

Integrated Genome Sequencing and Transcriptome Analysis Identifies Candidate Pathogenicity Genes from Ustilago crameri

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strain Isolates, Culture Conditions, and Genomic DNA and RNA Isolation
2.2. Genome Sequencing and Assembly
2.3. Gene Prediction and Annotation
2.4. Gene Function Annotation
2.5. Comparative Genomics Analysis
2.6. Transcriptome Expression
2.7. Secreted Proteins and Potential Effector Analysis
2.8. Quantitative Real-Time Reverse Transcription–Polymerase Chain Reaction
3. Results
3.1. Genome Sequencing and Assembly
3.2. Genome Annotation
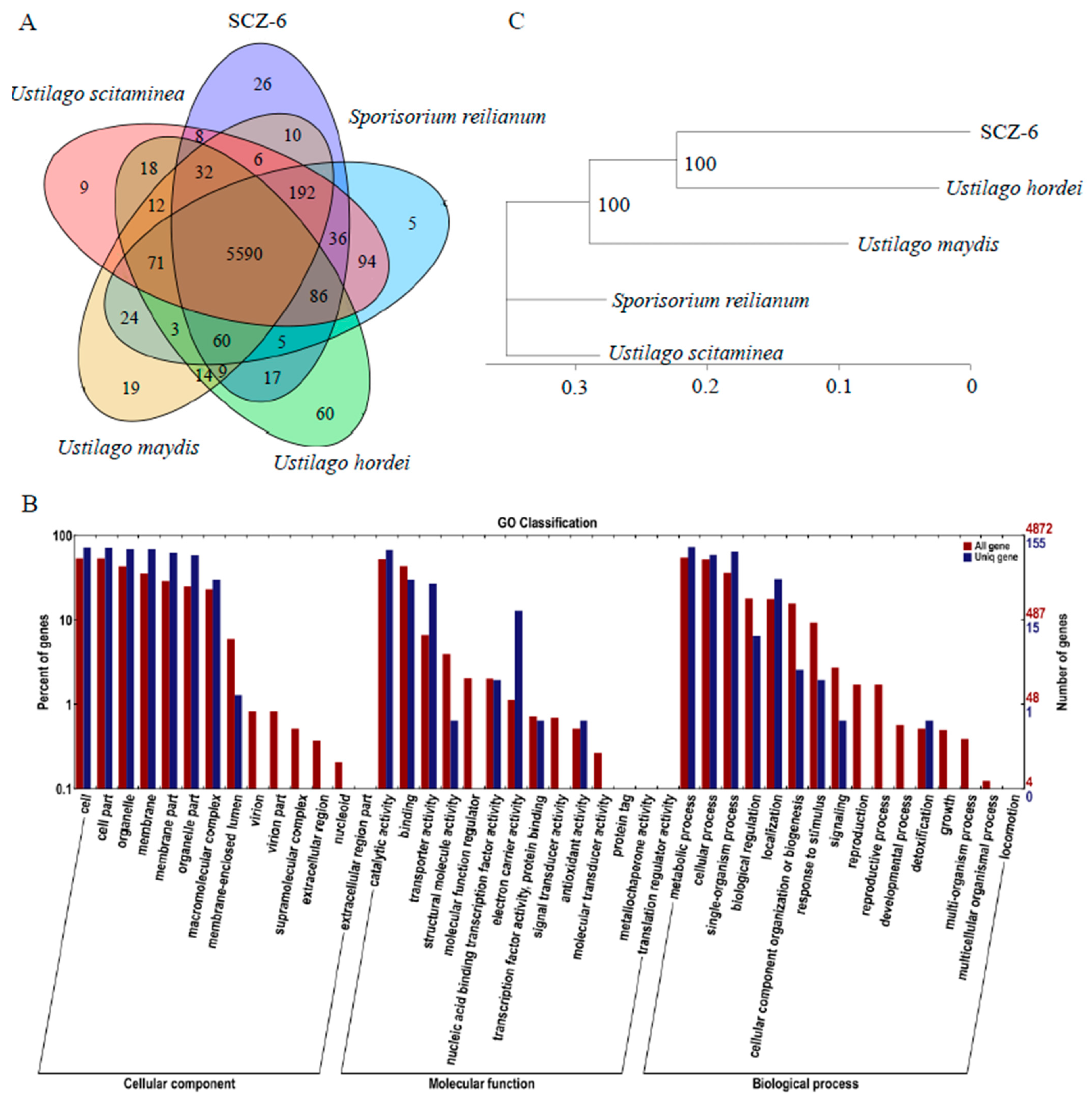
