
Leukocyte-Mediated Cardiac Repair after Myocardial Infarction in Non-Regenerative vs. Regenerative Systems


Abstract
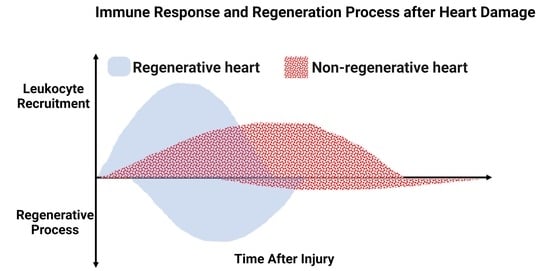
:
1. Introduction
2. Leukocytes Examined in Non-Regenerative and Regenerative Models
2.1. Neutrophils
2.2. Monocytes/Macrophages
2.3. T Lymphocytes
3. Leukocytes Currently Only Examined in Non-Regenerative Models
3.1. Eosinophils
3.2. Basophils
3.3. Dendritic Cells
3.4. Natural Killer Cells
3.5. B Lymphocytes
4. Leukocyte Interactions Examined in Non-Regenerative and Regenerative Models
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Centers for Disease Control and Prevention. Underlying Cause of Death, 1999–2018; CDC WONDER Online Database; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2018. Available online: https://wonder.cdc.gov/ucd-icd10.html (accessed on 12 March 2020).
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2021 Update: A Report from the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [Green Version]
- Elliott, M.R.; Koster, K.M.; Murphy, P.S. Efferocytosis Signaling in the Regulation of Macrophage Inflammatory Responses. J. Immunol. 2017, 198, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The extracellular matrix in myocardial injury, repair, and remodeling. J. Clin. Investig. 2017, 127, 1600–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, M.A.; Braunwald, E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation 1990, 81, 1161–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jessup, M.; Brozena, S. Heart failure. N. Engl. J. Med. 2003, 348, 2007–2018. [Google Scholar] [CrossRef]
- Kehat, I.; Molkentin, J.D. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation 2010, 122, 2727–2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabé-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; Dos Remedios, C.; et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015, 161, 1566–1575. [Google Scholar] [CrossRef] [Green Version]
- Senyo, S.E.; Steinhauser, M.L.; Pizzimenti, C.L.; Yang, V.K.; Cai, L.; Wang, M.; Wu, T.D.; Guerquin-Kern, J.L.; Lechene, C.P.; Lee, R.T. Mammalian heart renewal by pre-existing cardiomyocytes. Nature 2013, 493, 433–436. [Google Scholar] [CrossRef] [Green Version]
- Haubner, B.J.; Schneider, J.; Schweigmann, U.; Schuetz, T.; Dichtl, W.; Velik-Salchner, C.; Stein, J.I.; Penninger, J.M. Functional Recovery of a Human Neonatal Heart After Severe Myocardial Infarction. Circ. Res. 2016, 118, 216–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, N.T.; Sadek, H.A. Neonatal Heart Regeneration: Comprehensive Literature Review. Circulation 2018, 138, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.C.; Pereira, A.H.M.; Sadek, H.A. Mechanisms of Neonatal Heart Regeneration. Curr. Cardiol. Rep. 2020, 22, 33. [Google Scholar] [CrossRef] [PubMed]
- Eschenhagen, T.; Bolli, R.; Braun, T.; Field, L.J.; Fleischmann, B.K.; Frisén, J.; Giacca, M.; Hare, J.M.; Houser, S.; Lee, R.T.; et al. Cardiomyocyte Regeneration: A Consensus Statement. Circulation 2017, 136, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.J.; Rosenthal, N. Preparing the ground for tissue regeneration: From mechanism to therapy. Nat. Med. 2014, 20, 857–869. [Google Scholar] [CrossRef]
- Laflamme, M.A.; Murry, C.E. Heart regeneration. Nature 2011, 473, 326–335. [Google Scholar] [CrossRef] [Green Version]
- Beffagna, G. Zebrafish as a Smart Model to Understand Regeneration After Heart Injury: How Fish Could Help Humans. Front. Cardiovasc. Med. 2019, 6, 107. [Google Scholar] [CrossRef] [Green Version]
- Iribarne, M. Inflammation induces zebrafish regeneration. Neural Regen. Res. 2021, 16, 1693–1701. [Google Scholar] [CrossRef]
- Poss, K.D.; Wilson, L.G.; Keating, M.T. Heart regeneration in zebrafish. Science 2002, 298, 2188–2190. [Google Scholar] [CrossRef]
- Wang, J.; Panáková, D.; Kikuchi, K.; Holdway, J.E.; Gemberling, M.; Burris, J.S.; Singh, S.P.; Dickson, A.L.; Lin, Y.F.; Sabeh, M.K.; et al. The regenerative capacity of zebrafish reverses cardiac failure caused by genetic cardiomyocyte depletion. Development 2011, 138, 3421–3430. [Google Scholar] [CrossRef] [Green Version]
- González-Rosa, J.M.; Martín, V.; Peralta, M.; Torres, M.; Mercader, N. Extensive scar formation and regression during heart regeneration after cryoinjury in zebrafish. Development 2011, 138, 1663–1674. [Google Scholar] [CrossRef] [Green Version]
- Chablais, F.; Veit, J.; Rainer, G.; Jaźwińska, A. The zebrafish heart regenerates after cryoinjury-induced myocardial infarction. BMC Dev. Biol. 2011, 11, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnabel, K.; Wu, C.C.; Kurth, T.; Weidinger, G. Regeneration of cryoinjury induced necrotic heart lesions in zebrafish is associated with epicardial activation and cardiomyocyte proliferation. PLoS ONE 2011, 6, e18503. [Google Scholar] [CrossRef]
- Jopling, C.; Sleep, E.; Raya, M.; Martí, M.; Raya, A.; Izpisúa Belmonte, J.C. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature 2010, 464, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Holdway, J.E.; Werdich, A.A.; Anderson, R.M.; Fang, Y.; Egnaczyk, G.F.; Evans, T.; Macrae, C.A.; Stainier, D.Y.; Poss, K.D. Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature 2010, 464, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cao, J.; Dickson, A.L.; Poss, K.D. Epicardial regeneration is guided by cardiac outflow tract and Hedgehog signalling. Nature 2015, 522, 226–230. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Borikova, A.L.; Ben-Yair, R.; Guner-Ataman, B.; MacRae, C.A.; Lee, R.T.; Burns, C.G.; Burns, C.E. Notch signaling regulates cardiomyocyte proliferation during zebrafish heart regeneration. Proc. Natl. Acad. Sci. USA 2014, 111, 1403–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raya, A.; Koth, C.M.; Büscher, D.; Kawakami, Y.; Itoh, T.; Raya, R.M.; Sternik, G.; Tsai, H.J.; Rodríguez-Esteban, C.; Izpisúa-Belmonte, J.C. Activation of Notch signaling pathway precedes heart regeneration in zebrafish. Proc. Natl. Acad. Sci. USA 2003, 100 (Suppl. S1), 11889–11895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikuchi, K.; Holdway, J.E.; Major, R.J.; Blum, N.; Dahn, R.D.; Begemann, G.; Poss, K.D. Retinoic acid production by endocardium and epicardium is an injury response essential for zebrafish heart regeneration. Dev. Cell 2011, 20, 397–404. [Google Scholar] [CrossRef] [Green Version]
- Schindler, Y.L.; Garske, K.M.; Wang, J.; Firulli, B.A.; Firulli, A.B.; Poss, K.D.; Yelon, D. Hand2 elevates cardiomyocyte production during zebrafish heart development and regeneration. Development 2014, 141, 3112–3122. [Google Scholar] [CrossRef] [Green Version]
- Chablais, F.; Jazwinska, A. The regenerative capacity of the zebrafish heart is dependent on TGFβ signaling. Development 2012, 139, 1921–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Harrison, M.R.; Osorio, A.; Kim, J.; Baugh, A.; Duan, C.; Sucov, H.M.; Lien, C.L. Igf Signaling is Required for Cardiomyocyte Proliferation during Zebrafish Heart Development and Regeneration. PLoS ONE 2013, 8, e67266. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Wu, Q.; Zhang, Y.; Wiens, K.M.; Huang, Y.; Rubin, N.; Shimada, H.; Handin, R.I.; Chao, M.Y.; Tuan, T.L.; et al. PDGF signaling is required for epicardial function and blood vessel formation in regenerating zebrafish hearts. Proc. Natl. Acad. Sci. USA 2010, 107, 17206–17210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.H.; Jung, S.Y.; Yoo, S.Y.; Yoo, S.M.; Kim, D.Y.; Kang, S.; Baek, S.H.; Kwon, S.M. Regulation of ROS-independent ERK signaling rescues replicative cellular senescence in ex vivo expanded human c-kit-positive cardiac progenitor cells. Int. J. Cardiol. 2013, 169, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.; Suñé, G.; Faucherre, A.; Fabregat, C.; Izpisua Belmonte, J.C. Hypoxia induces myocardial regeneration in zebrafish. Circulation 2012, 126, 3017–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Gupta, V.; Karra, R.; Holdway, J.E.; Kikuchi, K.; Poss, K.D. Translational profiling of cardiomyocytes identifies an early Jak1/Stat3 injury response required for zebrafish heart regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 13416–13421. [Google Scholar] [CrossRef] [Green Version]
