
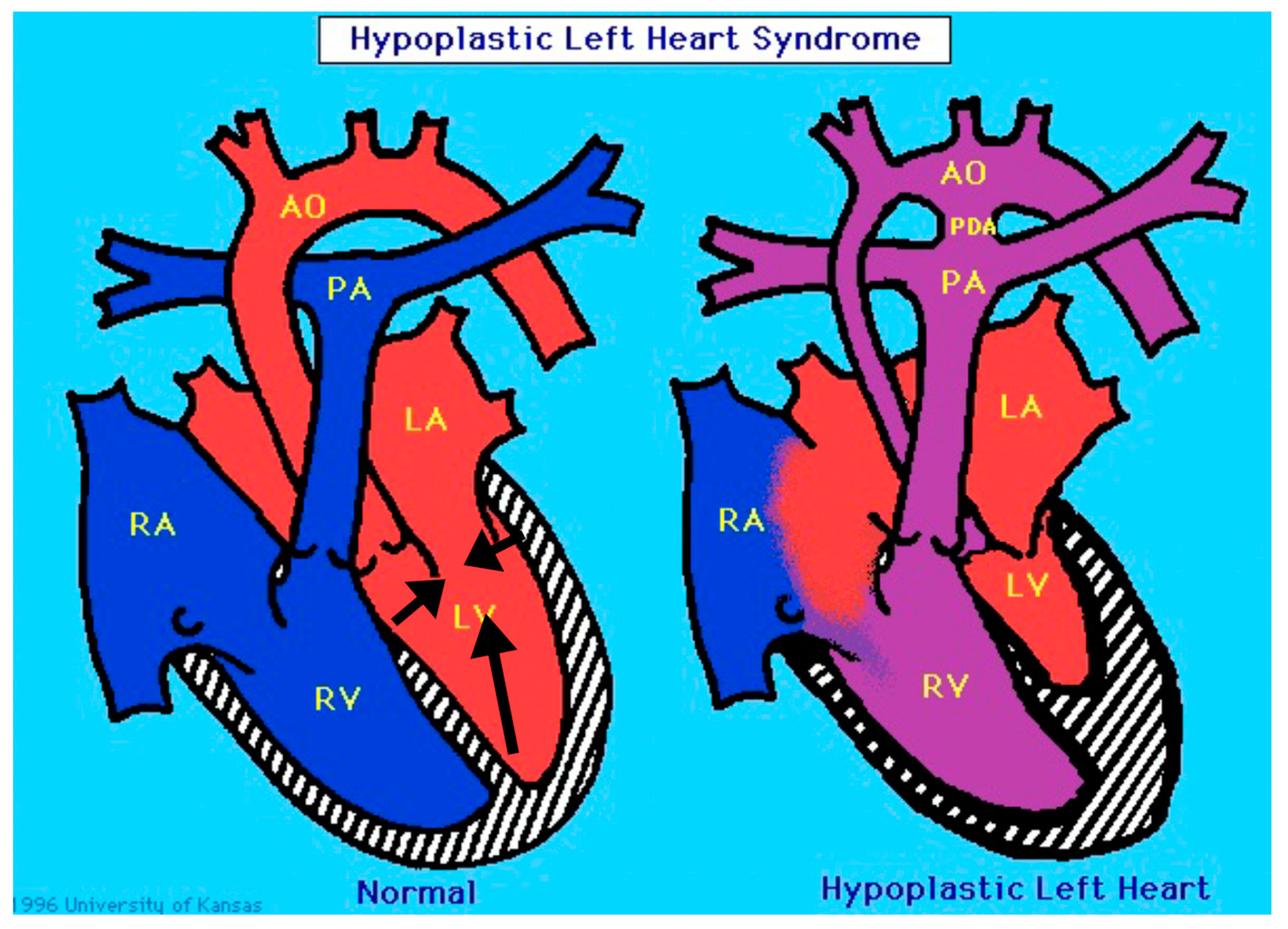
ETS1 and HLHS: Implications for the Role of the Endocardium

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
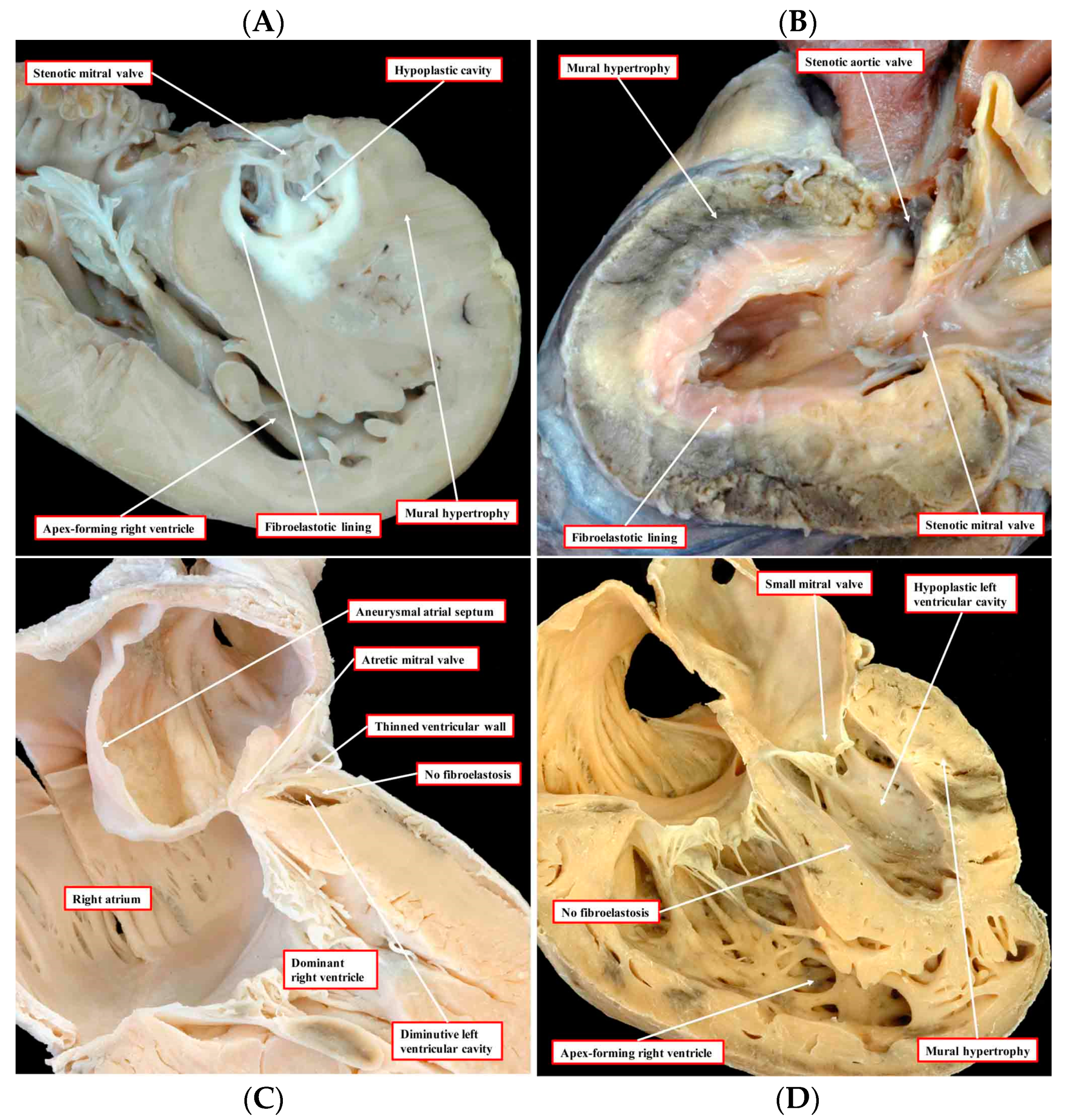
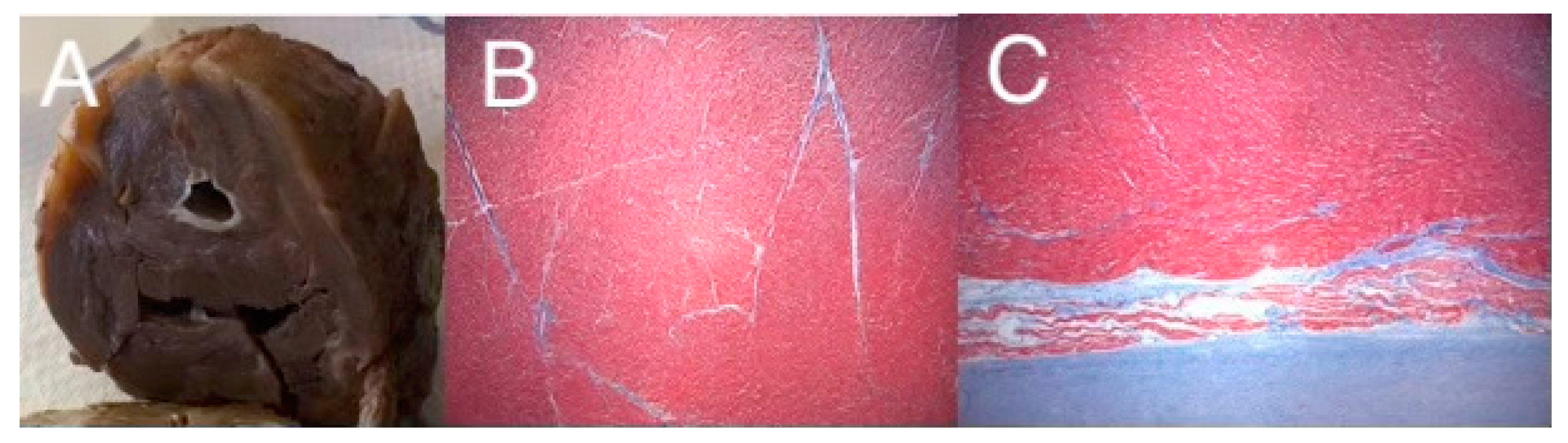
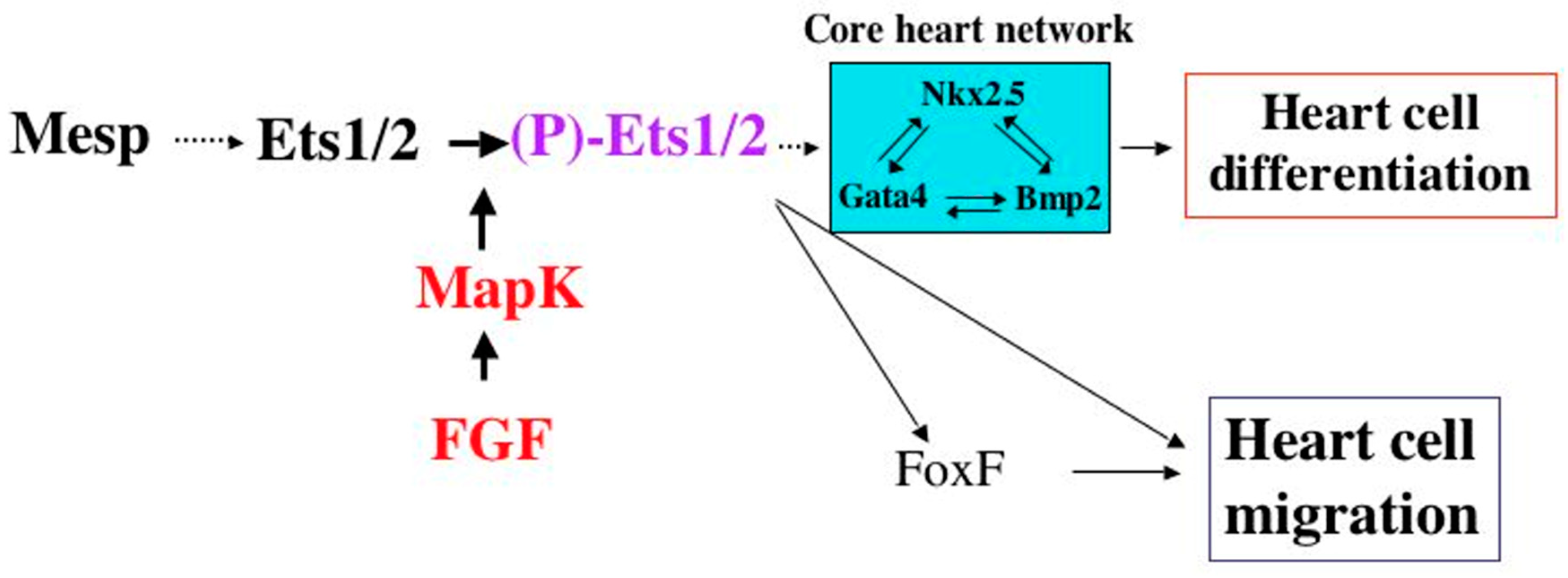
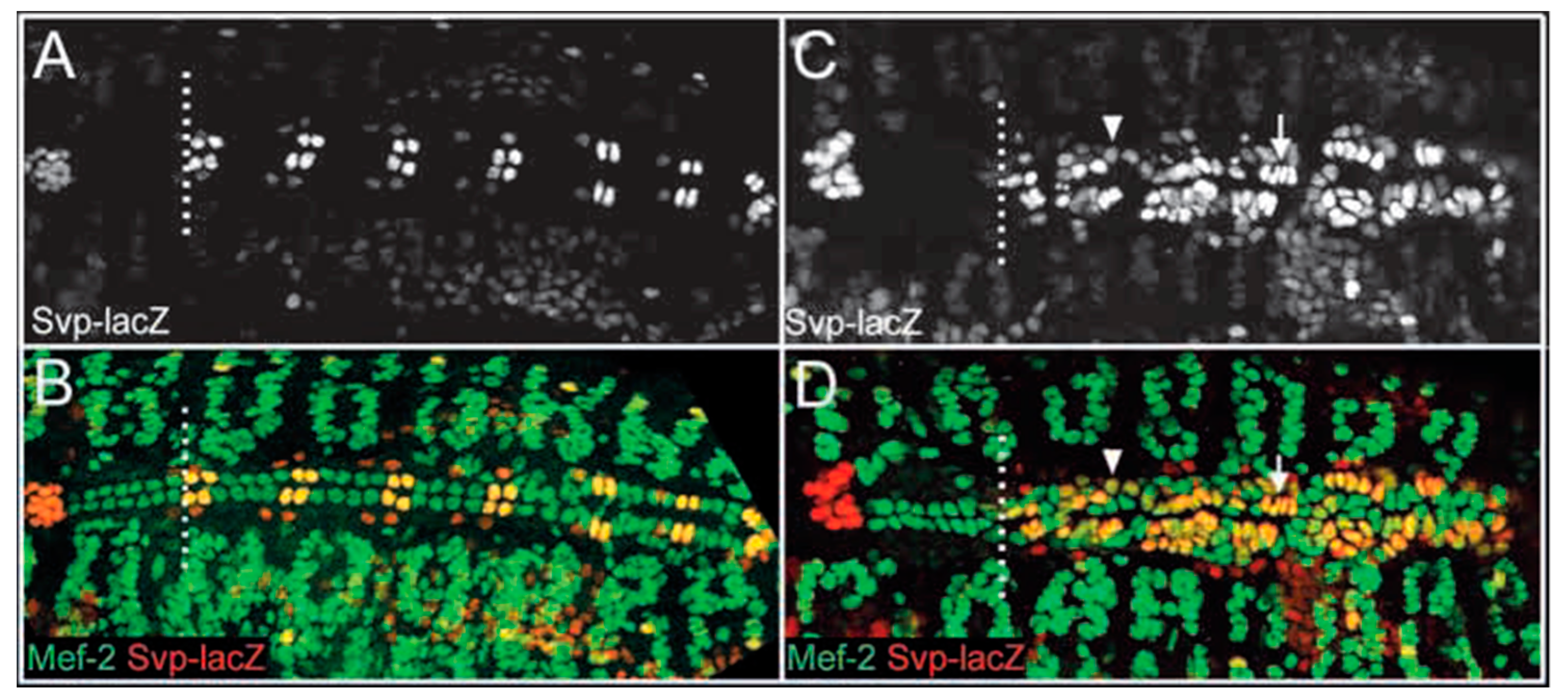
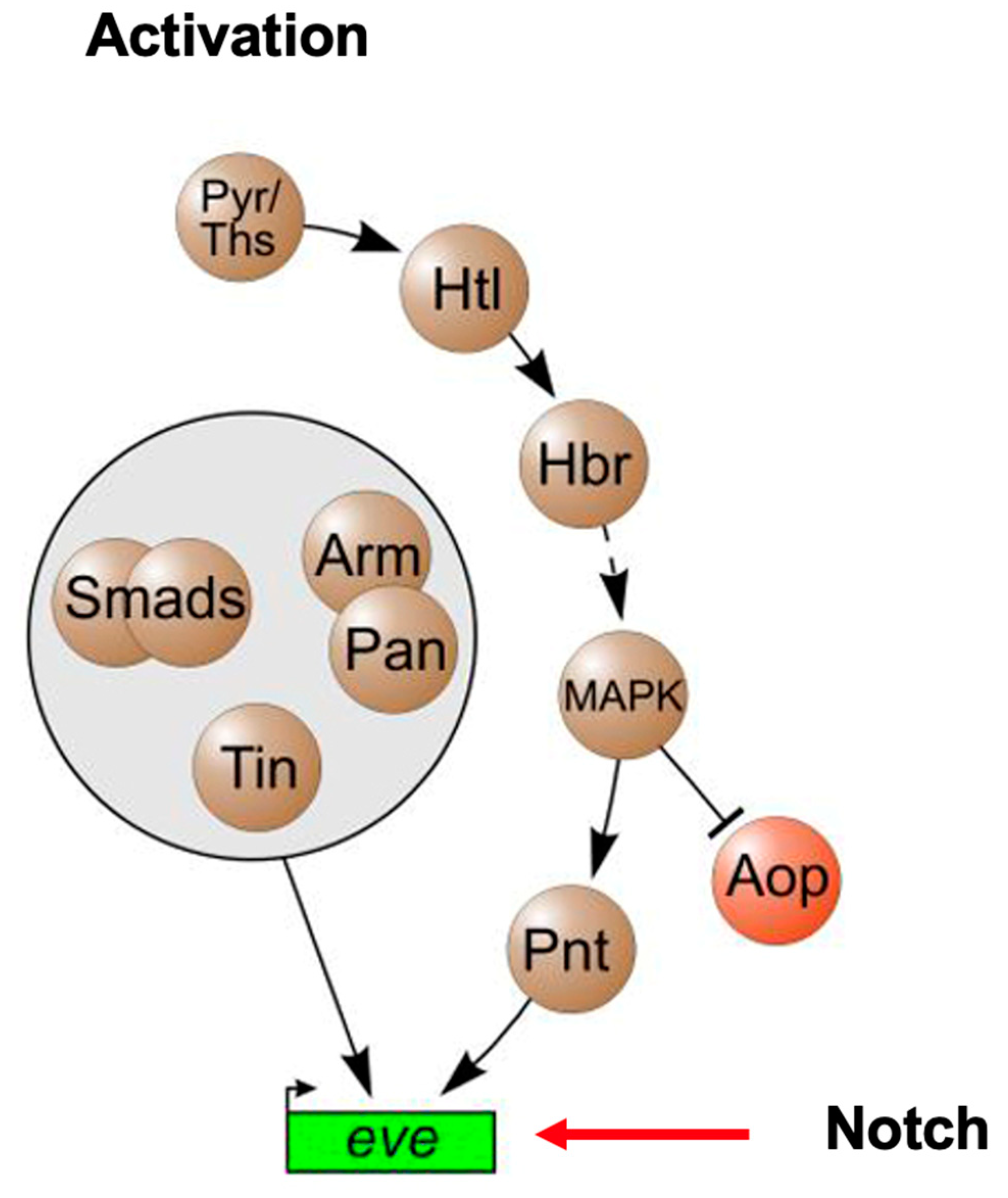
2. A Role for ETS1 in Heart Cell Fate Determination and the “Hypoplastic” Paradox: Reconciling Animal Models with Human HLHS
3. The Cardiac Myocyte and HLHS: Clinical Implications
4. Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grossfeld, P.; Nie, S.; Lin, L.; Wang, L.; Anderson, R.H. Hypoplastic Left Heart Syndrome: A New Paradigm for an Old Disease? J. Cardiovasc. Dev. Dis. 2019, 6, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crucean, A.; Alqahtani, A.; Barron, D.J.; Brawn, W.J.; Richardson, R.V.; O’Sullivan, J.; Anderson, R.H.; Henderson, D.J.; Chaudhry, B. Re-evaluation of hypoplastic left heart syndrome from a developmental and