; 8(5):47. https://doi.org/10.3390/jcdd8050047
Chicago/Turabian StylePeterson, Joshua C., Tim P. Kelder, Marie José T. H. Goumans, Monique R. M. Jongbloed, and Marco C. DeRuiter. 2021. "The Role of Cell Tracing and Fate Mapping Experiments in Cardiac Outflow Tract Development, New Opportunities through Emerging Technologies" Journal of Cardiovascular Development and Disease 8, no. 5: 47. https://doi.org/10.3390/jcdd8050047
APA StylePeterson, J. C., Kelder, T. P., Goumans, M. J. T. H., Jongbloed, M. R. M., & DeRuiter, M. C. (2021). The Role of Cell Tracing and Fate Mapping Experiments in Cardiac Outflow Tract Development, New Opportunities through Emerging Technologies. Journal of Cardiovascular Development and Disease, 8(5), 47. https://doi.org/10.3390/jcdd8050047