- Yin, V.P.; Lepilina, A.; Smith, A.; Poss, K.D. Regulation of zebrafish heart regeneration by miR-133. Dev. Biol. 2012, 365, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, K. Dedifferentiation, Transdifferentiation, and Proliferation: Mechanisms Underlying Cardiac Muscle Regeneration in Zebrafish. Curr. Pathobiol. Rep. 2015, 3, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient regenerative potential of the neonatal mouse heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef] [Green Version]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Johnson, B.A.; Grinsfelder, D.; Canseco, D.; Mammen, P.P.; Rothermel, B.A.; Olson, E.N.; Sadek, H.A. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc. Natl. Acad. Sci. USA 2013, 110, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Haubner, B.J.; Adamowicz-Brice, M.; Khadayate, S.; Tiefenthaler, V.; Metzler, B.; Aitman, T.; Penninger, J.M. Complete cardiac regeneration in a mouse model of myocardial infarction. Aging 2012, 4, 966–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.; D’Agostino, G.; Loo, S.J.; Wang, C.X.; Su, L.P.; Tan, S.H.; Tee, G.Z.; Pua, C.J.; Pena, E.M.; Cheng, R.B.; et al. Early Regenerative Capacity in the Porcine Heart. Circulation 2018, 138, 2798–2808. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Tan, J.; Yan, J.; Zhang, L.; Cai, X.; Wang, H.; Liu, F.; Ye, M.; Cai, C.L. Limited Regeneration Potential with Minimal Epicardial Progenitor Conversions in the Neonatal Mouse Heart after Injury. Cell Rep. 2019, 28, 190–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, A.; Baker, C.D.; Pritchett, E.M.; Burgos Villar, K.N.; Ashton, J.M.; Small, E.M. Characterizing Neonatal Heart Maturation, Regeneration, and Scar Resolution Using Spatial Transcriptomics. J. Cardiovasc. Dev. Dis. 2021, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Regulation of the inflammatory response in cardiac repair. Circ. Res. 2012, 110, 159–173. [Google Scholar] [CrossRef]
- Huang, W.C.; Yang, C.C.; Chen, I.H.; Liu, Y.M.; Chang, S.J.; Chuang, Y.J. Treatment of Glucocorticoids Inhibited Early Immune Responses and Impaired Cardiac Repair in Adult Zebrafish. PLoS ONE 2013, 8, e66613. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Nie, Y.; Lian, H.; Liu, R.; He, F.; Huang, H.; Hu, S. Acute inflammation stimulates a regenerative response in the neonatal mouse heart. Cell Res. 2015, 25, 1137–1151. [Google Scholar] [CrossRef] [Green Version]
- Kyne, L.; Hausdorff, J.M.; Knight, E.; Dukas, L.; Azhar, G.; Wei, J.Y. Neutrophilia and congestive heart failure after acute myocardial infarction. Am. Heart J. 2000, 139, 94–100. [Google Scholar] [CrossRef]
- Mocatta, T.J.; Pilbrow, A.P.; Cameron, V.A.; Senthilmohan, R.; Frampton, C.M.; Richards, A.M.; Winterbourn, C.C. Plasma concentrations of myeloperoxidase predict mortality after myocardial infarction. J. Am. Coll. Cardiol. 2007, 49, 1993–2000. [Google Scholar] [CrossRef] [Green Version]
- Arruda-Olson, A.M.; Reeder, G.S.; Bell, M.R.; Weston, S.A.; Roger, V.L. Neutrophilia predicts death and heart failure after myocardial infarction: A community-based study. Circ. Cardiovasc. Qual. Outcomes 2009, 2, 656–662. [Google Scholar] [CrossRef] [Green Version]
- Dogan, I.; Karaman, K.; Sonmez, B.; Celik, S.; Turker, O. Relationship between serum neutrophil count and infarct size in patients with acute myocardial infarction. Nucl. Med. Commun. 2009, 30, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Chia, S.; Nagurney, J.T.; Brown, D.F.; Raffel, O.C.; Bamberg, F.; Senatore, F.; Wackers, F.J.; Jang, I.K. Association of leukocyte and neutrophil counts with infarct size, left ventricular function and outcomes after percutaneous coronary intervention for ST-elevation myocardial infarction. Am. J. Cardiol. 2009, 103, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Guasti, L.; Dentali, F.; Castiglioni, L.; Maroni, L.; Marino, F.; Squizzato, A.; Ageno, W.; Gianni, M.; Gaudio, G.; Grandi, A.M.; et al. Neutrophils and clinical outcomes in patients with acute coronary syndromes and/or cardiac revascularisation. A systematic review on more than 34,000 subjects. Thromb. Haemost. 2011, 106, 591–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akpek, M.; Kaya, M.G.; Lam, Y.Y.; Sahin, O.; Elcik, D.; Celik, T.; Ergin, A.; Gibson, C.M. Relation of neutrophil/lymphocyte ratio to coronary flow to in-hospital major adverse cardiac events in patients with ST-elevated myocardial infarction undergoing primary coronary intervention. Am. J. Cardiol. 2012, 110, 621–627. [Google Scholar] [CrossRef]
- Zhang, S.; Diao, J.; Qi, C.; Jin, J.; Li, L.; Gao, X.; Gong, L.; Wu, W. Predictive value of neutrophil to lymphocyte ratio in patients with acute ST segment elevation myocardial infarction after percutaneous coronary intervention: A meta-analysis. BMC Cardiovasc. Disord. 2018, 18, 75. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Anzai, A.; Katsumata, Y.; Matsuhashi, T.; Ito, K.; Endo, J.; Yamamoto, T.; Takeshima, A.; Shinmura, K.; Shen, W.; et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J. Mol. Cell. Cardiol. 2013, 62, 24–35. [Google Scholar] [CrossRef]
- Rusinkevich, V.; Huang, Y.; Chen, Z.Y.; Qiang, W.; Wang, Y.G.; Shi, Y.F.; Yang, H.T. Temporal dynamics of immune response following prolonged myocardial ischemia/reperfusion with and without cyclosporine A. Acta Pharmacol. Sin. 2019, 40, 1168–1183. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Yabluchanskiy, A.; Lindsey, M.L. Neutrophil roles in left ventricular remodeling following myocardial infarction. Fibrogenes. Tissue Repair 2013, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y. Role of Neutrophils in Cardiac Injury and Repair Following Myocardial Infarction. Cells 2021, 10, 1676. [Google Scholar] [CrossRef]
- Bonaventura, A.; Montecucco, F.; Dallegri, F. Cellular recruitment in myocardial ischaemia/reperfusion injury. Eur. J. Clin. Investig. 2016, 46, 590–601. [Google Scholar] [CrossRef]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2017, 38, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavine, K.J.; Epelman, S.; Uchida, K.; Weber, K.J.; Nichols, C.G.; Schilling, J.D.; Ornitz, D.M.; Randolph, G.J.; Mann, D.L. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. USA 2014, 111, 16029–16034. [Google Scholar] [CrossRef] [Green Version]
- Aurora, A.B.; Porrello, E.R.; Tan, W.; Mahmoud, A.I.; Hill, J.A.; Bassel-Duby, R.; Sadek, H.A.; Olson, E.N. Macrophages are required for neonatal heart regeneration. J. Clin. Investig. 2014, 124, 1382–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, S.L.; Marín-Juez, R.; Moura, P.L.; Kuenne, C.; Lai, J.K.H.; Tsedeke, A.T.; Guenther, S.; Looso, M.; Stainier, D.Y. Reciprocal analyses in zebrafish and medaka reveal that harnessing the immune response promotes cardiac regeneration. eLife 2017, 6, e25605. [Google Scholar] [CrossRef]
- Xu, S.; Webb, S.E.; Lau, T.C.K.; Cheng, S.H. Matrix metalloproteinases (MMPs) mediate leukocyte recruitment during the inflammatory phase of zebrafish heart regeneration. Sci. Rep. 2018, 8, 7199. [Google Scholar] [CrossRef]
- Xu, S.; Xie, F.; Tian, L.; Manno, S.H.; Manno, F.A.M., 3rd; Cheng, S.H. Prolonged neutrophil retention in the wound impairs zebrafish heart regeneration after cryoinjury. Fish Shellfish Immunol. 2019, 94, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Bevan, L.; Lim, Z.W.; Venkatesh, B.; Riley, P.R.; Martin, P.; Richardson, R.J. Specific macrophage populations promote both cardiac scar deposition and subsequent resolution in adult zebrafish. Cardiovasc. Res. 2020, 116, 1357–1371. [Google Scholar] [CrossRef] [Green Version]
- Van der Laan, A.M.; Ter Horst, E.N.; Delewi, R.; Begieneman, M.P.; Krijnen, P.A.; Hirsch, A.; Lavaei, M.; Nahrendorf, M.; Horrevoets, A.J.; Niessen, H.W.; et al. Monocyte subset accumulation in the human heart following acute myocardial infarction and the role of the spleen as monocyte reservoir. Eur. Heart J. 2014, 35, 376–385. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, Y.; Anzai, T.; Yoshikawa, T.; Asakura, Y.; Takahashi, T.; Ishikawa, S.; Mitamura, H.; Ogawa, S. Prognostic significance of peripheral monocytosis after reperfused acute myocardial infarction:a possible role for left ventricular remodeling. J. Am. Coll. Cardiol. 2002, 39, 241–246. [Google Scholar] [CrossRef] [Green Version]
- Mariani, M.; Fetiveau, R.; Rossetti, E.; Poli, A.; Poletti, F.; Vandoni, P.; D’Urbano, M.; Cafiero, F.; Mariani, G.; Klersy, C.; et al. Significance of total and differential leucocyte count in patients with acute myocardial infarction treated with primary coronary angioplasty. Eur. Heart J. 2006, 27, 2511–2515. [Google Scholar] [CrossRef]