morphological perspective. Orphanet J. Rare Dis. 2017, 12, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, M.; Coldren, C.; Liang, X.; Mattina, T.; Goldmuntz, E.; Benson, D.W.; Ivy, D.; Perryman, M.B.; Garrett-Sinha, L.A.; Grossfeld, P. Deletion of ETS-1, a gene in the Jacobsen syndrome critical region, causes ventricular septal defects and abnormal ventricular morphology in mice. Hum. Mol. Genet. 2010, 19, 648–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, B.; Shi, W.; Levine, M. Uncoupling heart cell specification and migration in the simple chordate Ciona intestinalis. Development 2005, 132, 4811–4818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, A.D.; Shi, W.; Wilson, B.A.; Skeath, J.B. pannier and pointedP2 act sequentially to regulate Drosophila heart development. Development 2003, 130, 3015–3026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, S.; Bronner, M.E. Dual developmental role of transcriptional regulator Ets1 in Xenopus cardiac neural crest vs. heart mesoderm. Cardiovasc. Res. 2015, 106, 67–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohlmeyer, T.J.; Helmke, S.; Ge, S.; Lynch, J.; Brodsky, G.; Sederberg, J.H.; Robertson, A.D.; Minobe, W.; Bristow, M.R.; Perryman, M.B. Hypoplastic left heart syndrome myocytes are differentiated but possess a unique phenotype. Cardiovasc. Pathol. 2003, 12, 23–31. [Google Scholar] [CrossRef]
- Miao, Y.; Tian, L.; Martin, M.; Paige, S.L.; Galdos, F.X.; Li, J.; Klein, A.; Zhang, H.; Ma, N.; Wei, Y.; et al. Intrinsic Endocardial Defects Contribute to Hypoplastic Left Heart Syndrome. Cell Stem Cell 2020, 27, 574–589.e8. [Google Scholar] [CrossRef] [PubMed]
- Paige, S.L.; Galdos, F.X.; Lee, S.; Chin, E.T.; Ranjbarvaziri, S.; Feyen, D.A.M.; Darsha, A.K.; Xu, S.; Ryan, J.A.; Beck, A.L.; et al. Patient-Specific Induced Pluripotent Stem Cells Implicate Intrinsic Impaired Contractility in Hypoplastic Left Heart Syndrome. Circulation 2020, 142, 1605–1608. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Yoshida, M.; Tarui, S.; Hirata, M.; Nagai, Y.; Kasahara, S.; Naruse, K.; Ito, H.; Sano, S.; Oh, H. Directed differentiation of patient-specific induced pluripotent stem cells identifies the transcriptional repression and epigenetic modification of NKX2-5, HAND1, and NOTCH1 in hypoplastic left heart syndrome. PLoS ONE 2014, 9, e102796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Habibollah, S.; Tilgner, K.; Collin, J.; Barta, T.; Al-Aama, J.Y.; Tesarov, L.; Hussain, R.; Trafford, A.W.; Kirkwood, G.; et al. An induced pluripotent stem cell model of hypoplastic left heart syndrome (HLHS) reveals multiple expression and functional differences in HLHS-derived cardiac myocytes. Stem Cells Transl. Med. 2014, 3, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Hrstka, S.C.; Li, X.; Nelson, T.J.; Wanek Program Genetics Pipeline, G. NOTCH1-Dependent Nitric Oxide Signaling Deficiency in Hypoplastic Left Heart Syndrome Revealed Through Patient-Specific Phenotypes Detected in Bioengineered Cardiogenesis. Stem Cells 2017, 35, 1106–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theis, J.L.; Hrstka, S.C.; Evans, J.M.; O’Byrne, M.M.; de Andrade, M.; O’Leary, P.W.; Nelson, T.J.; Olson, T.M. Compound heterozygous NOTCH1 mutations underlie impaired cardiogenesis in a patient with hypoplastic left heart syndrome. Hum. Genet. 2015, 134, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Snider, P.; Simmons, O.; Wang, J.; Hoang, C.Q.; Conway, S.J. Ectopic Noggin in a Population of Nfatc1 Lineage Endocardial Progenitors Induces Embryonic Lethality. J. Cardiovasc. Dev. Dis. 2014, 1, 214–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- deAlmeida, A.; McQuinn, T.; Sedmera, D. Increased ventricular preload is compensated by myocyte proliferation in normal and hypoplastic fetal chick left ventricle. Circ. Res. 2007, 100, 1363–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pesevski, Z.; Kvasilova, A.; Stopkova, T.; Nanka, O.; Drobna Krejci, E.; Buffinton, C.; Kockova, R.; Eckhardt, A.; Sedmera, D. Endocardial Fibroelastosis is