- Tsujioka, H.; Imanishi, T.; Ikejima, H.; Kuroi, A.; Takarada, S.; Tanimoto, T.; Kitabata, H.; Okochi, K.; Arita, Y.; Ishibashi, K.; et al. Impact of heterogeneity of human peripheral blood monocyte subsets on myocardial salvage in patients with primary acute myocardial infarction. J. Am. Coll. Cardiol. 2009, 54, 130–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Laan, A.M.; Hirsch, A.; Robbers, L.F.; Nijveldt, R.; Lommerse, I.; Delewi, R.; van der Vleuten, P.A.; Biemond, B.J.; Zwaginga, J.J.; van der Giessen, W.J.; et al. A proinflammatory monocyte response is associated with myocardial injury and impaired functional outcome in patients with ST-segment elevation myocardial infarction: Monocytes and myocardial infarction. Am. Heart J. 2012, 163, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Ruparelia, N.; Godec, J.; Lee, R.; Chai, J.T.; Dall’Armellina, E.; McAndrew, D.; Digby, J.E.; Forfar, J.C.; Prendergast, B.D.; Kharbanda, R.K.; et al. Acute myocardial infarction activates distinct inflammation and proliferation pathways in circulating monocytes, prior to recruitment, and identified through conserved transcriptional responses in mice and humans. Eur. Heart J. 2015, 36, 1923–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajpai, G.; Schneider, C.; Wong, N.; Bredemeyer, A.; Hulsmans, M.; Nahrendorf, M.; Epelman, S.; Kreisel, D.; Liu, Y.; Itoh, A.; et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 2018, 24, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef] [Green Version]
- Bajpai, G.; Bredemeyer, A.; Li, W.; Zaitsev, K.; Koenig, A.L.; Lokshina, I.; Mohan, J.; Ivey, B.; Hsiao, H.M.; Weinheimer, C.; et al. Tissue Resident CCR2- and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ. Res. 2019, 124, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; Althagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 2019, 20, 29–39. [Google Scholar] [CrossRef]
- Sager, H.B.; Hulsmans, M.; Lavine, K.J.; Moreira, M.B.; Heidt, T.; Courties, G.; Sun, Y.; Iwamoto, Y.; Tricot, B.; Khan, O.F.; et al. Proliferation and Recruitment Contribute to Myocardial Macrophage Expansion in Chronic Heart Failure. Circ. Res. 2016, 119, 853–864. [Google Scholar] [CrossRef] [Green Version]
- Panizzi, P.; Swirski, F.K.; Figueiredo, J.L.; Waterman, P.; Sosnovik, D.E.; Aikawa, E.; Libby, P.; Pittet, M.; Weissleder, R.; Nahrendorf, M. Impaired infarct healing in atherosclerotic mice with Ly-6C(hi) monocytosis. J. Am. Coll. Cardiol. 2010, 55, 1629–1638. [Google Scholar] [CrossRef] [Green Version]
- Kaikita, K.; Hayasaki, T.; Okuma, T.; Kuziel, W.A.; Ogawa, H.; Takeya, M. Targeted deletion of CC chemokine receptor 2 attenuates left ventricular remodeling after experimental myocardial infarction. Am. J. Pathol. 2004, 165, 439–447. [Google Scholar] [CrossRef] [Green Version]
- Van Amerongen, M.J.; Harmsen, M.C.; van Rooijen, N.; Petersen, A.H.; van Luyn, M.J. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am. J. Pathol. 2007, 170, 818–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen-Chi, M.; Laplace-Builhe, B.; Travnickova, J.; Luz-Crawford, P.; Tejedor, G.; Phan, Q.T.; Duroux-Richard, I.; Levraud, J.P.; Kissa, K.; Lutfalla, G.; et al. Identification of polarized macrophage subsets in zebrafish. eLife 2015, 4, e07288. [Google Scholar] [CrossRef] [PubMed]
- De Preux Charles, A.S.; Bise, T.; Baier, F.; Marro, J.; Jaźwińska, A. Distinct effects of inflammation on preconditioning and regeneration of the adult zebrafish heart. Open Biol. 2016, 6, 160102. [Google Scholar] [CrossRef] [Green Version]
- Sanz-Morejón, A.; García-Redondo, A.B.; Reuter, H.; Marques, I.J.; Bates, T.; Galardi-Castilla, M.; Große, A.; Manig, S.; Langa, X.; Ernst, A.; et al. Wilms Tumor 1b Expression Defines a Pro-regenerative Macrophage Subtype and Is Required for Organ Regeneration in the Zebrafish. Cell Rep. 2019, 28, 1296–1306. [Google Scholar] [CrossRef] [Green Version]
- Boag, S.E.; Das, R.; Shmeleva, E.V.; Bagnall, A.; Egred, M.; Howard, N.; Bennaceur, K.; Zaman, A.; Keavney, B.; Spyridopoulos, I. T lymphocytes and fractalkine contribute to myocardial ischemia/reperfusion injury in patients. J. Clin. Investig. 2015, 125, 3063–3076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.; Zhou, W. Peripheral neutrophils and naive CD4 T cells predict the development of heart failure following acute myocardial infarction: A bioinformatic study. Rev. Port. Cardiol. 2021, 40, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, U.; Beyersdorf, N.; Weirather, J.; Podolskaya, A.; Bauersachs, J.; Ertl, G.; Kerkau, T.; Frantz, S. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation 2012, 125, 1652–1663. [Google Scholar] [CrossRef] [Green Version]
- Methe, H.; Brunner, S.; Wiegand, D.; Nabauer, M.; Koglin, J.; Edelman, E.R. Enhanced T-helper-1 lymphocyte activation patterns in acute coronary syndromes. J. Am. Coll. Cardiol. 2005, 45, 1939–1945. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Yu, X.; Ding, Y.J.; Fu, Q.Q.; Xie, J.J.; Tang, T.T.; Yao, R.; Chen, Y.; Liao, Y.H. The Th17/Treg imbalance in patients with acute coronary syndrome. Clin. Immunol. 2008, 127, 89–97. [Google Scholar] [CrossRef]
- Zhu, F.; Wang, Q.; Guo, C.; Wang, X.; Cao, X.; Shi, Y.; Gao, F.; Ma, C.; Zhang, L. IL-17 induces apoptosis of vascular endothelial cells—A potential mechanism for human acute coronary syndrome. Clin. Immunol. 2011, 141, 152–160. [Google Scholar] [CrossRef]
- Hashmi, S.; Zeng, Q.T. Role of interleukin-17 and interleukin-17-induced cytokines interleukin-6 and interleukin-8 in unstable coronary artery disease. Coron. Artery Dis. 2006, 17, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Z.; Wang, D.; Zhu, J.; Wang, Y. CD8+CD28+ T cells might mediate injury of cardiomyocytes in acute myocardial infarction. Mol. Immunol. 2018, 101, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Tae Yu, H.; Youn, J.C.; Lee, J.; Park, S.; Chi, H.S.; Lee, J.; Choi, C.; Park, S.; Choi, D.; Ha, J.W.; et al. Characterization of CD8+CD57+ T cells in patients with acute myocardial infarction. Cell. Mol. Immunol. 2015, 12, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Forte, E.; Perkins, B.; Sintou, A.; Kalkat, H.S.; Papanikolaou, A.; Jenkins, C.; Alsubaie, M.; Chowdhury, R.A.; Duffy, T.M.; Skelly, D.A.; et al. Cross-Priming Dendritic Cells Exacerbate Immunopathology After Ischemic Tissue Damage in the Heart. Circulation 2021, 143, 821–836. [Google Scholar] [CrossRef]
- Tang, T.T.; Zhu, Y.C.; Dong, N.G.; Zhang, S.; Cai, J.; Zhang, L.X.; Han, Y.; Xia, N.; Nie, S.F.; Zhang, M.; et al. Pathologic T-cell response in ischaemic failing hearts elucidated by T-cell receptor sequencing and phenotypic characterization. Eur. Heart J. 2019, 40, 3924–3933. [Google Scholar] [CrossRef]
- Okamoto, N.; Noma, T.; Ishihara, Y.; Miyauchi, Y.; Takabatake, W.; Oomizu, S.; Yamaoka, G.; Ishizawa, M.; Namba, T.; Murakami, K.; et al. Prognostic value of circulating regulatory T cells for worsening heart failure in heart failure patients with reduced ejection fraction. Int. Heart J. 2014, 55, 271–277. [Google Scholar] [CrossRef] [Green Version]
- Tang, T.T.; Ding, Y.J.; Liao, Y.H.; Yu, X.; Xiao, H.; Xie, J.J.; Yuan, J.; Zhou, Z.H.; Liao, M.Y.; Yao, R.; et al. Defective circulating CD4CD25+Foxp3+CD127(low) regulatory T-cells in patients with chronic heart failure. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2010, 25, 451–458. [Google Scholar] [CrossRef]
- Yang, Z.; Day, Y.J.; Toufektsian, M.C.; Xu, Y.; Ramos, S.I.; Marshall, M.A.; French, B.A.; Linden, J. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation 2006, 114, 2056–2064. [Google Scholar] [CrossRef] [Green Version]
- Weirather, J.; Hofmann, U.D.; Beyersdorf, N.; Ramos, G.C.; Vogel, B.; Frey, A.; Ertl, G.; Kerkau, T.; Frantz, S. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ. Res. 2014, 115, 55–67. [Google Scholar] [CrossRef]
- Curato, C.; Slavic, S.; Dong, J.; Skorska, A.; Altarche-Xifró, W.; Miteva, K.; Kaschina, E.; Thiel, A.; Imboden, H.; Wang, J.; et al. Identification of Noncytotoxic and IL-10–Producing CD8+ AT2R+ T Cell Population in Response to Ischemic Heart Injury. J. Immunol. 2010, 185, 6286. [Google Scholar] [CrossRef] [Green Version]
- Bansal, S.S.; Ismahil, M.A.; Goel, M.; Patel, B.; Hamid, T.; Rokosh, G.; Prabhu, S.D. Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure. Circ. Heart Fail. 2017, 10, e003688. [Google Scholar] [CrossRef] [Green Version]
- Santos-Zas, I.; Lemarié, J.; Zlatanova, I.; Cachanado, M.; Seghezzi, J.C.; Benamer, H.; Goube, P.; Vandestienne, M.; Cohen, R.; Ezzo, M.; et al. Cytotoxic CD8(+) T cells promote granzyme B-dependent adverse post-ischemic cardiac remodeling. Nat. Commun. 2021, 12, 1483. [Google Scholar] [CrossRef] [PubMed]
- Komai, K.; Ito, M.; Nomura, S.; Shichino, S.; Katoh, M.; Yamada, S.; Ko, T.; Iizuka-Koga, M.; Nakatsukasa, H.; Yoshimura, A. Single-Cell Analysis Revealed the Role of CD8(+) Effector T Cells in Preventing Cardioprotective Macrophage Differentiation in the Early Phase of Heart Failure. Front. Immunol. 2021, 12, 763647. [Google Scholar] [CrossRef] [PubMed]
- Ilatovskaya, D.V.; Pitts, C.; Clayton, J.; Domondon, M.; Troncoso, M.; Pippin, S.; DeLeon-Pennell, K.Y. CD8(+) T-cells negatively regulate inflammation post-myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H581–H596. [Google Scholar] [CrossRef]
- Liao, Y.-H.; Xia, N.; Zhou, S.-F.; Tang, T.-T.; Yan, X.-X.; Lv, B.-J.; Nie, S.-F.; Wang, J.; Iwakura, Y.; Xiao, H.; et al. Interleukin-17A Contributes to Myocardial Ischemia/Reperfusion Injury by Regulating Cardiomyocyte Apoptosis and Neutrophil Infiltration. J. Am. Coll. Cardiol. 2012, 59, 420–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X.; Shichita, T.; Katsumata, Y.; Matsuhashi, T.; Ito, H.; Ito, K.; Anzai, A.; Endo, J.; Tamura, Y.; Kimura, K.; et al. Deleterious Effect of the IL-23/IL-17A Axis and γδT cells on Left Ventricular Remodeling After Myocardial Infarction. Circulation 2012, 1, e004408. [Google Scholar] [CrossRef] [Green Version]
- Rieckmann, M.; Delgobo, M.; Gaal, C.; Büchner, L.; Steinau, P.; Reshef, D.; Gil-Cruz, C.; Horst, E.N.T.; Kircher, M.; Reiter, T.; et al. Myocardial infarction triggers cardioprotective antigen-specific T helper cell responses. J. Clin. Investig. 2019, 129, 4922–4936. [Google Scholar] [CrossRef] [Green Version]
- Saxena, A.; Dobaczewski, M.; Rai, V.; Haque, Z.; Chen, W.; Li, N.; Frangogiannis, N.G. Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function. Am. J. Physiol.-Heart Circ. Physiol. 2014, 307, H1233–H1242. [Google Scholar] [CrossRef]
- Zouggari, Y.; Ait-Oufella, H.; Waeckel, L.; Vilar, J.; Loinard, C.; Cochain, C.; Récalde, A.; Duriez, M.; Levy, B.I.; Lutgens, E.; et al. Regulatory T cells modulate postischemic neovascularization. Circulation 2009, 120, 1415–1425. [Google Scholar] [CrossRef] [Green Version]
- Dobaczewski, M.; Xia, Y.; Bujak, M.; Gonzalez-Quesada, C.; Frangogiannis, N.G. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am. J. Pathol. 2010, 176, 2177–2187. [Google Scholar] [CrossRef]
- Matsumoto, K.; Ogawa, M.; Suzuki, J.; Hirata, Y.; Nagai, R.; Isobe, M. Regulatory T lymphocytes attenuate myocardial infarction-induced ventricular remodeling in mice. Int. Heart J. 2011, 52, 382–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, T.T.; Yuan, J.; Zhu, Z.F.; Zhang, W.C.; Xiao, H.; Xia, N.; Yan, X.X.; Nie, S.F.; Liu, J.; Zhou, S.F.; et al. Regulatory T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res. Cardiol. 2012, 107, 232. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Jiao, J.; Tang, T.-T.; Lv, B.-J.; Lu, Y.-Z.; Wang, K.-J.; Zhu, Z.-F.; Mao, X.-B.; Nie, S.-F.; Wang, Q.; et al. Activated regulatory T-cells attenuate myocardial ischaemia/reperfusion injury through a CD39-dependent mechanism. Clin. Sci. 2015, 128, 679–693. [Google Scholar] [CrossRef]
- Zacchigna, S.; Martinelli, V.; Moimas, S.; Colliva, A.; Anzini, M.; Nordio, A.; Costa, A.; Rehman, M.; Vodret, S.; Pierro, C.; et al. Paracrine effect of regulatory T cells promotes cardiomyocyte proliferation during pregnancy and after myocardial infarction. Nat. Commun. 2018, 9, 2432. [Google Scholar] [CrossRef]
- Li, J.; Yang, K.Y.; Tam, R.C.Y.; Chan, V.W.; Lan, H.Y.; Hori, S.; Zhou, B.; Lui, K.O. Regulatory T-cells regulate neonatal heart regeneration by potentiating cardiomyocyte proliferation in a paracrine manner. Theranostics 2019, 9, 4324–4341. [Google Scholar] [CrossRef]
- Li, J.; Liang, C.; Yang, K.Y.; Huang, X.; Han, M.Y.; Li, X.; Chan, V.W.; Chan, K.S.; Liu, D.; Huang, Z.P.; et al. Specific ablation of CD4(+) T-cells promotes heart regeneration in juvenile mice. Theranostics 2020, 10, 8018–8035. [Google Scholar] [CrossRef]
- Hui, S.P.; Sheng, D.Z.; Sugimoto, K.; Gonzalez-Rajal, A.; Nakagawa, S.; Hesselson, D.; Kikuchi, K. Zebrafish Regulatory T Cells Mediate Organ-Specific Regenerative Programs. Dev. Cell 2017, 43, 659–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rios-Navarro, C.; Gavara, J.; Vidal, V.; Bonanad, C.; Racugno, P.; Bayes-Genis, A.; Miñana, G.; Husser, O.; Oltra, R.; Nuñez, J.; et al. Characterization and implications of the dynamics of eosinophils in blood and in the infarcted myocardium after coronary reperfusion. PLoS ONE 2018, 13, e0206344. [Google Scholar] [CrossRef]
- Shah, A.D.; Denaxas, S.; Nicholas, O.; Hingorani, A.D.; Hemingway, H. Low eosinophil and low lymphocyte counts and the incidence of 12 cardiovascular diseases: A CALIBER cohort study. Open Heart 2016, 3, e000477. [Google Scholar] [CrossRef] [Green Version]
- Konishi, T.; Funayama, N.; Yamamoto, T.; Morita, T.; Hotta, D.; Nishihara, H.; Tanaka, S. Prognostic Value of Eosinophil to Leukocyte Ratio in Patients with ST-Elevation Myocardial Infarction Undergoing Primary Percutaneous Coronary Intervention. J. Atheroscler. Thromb. 2017, 24, 827–840. [Google Scholar] [CrossRef] [Green Version]
- Shiyovich, A.; Gilutz, H.; Plakht, Y. White Blood Cell Subtypes Are Associated with a Greater Long-Term Risk of Death after Acute Myocardial Infarction. Tex. Heart Inst. J. 2017, 44, 176–188. [Google Scholar] [CrossRef] [Green Version]
- Toor, I.S.; Jaumdally, R.; Lip, G.Y.; Millane, T.; Varma, C. Eosinophil count predicts mortality following percutaneous coronary intervention. Thromb. Res. 2012, 130, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Toor, I.S.; Rückerl, D.; Mair, I.; Ainsworth, R.; Meloni, M.; Spiroski, A.M.; Benezech, C.; Felton, J.M.; Thomson, A.; Caporali, A.; et al. Eosinophil Deficiency Promotes Aberrant Repair and Adverse Remodeling Following Acute Myocardial Infarction. JACC Basic Transl. Sci. 2020, 5, 665–681. [Google Scholar] [CrossRef]
- Liu, J.; Yang, C.; Liu, T.; Deng, Z.; Fang, W.; Zhang, X.; Li, J.; Huang, Q.; Liu, C.; Wang, Y.; et al. Eosinophils improve cardiac function after myocardial infarction. Nat. Commun. 2020, 11, 6396. [Google Scholar] [CrossRef] [PubMed]
- Sicklinger, F.; Meyer, I.S.; Li, X.; Radtke, D.; Dicks, S.; Kornadt, M.P.; Mertens, C.; Meier, J.K.; Lavine, K.J.; Zhang, Y.; et al. Basophils balance healing after myocardial infarction via IL-4/IL-13. J. Clin. Investig. 2021, 131, e136778. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, D.; Betge, S.; Windisch, A.; Pistulli, R.; Rohm, I.; Fritzenwanger, M.; Jung, C.; Schubert, K.; Theis, B.; Petersen, I.; et al. Recruitment of circulating dendritic cell precursors into the infarcted myocardium and pro-inflammatory response in acute myocardial infarction. Clin. Sci. 2012, 123, 387–398. [Google Scholar] [CrossRef] [Green Version]
- Carvalheiro, T.; Velada, I.; Valado, A.; Mendes, F.; Martinho, A.; António, N.; Gonçalves, L.; Providência, L.; Pais, M.L.; Paiva, A. Phenotypic and functional alterations on inflammatory peripheral blood cells after acute myocardial infarction. J. Cardiovasc. Transl. Res. 2012, 5, 309–320. [Google Scholar] [CrossRef]
- Fukui, D.; Yasukawa, H.; Sugi, Y.; Oba, T.; Nagata, T.; Kyogoku, S.; Futamata, N.; Yokoyama, T.; Yokoyama, S.; Kai, H.; et al. Transient reduction and activation of circulating dendritic cells in patients with acute myocardial infarction. Int. J. Cardiol. 2012, 160, 216–219. [Google Scholar] [CrossRef]
- Yilmaz, A.; Weber, J.; Cicha, I.; Stumpf, C.; Klein, M.; Raithel, D.; Daniel, W.G.; Garlichs, C.D. Decrease in circulating myeloid dendritic cell precursors in coronary artery disease. J. Am. Coll. Cardiol. 2006, 48, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, A.; Dietel, B.; Cicha, I.; Schubert, K.; Hausmann, R.; Daniel, W.G.; Garlichs, C.D.; Stumpf, C. Emergence of dendritic cells in the myocardium after acute myocardial infarction—Implications for inflammatory myocardial damage. Int. J. Biomed. Sci. IJBS 2010, 6, 27–36. [Google Scholar]
- Nagai, T.; Honda, S.; Sugano, Y.; Matsuyama, T.A.; Ohta-Ogo, K.; Asaumi, Y.; Ikeda, Y.; Kusano, K.; Ishihara, M.; Yasuda, S.; et al. Decreased myocardial dendritic cells is associated with impaired reparative fibrosis and development of cardiac rupture after myocardial infarction in humans. J. Am. Heart Assoc. 2014, 3, e000839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Yu, Z.X.; Fujita, S.; Yamaguchi, M.L.; Ferrans, V.J. Interstitial dendritic cells of the rat heart. Quantitative and ultrastructural changes in experimental myocardial infarction. Circulation 1993, 87, 909–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naito, K.; Anzai, T.; Sugano, Y.; Maekawa, Y.; Kohno, T.; Yoshikawa, T.; Matsuno, K.; Ogawa, S. Differential effects of GM-CSF and G-CSF on infiltration of dendritic cells during early left ventricular remodeling after myocardial infarction. J. Immunol. 2008, 181, 5691–5701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maekawa, Y.; Mizue, N.; Chan, A.; Shi, Y.; Liu, Y.; Dawood, S.; Chen, M.; Dawood, F.; de Couto, G.; Li, G.H.; et al. Survival and cardiac remodeling after myocardial infarction are critically dependent on the host innate immune interleukin-1 receptor-associated kinase-4 signaling: A regulator of bone marrow-derived dendritic cells. Circulation 2009, 120, 1401–1414. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Jeong, S.J.; Kim, S.; Chalifour, L.; Yun, T.J.; Miah, M.A.; Li, B.; Majdoubi, A.; Sabourin, A.; Keler, T.; et al. Conventional Dendritic Cells Impair Recovery after Myocardial Infarction. J. Immunol. 2018, 201, 1784–1798. [Google Scholar] [CrossRef]