Secondary to Hemodynamic Alterations in the Chick Embryonic Model of Hypoplastic Left Heart Syndrome. Dev. Dyn. 2018, 247, 509–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, A.; DeYoung, T.; Cahill, L.S.; Yee, Y.; Debebe, S.K.; Botelho, O.; Seed, M.; Chaturvedi, R.R.; Sled, J.G. A mouse model of hypoplastic left heart syndrome demonstrating left heart hypoplasia and retrograde aortic arch flow. Dis. Models Mech. 2021, 14, dmm049077. [Google Scholar] [CrossRef] [PubMed]
- Tworetzky, W.; Wilkins-Haug, L.; Jennings, R.W.; van der Velde, M.E.; Marshall, A.C.; Marx, G.R.; Colan, S.D.; Benson, C.B.; Lock, J.E.; Perry, S.B. Balloon dilation of severe aortic stenosis in the fetus: Potential for prevention of hypoplastic left heart syndrome: Candidate selection, technique, and results of successful intervention. Circulation 2004, 110, 2125–2131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryantsev, A.; Cripps, R.M. Cardiac gene regulatory networks in Drosophila. Biochim. Biophys. Acta 2009, 1789, 343–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartenstein, A.Y.; Rugendorff, A.; Tepass, U.; Hartenstein, V. The function of the neurogenic genes during epithelial development in the Drosophila embryo. Development 1992, 116, 1203–1220. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grossfeld, P. ETS1 and HLHS: Implications for the Role of the Endocardium. J. Cardiovasc. Dev. Dis. 2022, 9, 219. https://doi.org/10.3390/jcdd9070219
Grossfeld P. ETS1 and HLHS: Implications for the Role of the Endocardium. Journal of Cardiovascular Development and Disease. 2022; 9(7):219. https://doi.org/10.3390/jcdd9070219
Chicago/Turabian StyleGrossfeld, Paul. 2022. "ETS1 and HLHS: Implications for the Role of the Endocardium" Journal of Cardiovascular Development and Disease 9, no. 7: 219. https://doi.org/10.3390/jcdd9070219
APA StyleGrossfeld, P. (2022). ETS1 and HLHS: Implications for the Role of the Endocardium. Journal of Cardiovascular Development and Disease, 9(7), 219. https://doi.org/10.3390/jcdd9070219