- Van der Borght, K.; Scott, C.L.; Nindl, V.; Bouché, A.; Martens, L.; Sichien, D.; Van Moorleghem, J.; Vanheerswynghels, M.; De Prijck, S.; Saeys, Y.; et al. Myocardial Infarction Primes Autoreactive T Cells through Activation of Dendritic Cells. Cell Rep. 2017, 18, 3005–3017. [Google Scholar] [CrossRef] [Green Version]
- Lai, L.; Zhang, A.; Yang, B.; Charles, E.J.; Kron, I.L.; Yang, Z. Plasmacytoid Dendritic Cells Mediate Myocardial Ischemia/Reperfusion Injury by Secreting Type I Interferons. J. Am. Heart Assoc. 2021, 10, e020754. [Google Scholar] [CrossRef]
- Anzai, A.; Anzai, T.; Nagai, S.; Maekawa, Y.; Naito, K.; Kaneko, H.; Sugano, Y.; Takahashi, T.; Abe, H.; Mochizuki, S.; et al. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation 2012, 125, 1234–1245. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Dembowsky, K.; Chevalier, E.; Stüve, P.; Korf-Klingebiel, M.; Lochner, M.; Napp, L.C.; Frank, H.; Brinkmann, E.; Kanwischer, A.; et al. C-X-C Motif Chemokine Receptor 4 Blockade Promotes Tissue Repair After Myocardial Infarction by Enhancing Regulatory T Cell Mobilization and Immune-Regulatory Function. Circulation 2019, 139, 1798–1812. [Google Scholar] [CrossRef]
- Szodoray, P.; Timar, O.; Veres, K.; Der, H.; Szomjak, E.; Lakos, G.; Aleksza, M.; Nakken, B.; Szegedi, G.; Soltesz, P. TH1/TH2 imbalance, measured by circulating and intracytoplasmic inflammatory cytokines—Immunological alterations in acute coronary syndrome and stable coronary artery disease. Scand. J. Immunol. 2006, 64, 336–344. [Google Scholar] [CrossRef] [Green Version]
- Laskarin, G.; Persic, V.; Ruzic, A.; Miletic, B.; Rakic, M.; Samsa, D.T.; Raljevic, D.; Pejcinovic, V.P.; Miskulin, R.; Rukavina, D. Perforin-mediated cytotoxicity in non-ST elevation myocardial infarction. Scand. J. Immunol. 2011, 74, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Rodríguez, A.C.; Marín-Jáuregui, L.S.; Martínez-Shio, E.; Hernández Castro, B.; González-Amaro, R.; Escobedo-Uribe, C.D.; Monsiváis-Urenda, A.E. Altered NK cell receptor repertoire and function of natural killer cells in patients with acute myocardial infarction: A three-month follow-up study. Immunobiology 2020, 225, 151909. [Google Scholar] [CrossRef] [PubMed]
- Klarlund, K.; Pedersen, B.K.; Theander, T.G.; Andersen, V. Depressed natural killer cell activity in acute myocardial infarction. Clin. Exp. Immunol. 1987, 70, 209–216. [Google Scholar] [PubMed]
- Jonasson, L.; Backteman, K.; Ernerudh, J. Loss of natural killer cell activity in patients with coronary artery disease. Atherosclerosis 2005, 183, 316–321. [Google Scholar] [CrossRef]
- Ayach, B.B.; Yoshimitsu, M.; Dawood, F.; Sun, M.; Arab, S.; Chen, M.; Higuchi, K.; Siatskas, C.; Lee, P.; Lim, H.; et al. Stem cell factor receptor induces progenitor and natural killer cell-mediated cardiac survival and repair after myocardial infarction. Proc. Natl. Acad. Sci. USA 2006, 103, 2304–2309. [Google Scholar] [CrossRef] [Green Version]
- Bouchentouf, M.; Forner, K.A.; Cuerquis, J.; Michaud, V.; Zheng, J.; Paradis, P.; Schiffrin, E.L.; Galipeau, J. Induction of cardiac angiogenesis requires killer cell lectin-like receptor 1 and α4β7 integrin expression by NK cells. J. Immunol. 2010, 185, 7014–7025. [Google Scholar] [CrossRef] [Green Version]
- Bouchentouf, M.; Williams, P.; Forner, K.A.; Cuerquis, J.; Michaud, V.; Paradis, P.; Schiffrin, E.L.; Galipeau, J. Interleukin-2 enhances angiogenesis and preserves cardiac function following myocardial infarction. Cytokine 2011, 56, 732–738. [Google Scholar] [CrossRef]
- Luger, D.; Lipinski, M.J.; Westman, P.C.; Glover, D.K.; Dimastromatteo, J.; Frias, J.C.; Albelda, M.T.; Sikora, S.; Kharazi, A.; Vertelov, G.; et al. Intravenously Delivered Mesenchymal Stem Cells: Systemic Anti-Inflammatory Effects Improve Left Ventricular Dysfunction in Acute Myocardial Infarction and Ischemic Cardiomyopathy. Circ. Res. 2017, 120, 1598–1613. [Google Scholar] [CrossRef]
- Noutsias, M.; Pauschinger, M.; Schultheiss, H.-P.; Kühl, U. Phenotypic characterization of infiltrates in dilated cardiomyopathy—Diagnostic significance of T-lymphocytes and macrophages in inflammatory cardiomyopathy. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2002, 8, 478–487. [Google Scholar]
- Adamo, L.; Rocha-Resende, C.; Lin, C.Y.; Evans, S.; Williams, J.; Dun, H.; Li, W.; Mpoy, C.; Andhey, P.S.; Rogers, B.E.; et al. Myocardial B cells are a subset of circulating lymphocytes with delayed transit through the heart. JCI Insight 2020, 5, e134700. [Google Scholar] [CrossRef] [Green Version]
- Zouggari, Y.; Ait-Oufella, H.; Bonnin, P.; Simon, T.; Sage, A.P.; Guérin, C.; Vilar, J.; Caligiuri, G.; Tsiantoulas, D.; Laurans, L.; et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat. Med. 2013, 19, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Horckmans, M.; Bianchini, M.; Santovito, D.; Megens, R.T.A.; Springael, J.Y.; Negri, I.; Vacca, M.; Di Eusanio, M.; Moschetta, A.; Weber, C.; et al. Pericardial Adipose Tissue Regulates Granulopoiesis, Fibrosis, and Cardiac Function After Myocardial Infarction. Circulation 2018, 137, 948–960. [Google Scholar] [CrossRef] [PubMed]
- Mo, F.; Luo, Y.; Yan, Y.; Li, J.; Lai, S.; Wu, W. Are activated B cells involved in the process of myocardial fibrosis after acute myocardial infarction? An in vivo experiment. BMC Cardiovasc. Disord. 2021, 21, 5. [Google Scholar] [CrossRef] [PubMed]
- Goodchild, T.T.; Robinson, K.A.; Pang, W.; Tondato, F.; Cui, J.; Arrington, J.; Godwin, L.; Ungs, M.; Carlesso, N.; Weich, N.; et al. Bone marrow-derived B cells preserve ventricular function after acute myocardial infarction. JACC Cardiovasc. Interv. 2009, 2, 1005–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Dalal, R.; Cao, C.D.; Postoak, J.L.; Yang, G.; Zhang, Q.; Wang, Z.; Lal, H.; Van Kaer, L. IL-10-producing B cells are enriched in murine pericardial adipose tissues and ameliorate the outcome of acute myocardial infarction. Proc. Natl. Acad. Sci. USA 2019, 116, 21673–21684. [Google Scholar] [CrossRef] [Green Version]
- Jiao, J.; He, S.; Wang, Y.; Lu, Y.; Gu, M.; Li, D.; Tang, T.; Nie, S.; Zhang, M.; Lv, B.; et al. Regulatory B cells improve ventricular remodeling after myocardial infarction by modulating monocyte migration. Basic Res. Cardiol. 2021, 116, 46. [Google Scholar] [CrossRef]
- Bonaventura, A.; Vecchié, A.; Abbate, A.; Montecucco, F. Neutrophil Extracellular Traps and Cardiovascular Diseases: An Update. Cells 2020, 9, 231. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Yabluchanskiy, A.; Iyer, R.P.; Cannon, P.L.; Flynn, E.R.; Jung, M.; Henry, J.; Cates, C.A.; Deleon-Pennell, K.Y.; Lindsey, M.L. Temporal neutrophil polarization following myocardial infarction. Cardiovasc. Res. 2016, 110, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Van Furth, R.; Cohn, Z.A. The origin and kinetics of mononuclear phagocytes. J. Exp. Med. 1968, 128, 415–435. [Google Scholar] [CrossRef]
- Swirski, F.K.; Nahrendorf, M.; Etzrodt, M.; Wildgruber, M.; Cortez-Retamozo, V.; Panizzi, P.; Figueiredo, J.L.; Kohler, R.H.; Chudnovskiy, A.; Waterman, P.; et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 2009, 325, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Serbina, N.V.; Pamer, E.G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006, 7, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Mouton, A.J.; Lindsey, M.L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. J. Lab. Clin. Med. 2018, 191, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Yona, S.; Kim, K.W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Da Silva, N.; Panizzi, P.; van der Laan, A.M.; et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 2014, 115, 284–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molawi, K.; Wolf, Y.; Kandalla, P.K.; Favret, J.; Hagemeyer, N.; Frenzel, K.; Pinto, A.R.; Klapproth, K.; Henri, S.; Malissen, B.; et al. Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med. 2014, 211, 2151–2158. [Google Scholar] [CrossRef]
- Simões, F.C.; Cahill, T.J.; Kenyon, A.; Gavriouchkina, D.; Vieira, J.M.; Sun, X.; Pezzolla, D.; Ravaud, C.; Masmanian, E.; Weinberger, M.; et al. Macrophages directly contribute collagen to scar formation during zebrafish heart regeneration and mouse heart repair. Nat. Commun. 2020, 11, 600. [Google Scholar] [CrossRef] [Green Version]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, H.; Pei, J.; Hu, S.; Nie, Y. Transplantation of murine neonatal cardiac macrophage improves adult cardiac repair. Cell. Mol. Immunol. 2021, 18, 492–494. [Google Scholar] [CrossRef]
- Van Rooijen, N.; Hendrikx, E. Liposomes for specific depletion of macrophages from organs and tissues. Methods Mol. Biol. 2010, 605, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Gorczyca, W.; Sun, Z.Y.; Cronin, W.; Li, X.; Mau, S.; Tugulea, S. Immunophenotypic pattern of myeloid populations by flow cytometry analysis. Methods Cell Biol. 2011, 103, 221–266. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Feng, J.; Li, Y.; Pei, J.; Hu, S.; Nie, Y. Transplantation of Neonatal Mouse Cardiac Macrophages into Adult Mice. J. Vis. Exp. JoVE 2021, e62108. [Google Scholar] [CrossRef]
- Morales, R.A.; Allende, M.L. Peripheral Macrophages Promote Tissue Regeneration in Zebrafish by Fine-Tuning the Inflammatory Response. Front. Immunol. 2019, 10, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrie, T.A.; Strand, N.S.; Yang, C.T.; Rabinowitz, J.S.; Moon, R.T. Macrophages modulate adult zebrafish tail fin regeneration. Development 2014, 141, 2581–2591. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [Green Version]
- Nahrendorf, M.; Swirski, F.K. Abandoning M1/M2 for a Network Model of Macrophage Function. Circ. Res. 2016, 119, 414–417. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [Green Version]
- De Couto, G. Macrophages in cardiac repair: Environmental cues and therapeutic strategies. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Duncan, S.E.; Gao, S.; Sarhene, M.; Coffie, J.W.; Linhua, D.; Bao, X.; Jing, Z.; Li, S.; Guo, R.; Su, J.; et al. Macrophage Activities in Myocardial Infarction and Heart Failure. Cardiol. Res. Pract. 2020, 2020, 4375127. [Google Scholar] [CrossRef]
- Moyse, B.R.; Richardson, R.J. A Population of Injury-Responsive Lymphoid Cells Expresses mpeg1.1 in the Adult Zebrafish Heart. ImmunoHorizons 2020, 4, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, G.; Gomez, E.; Lyer, S.; Rovira, M.; Miserocchi, M.; Langenau, D.M.; Bertrand, J.Y.; Wittamer, V. The macrophage-expressed gene (mpeg) 1 identifies a subpopulation of B cells in the adult zebrafish. J. Leukoc. Biol. 2020, 107, 431–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peet, C.; Ivetic, A.; Bromage, D.I.; Shah, A.M. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc. Res. 2020, 116, 1101–1112. [Google Scholar] [CrossRef] [Green Version]
- Kumar, B.V.; Connors, T.J.; Farber, D.L. Human T Cell Development, Localization, and Function throughout Life. Immunity 2018, 48, 202–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Casey, K.A.; Fraser, K.A.; Schenkel, J.M.; Moran, A.; Abt, M.C.; Beura, L.K.; Lucas, P.J.; Artis, D.; Wherry, E.J.; Hogquist, K.; et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J. Immunol. 2012, 188, 4866–4875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, U.; Frantz, S. Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction. Circ. Res. 2015, 116, 354–367. [Google Scholar] [CrossRef] [Green Version]
- Fishbein, M.C.; Maclean, D.; Maroko, P.R. The histopathologic evolution of myocardial infarction. Chest 1978, 73, 843–849. [Google Scholar] [CrossRef] [Green Version]
- Abbate, A.; Bussani, R.; Liuzzo, G.; Biondi-Zoccai, G.G.; Barresi, E.; Mellone, P.; Sinagra, G.; Dobrina, A.; De Giorgio, F.; Sharma, R.; et al. Sudden coronary death, fatal acute myocardial infarction and widespread coronary and myocardial inflammation. Heart 2008, 94, 737–742. [Google Scholar] [CrossRef]
- Langenau, D.M.; Zon, L.I. The zebrafish: A new model of T-cell and thymic development. Nat. Rev. Immunol. 2005, 5, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Sharir, R.; Semo, J.; Shimoni, S.; Ben-Mordechai, T.; Landa-Rouben, N.; Maysel-Auslender, S.; Shaish, A.; Entin-Meer, M.; Keren, G.; George, J. Experimental myocardial infarction induces altered regulatory T cell hemostasis, and adoptive transfer attenuates subsequent remodeling. PLoS ONE 2014, 9, e113653. [Google Scholar] [CrossRef] [PubMed]
- Varda-Bloom, N.; Leor, J.; Ohad, D.G.; Hasin, Y.; Amar, M.; Fixler, R.; Battler, A.; Eldar, M.; Hasin, D. Cytotoxic T lymphocytes are activated following myocardial infarction and can recognize and kill healthy myocytes in vitro. J. Mol. Cell. Cardiol. 2000, 32, 2141–2149. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Miyahara, Y.; Guo, Z.; Khattar, M.; Stepkowski, S.M.; Chen, W. “Defaul” generation of neonatal regulatory T cells. J. Immunol. 2010, 185, 71–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.-Y.; Xiong, Y.-Y.; Lu, X.-T.; Yang, Y.-J. Regulation of Type 2 Immunity in Myocardial Infarction. Front. Immunol. 2019, 10, 62. [Google Scholar] [CrossRef]
- Kasheta, M.; Painter, C.A.; Moore, F.E.; Lobbardi, R.; Bryll, A.; Freiman, E.; Stachura, D.; Rogers, A.B.; Houvras, Y.; Langenau, D.M.; et al. Identification and characterization of T reg-like cells in zebrafish. J. Exp. Med. 2017, 214, 3519–3530. [Google Scholar] [CrossRef]
- Dee, C.T.; Nagaraju, R.T.; Athanasiadis, E.I.; Gray, C.; Fernandez Del Ama, L.; Johnston, S.A.; Secombes, C.J.; Cvejic, A.; Hurlstone, A.F. CD4-Transgenic Zebrafish Reveal Tissue-Resident Th2- and Regulatory T Cell-like Populations and Diverse Mononuclear Phagocytes. J. Immunol. 2016, 197, 3520–3530. [Google Scholar] [CrossRef]
- Stacy, N.I.; Ackerman, S.J. A tribute to eosinophils from a comparative and evolutionary perspective. J. Allergy Clin. Immunol. 2021, 147, 1115–1116. [Google Scholar] [CrossRef]
- Heredia, J.E.; Mukundan, L.; Chen, F.M.; Mueller, A.A.; Deo, R.C.; Locksley, R.M.; Rando, T.A.; Chawla, A. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 2013, 153, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Balla, K.M.; Lugo-Villarino, G.; Spitsbergen, J.M.; Stachura, D.L.; Hu, Y.; Bañuelos, K.; Romo-Fewell, O.; Aroian, R.V.; Traver, D. Eosinophils in the zebrafish: Prospective isolation, characterization, and eosinophilia induction by helminth determinants. Blood 2010, 116, 3944–3954. [Google Scholar] [CrossRef] [Green Version]
- Wan, F.; Hu, C.B.; Ma, J.X.; Gao, K.; Xiang, L.X.; Shao, J.Z. Characterization of γδ T Cells from Zebrafish Provides Insights into Their Important Role in Adaptive Humoral Immunity. Front. Immunol. 2016, 7, 675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, M.; Giladi, A.; Gorki, A.D.; Solodkin, D.G.; Zada, M.; Hladik, A.; Miklosi, A.; Salame, T.M.; Halpern, K.B.; David, E.; et al. Lung Single-Cell Signaling Interaction Map Reveals Basophil Role in Macrophage Imprinting. Cell 2018, 175, 1031–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piliponsky, A.M.; Shubin, N.J.; Lahiri, A.K.; Truong, P.; Clauson, M.; Niino, K.; Tsuha, A.L.; Nedospasov, S.A.; Karasuyama, H.; Reber, L.L.; et al. Basophil-derived tumor necrosis factor can enhance survival in a sepsis model in mice. Nat. Immunol. 2019, 20, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Hogan, S.P.; Rosenberg, H.F.; Moqbel, R.; Phipps, S.; Foster, P.S.; Lacy, P.; Kay, A.B.; Rothenberg, M.E. Eosinophils: Biological properties and role in health and disease. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2008, 38, 709–750. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, J.B.; Robinowitz, M.; McAllister, H.A.; Virmani, R. Association of eosinophils with cardiac rupture. Hum. Pathol. 1985, 16, 562–568. [Google Scholar] [CrossRef]
- Goh, Y.P.; Henderson, N.C.; Heredia, J.E.; Red Eagle, A.; Odegaard, J.I.; Lehwald, N.; Nguyen, K.D.; Sheppard, D.; Mukundan, L.; Locksley, R.M.; et al. Eosinophils secrete IL-4 to facilitate liver regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 9914–9919. [Google Scholar] [CrossRef] [Green Version]
- Ohnmacht, C.; Schwartz, C.; Panzer, M.; Schiedewitz, I.; Naumann, R.; Voehringer, D. Basophils orchestrate chronic allergic dermatitis and protective immunity against helminths. Immunity 2010, 33, 364–374. [Google Scholar] [CrossRef] [Green Version]
- Schiechl, G.; Hermann, F.J.; Rodriguez Gomez, M.; Kutzi, S.; Schmidbauer, K.; Talke, Y.; Neumayer, S.; Goebel, N.; Renner, K.; Brühl, H.; et al. Basophils Trigger Fibroblast Activation in Cardiac Allograft Fibrosis Development. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2016, 16, 2574–2588. [Google Scholar] [CrossRef]
- Choo, E.H.; Lee, J.H.; Park, E.H.; Park, H.E.; Jung, N.C.; Kim, T.H.; Koh, Y.S.; Kim, E.; Seung, K.B.; Park, C.; et al. Infarcted Myocardium-Primed Dendritic Cells Improve Remodeling and Cardiac Function After Myocardial Infarction by Modulating the Regulatory T Cell and Macrophage Polarization. Circulation 2017, 135, 1444–1457. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Y.; Yuan, J.; Gao, W.; Zhong, X.; Yao, K.; Lin, L.; Ge, J. Dendritic cell-derived exosomal miR-494-3p promotes angiogenesis following myocardial infarction. Int. J. Mol. Med. 2021, 47, 315–325. [Google Scholar] [CrossRef]
- Odaka, T.; Suetake, H.; Maeda, T.; Miyadai, T. Teleost Basophils Have IgM-Dependent and Dual Ig-Independent Degranulation Systems. J. Immunol. 2018, 200, 2767–2776. [Google Scholar] [CrossRef] [PubMed]
- Voehringer, D. Protective and pathological roles of mast cells and basophils. Nat. Rev. Immunol. 2013, 13, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Balan, S.; Saxena, M.; Bhardwaj, N. Dendritic cell subsets and locations. Int. Rev. Cell Mol. Biol. 2019, 348, 1–68. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yuan, J.; Hu, J.; Gao, W.; Zou, Y.; Ge, J. ACE inhibitor suppresses cardiac remodeling after myocardial infarction by regulating dendritic cells and AT(2) receptor-mediated mechanism in mice. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 114, 108660. [Google Scholar] [CrossRef]
- Ma, Y.; Ma, L.; Ma, J.; Wu, R.; Zou, Y.; Ge, J. Hyperlipidemia inhibits the protective effect of lisinopril after myocardial infarction via activation of dendritic cells. J. Cell. Mol. Med. 2020, 24, 4082–4091. [Google Scholar] [CrossRef] [Green Version]
- Zhu, R.; Sun, H.; Yu, K.; Zhong, Y.; Shi, H.; Wei, Y.; Su, X.; Xu, W.; Luo, Q.; Zhang, F.; et al. Interleukin-37 and Dendritic Cells Treated with Interleukin-37 Plus Troponin I Ameliorate Cardiac Remodeling After Myocardial Infarction. J. Am. Heart Assoc. 2016, 5, e004406. [Google Scholar] [CrossRef]
- Wei, Y.; Lan, Y.; Zhong, Y.; Yu, K.; Xu, W.; Zhu, R.; Sun, H.; Ding, Y.; Wang, Y.; Zeng, Q. Interleukin-38 alleviates cardiac remodelling after myocardial infarction. J. Cell. Mol. Med. 2020, 24, 371–384. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Gao, W.; Yuan, J.; Wu, C.; Yao, K.; Zhang, L.; Ma, L.; Zhu, J.; Zou, Y.; Ge, J. Exosomes derived from dendritic cells improve cardiac function via activation of CD4(+) T lymphocytes after myocardial infarction. J. Mol. Cell. Cardiol. 2016, 91, 123–133. [Google Scholar] [CrossRef]
- Zhang, Y.; Cai, Z.; Shen, Y.; Lu, Q.; Gao, W.; Zhong, X.; Yao, K.; Yuan, J.; Liu, H. Hydrogel-load exosomes derived from dendritic cells improve cardiac function via Treg cells and the polarization of macrophages following myocardial infarction. J. Nanobiotechnol. 2021, 19, 271. [Google Scholar] [CrossRef]
- Lugo-Villarino, G.; Balla, K.M.; Stachura, D.L.; Bañuelos, K.; Werneck, M.B.; Traver, D. Identification of dendritic antigen-presenting cells in the zebrafish. Proc. Natl. Acad. Sci. USA 2010, 107, 15850–15855. [Google Scholar] [CrossRef] [Green Version]
- Wittamer, V.; Bertrand, J.Y.; Gutschow, P.W.; Traver, D. Characterization of the mononuclear phagocyte system in zebrafish. Blood 2011, 117, 7126–7135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, T.; Zhu, L.Y.; Nie, L.; Shi, W.; Dong, W.R.; Xiang, L.X.; Shao, J.Z. Characterization of surface phenotypic molecules of teleost dendritic cells. Dev. Comp. Immunol. 2015, 49, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Kiessling, R.; Klein, E.; Wigzell, H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur. J. Immunol. 1975, 5, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Herberman, R.B.; Nunn, M.E.; Lavrin, D.H. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I. Distribution of reactivity and specificity. Int. J. Cancer 1975, 16, 216–229. [Google Scholar] [CrossRef]
- Fauriat, C.; Long, E.O.; Ljunggren, H.G.; Bryceson, Y.T. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood 2010, 115, 2167–2176. [Google Scholar] [CrossRef] [Green Version]
- Vivier, E.; Nunès, J.A.; Vély, F. Natural killer cell signaling pathways. Science 2004, 306, 1517–1519. [Google Scholar] [CrossRef]
- Zitti, B.; Bryceson, Y.T. Natural killer cells in inflammation and autoimmunity. Cytokine Growth Factor Rev. 2018, 42, 37–46. [Google Scholar] [CrossRef]
- Yan, W.; Zhou, L.; Wen, S.; Duan, Q.; Huang, F.; Tang, Y.; Liu, X.; Chai, Y.; Wang, L. Differential loss of natural killer cell activity in patients with acute myocardial infarction and stable angina pectoris. Int. J. Clin. Exp. Pathol. 2015, 8, 14667–14675. [Google Scholar]
- Pereiro, P.; Varela, M.; Diaz-Rosales, P.; Romero, A.; Dios, S.; Figueras, A.; Novoa, B. Zebrafish Nk-lysins: First insights about their cellular and functional diversification. Dev. Comp. Immunol. 2015, 51, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Moore, F.E.; Garcia, E.G.; Lobbardi, R.; Jain, E.; Tang, Q.; Moore, J.C.; Cortes, M.; Molodtsov, A.; Kasheta, M.; Luo, C.C.; et al. Single-cell transcriptional analysis of normal, aberrant, and malignant hematopoiesis in zebrafish. J. Exp. Med. 2016, 213, 979–992. [Google Scholar] [CrossRef] [Green Version]
- Carmona, S.J.; Teichmann, S.A.; Ferreira, L.; Macaulay, I.C.; Stubbington, M.J.; Cvejic, A.; Gfeller, D. Single-cell transcriptome analysis of fish immune cells provides insight into the evolution of vertebrate immune cell types. Genome Res. 2017, 27, 451–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Q.; Iyer, S.; Lobbardi, R.; Moore, J.C.; Chen, H.; Lareau, C.; Hebert, C.; Shaw, M.L.; Neftel, C.; Suva, M.L.; et al. Dissecting hematopoietic and renal cell heterogeneity in adult zebrafish at single-cell resolution using RNA sequencing. J. Exp. Med. 2017, 214, 2875–2887. [Google Scholar] [CrossRef] [PubMed]
- Muire, P.J.; Hanson, L.A.; Wills, R.; Petrie-Hanson, L. Differential gene expression following TLR stimulation in rag1-/- mutant zebrafish tissues and morphological descriptions of lymphocyte-like cell populations. PLoS ONE 2017, 12, e0184077. [Google Scholar] [CrossRef] [Green Version]
- Kumrić, M.; Kurir, T.T.; Borovac, J.A.; Božić, J. The Role of Natural Killer (NK) Cells in Acute Coronary Syndrome: A Comprehensive Review. Biomolecules 2020, 10, 1514. [Google Scholar] [CrossRef]
- Porsch, F.; Mallat, Z.; Binder, C.J. Humoral immunity in atherosclerosis and myocardial infarction: From B cells to antibodies. Cardiovasc. Res. 2021, 117, 2544–2562. [Google Scholar] [CrossRef]
- Heinrichs, M.; Ashour, D.; Siegel, J.; Büchner, L.; Wedekind, G.; Heinze, K.G.; Arampatzi, P.; Saliba, A.E.; Cochain, C.; Hofmann, U.; et al. The healing myocardium mobilizes a distinct B-cell subset through a CXCL13-CXCR5-dependent mechanism. Cardiovasc. Res. 2021, 117, 2664–2676. [Google Scholar] [CrossRef]
- Wang, Z.; Cui, M.; Shah, A.M.; Ye, W.; Tan, W.; Min, Y.L.; Botten, G.A.; Shelton, J.M.; Liu, N.; Bassel-Duby, R.; et al. Mechanistic basis of neonatal heart regeneration revealed by transcriptome and histone modification profiling. Proc. Natl. Acad. Sci. USA 2019, 116, 18455–18465. [Google Scholar] [CrossRef] [Green Version]
- Page, D.M.; Wittamer, V.; Bertrand, J.Y.; Lewis, K.L.; Pratt, D.N.; Delgado, N.; Schale, S.E.; McGue, C.; Jacobsen, B.H.; Doty, A.; et al. An evolutionarily conserved program of B-cell development and activation in zebrafish. Blood 2013, 122, e1–e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamo, L.; Rocha-Resende, C.; Mann, D.L. The Emerging Role of B Lymphocytes in Cardiovascular Disease. Annu. Rev. Immunol. 2020, 38, 99–121. [Google Scholar] [CrossRef]
- Ryan, R.; Moyse, B.R.; Richardson, R.J. Zebrafish cardiac regeneration-looking beyond cardiomyocytes to a complex microenvironment. Histochem. Cell Biol. 2020, 154, 533–548. [Google Scholar] [CrossRef]
- Talman, V.; Kivelä, R. Cardiomyocyte—Endothelial Cell Interactions in Cardiac Remodeling and Regeneration. Front. Cardiovasc. Med. 2018, 5, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Poss, K.D. The epicardium as a hub for heart regeneration. Nat. Rev. Cardiol. 2018, 15, 631–647. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Leukocyte | Humapeak 3 Days with Permanent Ligation; Days 1ns | Adult Rodents | Neonatal Mice | Adult Zebrafish |
|---|---|---|---|---|
| Neutrophils | Detrimental: Neutrophilia associated with increased infarct size, heart failure, death [49,50,51,52,53,54,55,56]. | Timing: Days 1–14, peak 3 days with permanent ligation; days 1–7, peak day 1 with reperfusion [57,58]. Detrimental: CM death and adverse cardiac remodeling [59,60,61]. Beneficial: Clear dead cells/debris, angiogenesis, inflammation resolution [60]; reparative macrophage polarization [62]. | Timing: Overall larger neutrophil recruitment in P1 neonates vs. P14 juveniles [63,64]. | Timing: From 6 h to 7 days; peaks at day 1, drops days 3–7, basal levels day 14 after cryoinjury [65,66,67,68]. Detrimental: Delayed removal extends inflammatory phase and inhibits regeneration with scar retention, lowered CM proliferation [65,67]. Beneficial: Support angiogenesis [67]. |
| Monocytes/ Macrophages | Timing: At infarct border region (12 h-5 days post-MI); and infarct (5–14 days post-MI) [69]. Detrimental: Pro-inflammatory monocytes and adverse recovery [70,71,72,73,74]; pro-inflammatory monocyte-derived CCR2+ macrophages accumulate in heart failure patients with LV remodeling [75]. | Timing: Biphasic with days 1–3 pro-inflammatory Ly6Chi cells; day 5 onward reparative Ly6Clo cells; remain at 14 days after injury [57,76]. Detrimental: CCR2+ macrophages persist in myocardium for months, role in ventricular remodeling [77,78,79,80,81]. Beneficial: Clear cell debris/dead cells and neutrophils; angiogenesis; and collagen deposition [82]; early phase for cell clearance; late phase for tissue granulation/collagen deposition [76]; tissue-resident CCR2- cardioprotective [77,78,79,80,81]. | Timing: Biphasic with pro-inflammatory and then anti-inflammatory cells; regenerative P1 higher levels than non-regenerative P14 juveniles [64]. Beneficial: Depletion abolishes regeneration with scar retention, lowered angiogenesis, and cardiac remodeling [63,64]; neonates expand tissue-resident CCR2- population and do not recruit CCR2+ monocytes after injury [63]. | Timing: Biphasic recruitment of pro-inflammatory TNFα+ (1–3 dpi) and anti-inflammatory TNFα- (3–7 dpi) after injury; returns to basal levels by 14 dpi [68,83]. Beneficial: Depletion or delayed mobilization inhibits neutrophil clearance, CM proliferation, scar resolution, and angiogenesis [65,66,67,68,84]; reparative wt1b+ macrophages support CM proliferation [85]. |
| T lymphocytes | Timing: Decrease in CD4+ and CD8+ T cells immediately after AMI with increase in Tregs 4–6 days later [86,87,88]; increase in Th1 and Th17 CD4+ T cells with decrease in Tregs [89,90,91,92]. Detrimental: CD8+ CD28+ T cells lowered cardiac function [93]; CD8 + CD57+ T cells correlate with mortality [94]; heart failure patients enriched with CD4+ Th1 and CD8+ T cells [95,96]. Beneficial: Low Treg levels associated with LV remodeling, and lowered survival [97,98]. | Timing: T cells mobilize to injured heart from days 1 to 14, peaking at day 7 post-injury with permanent ligation, day 3 with reperfusion [57,88,99,100,101,102]; remain for weeks after injury [102]. Detrimental: CD4+ cells aggravate injury [99]; CD8+ cells enhance inflammation and CM apoptosis [103,104,105]; γδ cells promote inflammation, CM death, fibrosis, adverse remodeling [106,107]. Beneficial: CD4+ for collagen deposition, neovascularization, cardioprotective phenotype [88,108]; Tregs in wound recovery, resolve inflammation [100,109,110,111,112,113,114,115]; CD8+ to prevent cardiac rupture [104,105]. | Timing: T cells mobilize from days 1 to 14, peak at day 7 post-injury, return to basal levels day 14 in P3 mice; significantly higher CD4+ T cells in P8 juveniles vs. P3 neonates, with T-cell persistance high through day 14 [116]. Detrimental: Depletion of CD4+ T cells in P8 juveniles facilitated regeneration with CM proliferation and reduced fibrosis [117]. Beneficial: Tregs support regeneration in neonates with CM proliferation and reduced cardiac fibrosis [116]. | Timing: T cells mobilize to cardiac wound from day 1, peak at 7 dpi, resolved by 14 dpi [68,118]. Beneficial: Depletion of Tregs during cardiac cryoinjury led to thinner myocardial walls, persistent collagenous scar, lowered CM proliferation, and macrophage polarization towards classical inflammatory phenotype [118]. |
| Leukocyte | Humans | Adult Rodents |
|---|---|---|
| Eosinophils | Detrimental: Lower peripheral blood levels associated with increased risk for MACE and death [119,120,121,122,123,124]. Beneficial: Enhanced levels cardioprotective: limit CM death, fibrosis, and inflammation [125]; protection 6 months post-reperfusion [123]. | Timing: Mobilize from days 1 to 7, peak at day 4 post-injury, lower at day 7 [125]. Beneficial: Depletion causes severe cardiac dysfunction after MI with larger infarct, increased cell death and collagen deposition [124,125]. |
| Basophils | Timing: Peak at 96 h post-MI [126]. Detrimental: Both high and low counts associated with higher mortality [122]. Beneficial: Lower numbers 1 week post-MI associated with larger infarct and adverse cardiac outcomes [126]. | Timing: Mobilize from days 1 to 7, peak between days 3 and 7 post-injury, return to baseline by day 14; infiltrated infarcted hearts [126]. Beneficial: Depletion leads to severe cardiac dysfunction; enhanced pro-inflammatory Ly6Chi monocytes and lowered anti-inflammatory macrophages [126]. |
| Dendritic Cells | Timing/Localization: Decrease in patients with AMI; increase in activated DCs [127,128,129,130]; significant elevation in DCs in infarcted myocardium; interaction of infiltrated DCs with T cells [127,131]. Beneficial: Cardioprotective, lower amounts of DCs in infarcted myocardium associated with enhanced macrophages, limited fibrosis, and cardiac rupture [132]. | Timing: Accumulate from day 1 post-injury, peak at day 7, and lowered at day 14 with permanent ligation; peaked day 3 with reperfusion and lowered by day 7 [57,133]. Detrimental: Mature DCs worsen ventricular remodeling [134]; prevention of DC mobilization from bone marrow improved cardiac function [135]; depletion of cDCs limited inflammatory response with lowered neutrophil, macrophage, and T-cell infiltration and reduced adverse remodeling [136]; cross-priming cytotoxic T cells for perpetuation of myocardial damage [95,137]; pDCs release IFN-γ and further damaging inflammatory responses [138]. Beneficial: DC depletion led to reduced cardiac function and adverse remodeling; required for recruitment of anti-inflammatory reparative macrophages [139]; DC depletion led to larger infarcts, severe cardiac remodeling, and dysfunction [140]. |
| Natural Killer Cells | Timing/Localization: Either elevated or lowered levels in peripheral blood [141,142,143]; infiltrate myocardium [142]; defective functionalities, reduction in cytotoxicity after AMI [143,144,145]. Beneficial: IL-10+ NK cells 72 h post-MI; reduced IL-10+ NK cells associated with enhanced functional recovery 3 months post-MI [143]. | Timing: Mobilize from days 1 to 7, peak day 7 with permanent ligation, and lowered at day 14; peaked day 3 with reperfusion, lowered day 7 [58,59,146]. Beneficial: Mobilization to infarct improves cardiac function and remodeling [146]; IL-2-activated NK cells support angiogenesis and reduce fibrosis [147,148]. Detrimental: NK depletion before injury led to reduced infarct and limited adverse remodeling [149]. |
| B lymphocytes | Timing/Localization: B cells present in non-ischemic hearts [150]; decrease after AMI and increase after reperfusion [86]; intravascular B cells that associate with cardiac endothelium [151]. | Timing: Mobilize from days 1 to 7, peak at day 7 post-injury, and lowered at day 14 with permanent ligation; peaked at day 3 and lowered day 7 with reperfusion [57]. Detrimental: B2 cell depletion limits cardiac injury, prevents adverse remodeling, and enhances cardiac function; activated B2 cells secrete Ccl7 for pro-inflammatory Ly6Chi monocytes and to extend inflammation [152]; enhanced B cells associated with increased cardiac fibrosis and remodeling; GM-CSF-producing B cells promote DC and T-cell expansion in PAT and neutrophil infiltration to infarct [153]; B-cell knockout system lowered pro-inflammatory cytokine levels, ventricular remodeling, fibrosis [154]. Beneficial: B-cell injections improved cardiac function, reduced apoptosis [155]; IL-10-producing B1a cells required for inflammation resolution [156]; Breg adoptive transfer enhances cardiac function through IL-10 secretion to limit mobilization of CCR2+ Ly6Chi monocytes to heart [157]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peterson, E.A.; Sun, J.; Wang, J. Leukocyte-Mediated Cardiac Repair after Myocardial Infarction in Non-Regenerative vs. Regenerative Systems. J. Cardiovasc. Dev. Dis. 2022, 9, 63. https://doi.org/10.3390/jcdd9020063
Peterson EA, Sun J, Wang J. Leukocyte-Mediated Cardiac Repair after Myocardial Infarction in Non-Regenerative vs. Regenerative Systems. Journal of Cardiovascular Development and Disease. 2022; 9(2):63. https://doi.org/10.3390/jcdd9020063
Chicago/Turabian StylePeterson, Elizabeth Anne, Jisheng Sun, and Jinhu Wang. 2022. "Leukocyte-Mediated Cardiac Repair after Myocardial Infarction in Non-Regenerative vs. Regenerative Systems" Journal of Cardiovascular Development and Disease 9, no. 2: 63. https://doi.org/10.3390/jcdd9020063
APA StylePeterson, E. A., Sun, J., & Wang, J. (2022). Leukocyte-Mediated Cardiac Repair after Myocardial Infarction in Non-Regenerative vs. Regenerative Systems. Journal of Cardiovascular Development and Disease, 9(2), 63. https://doi.org/10.3390/jcdd9020063