3.3. Comparative Genomics of Five Smut Fungi
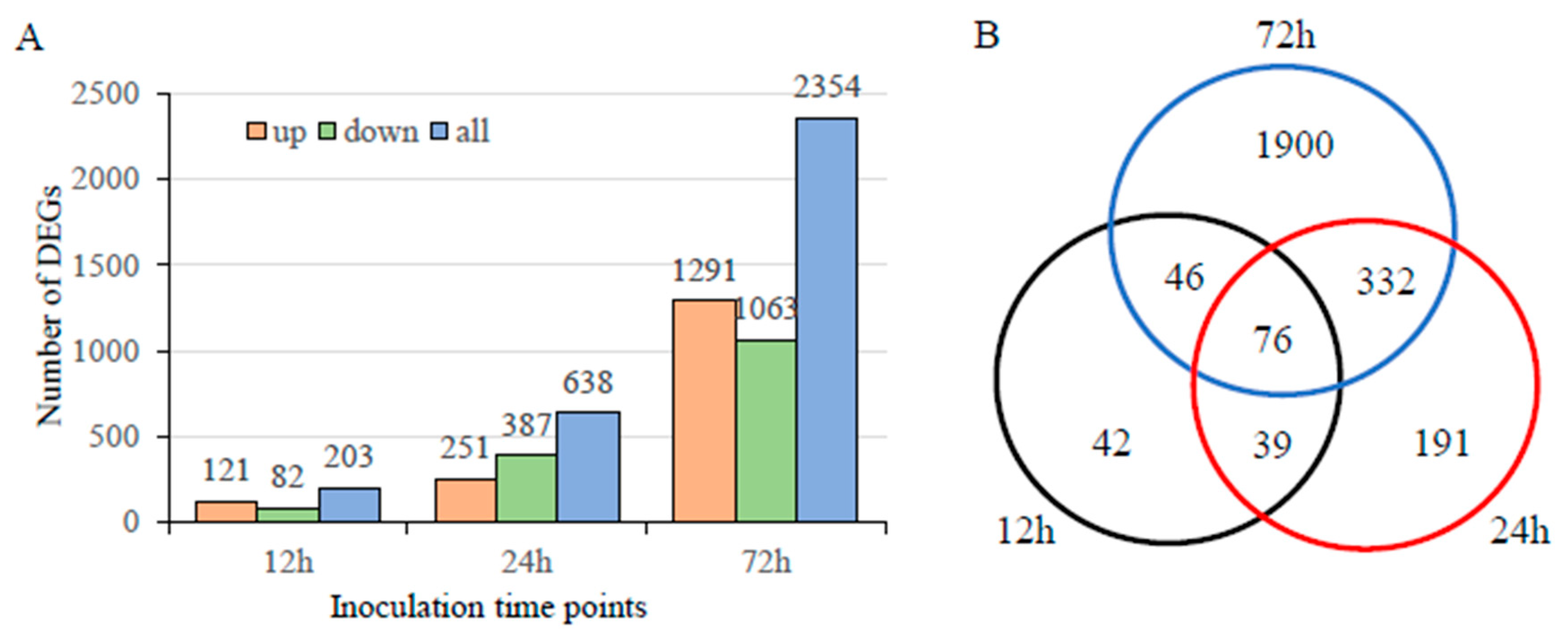
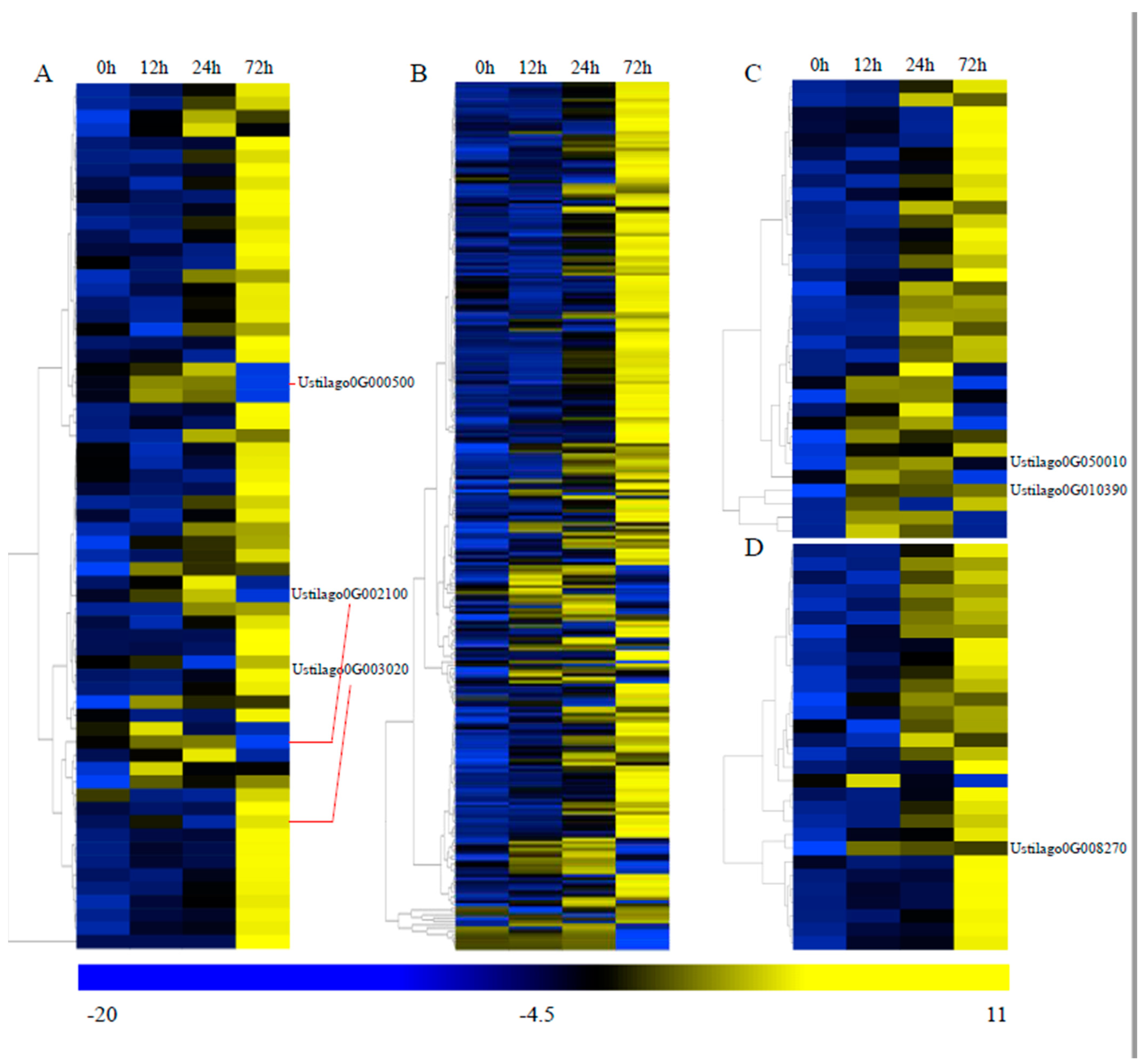
3.4. Transcriptome Analysis during Infection
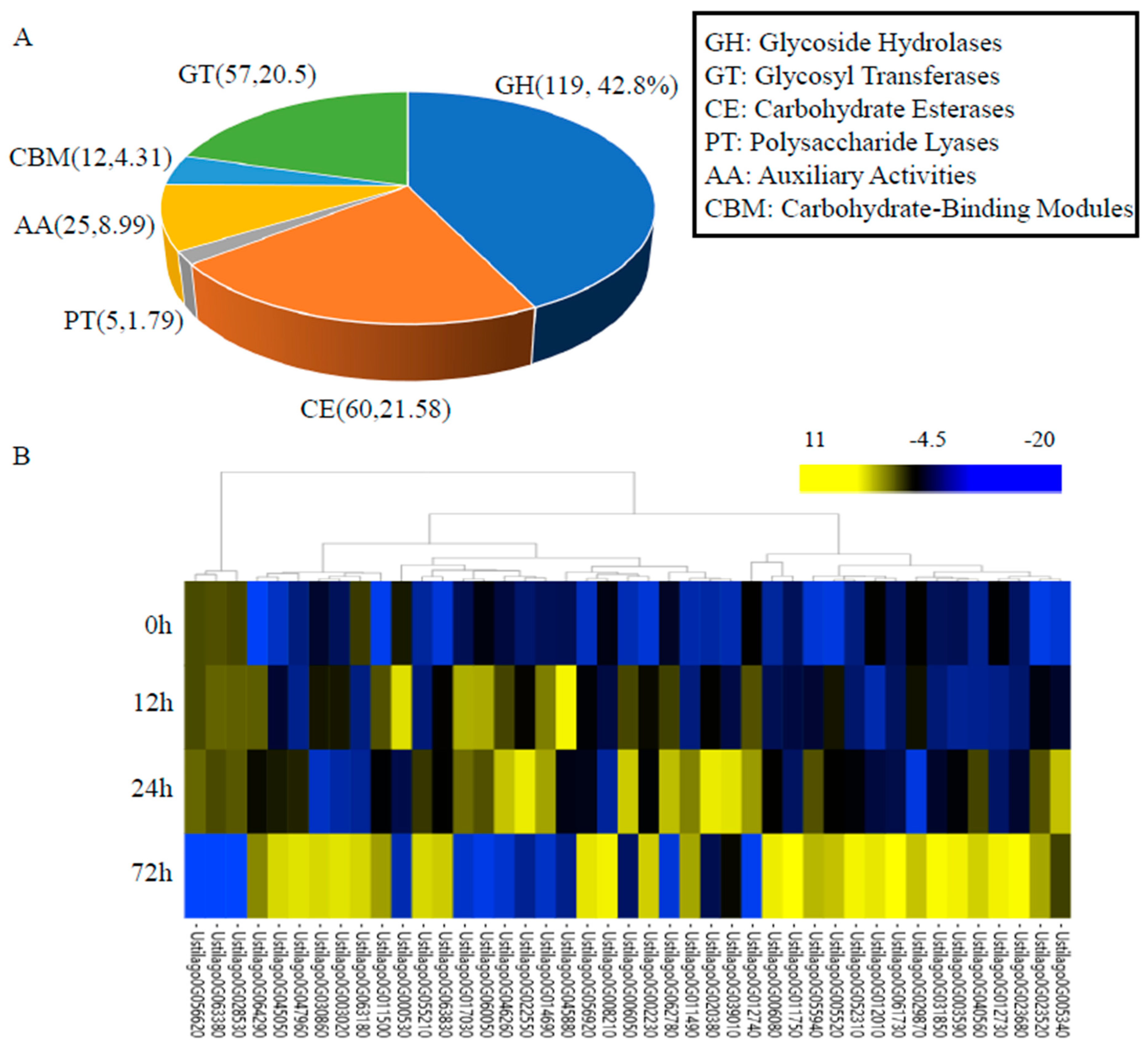
3.5. Carbohydrate-Active Enzymes
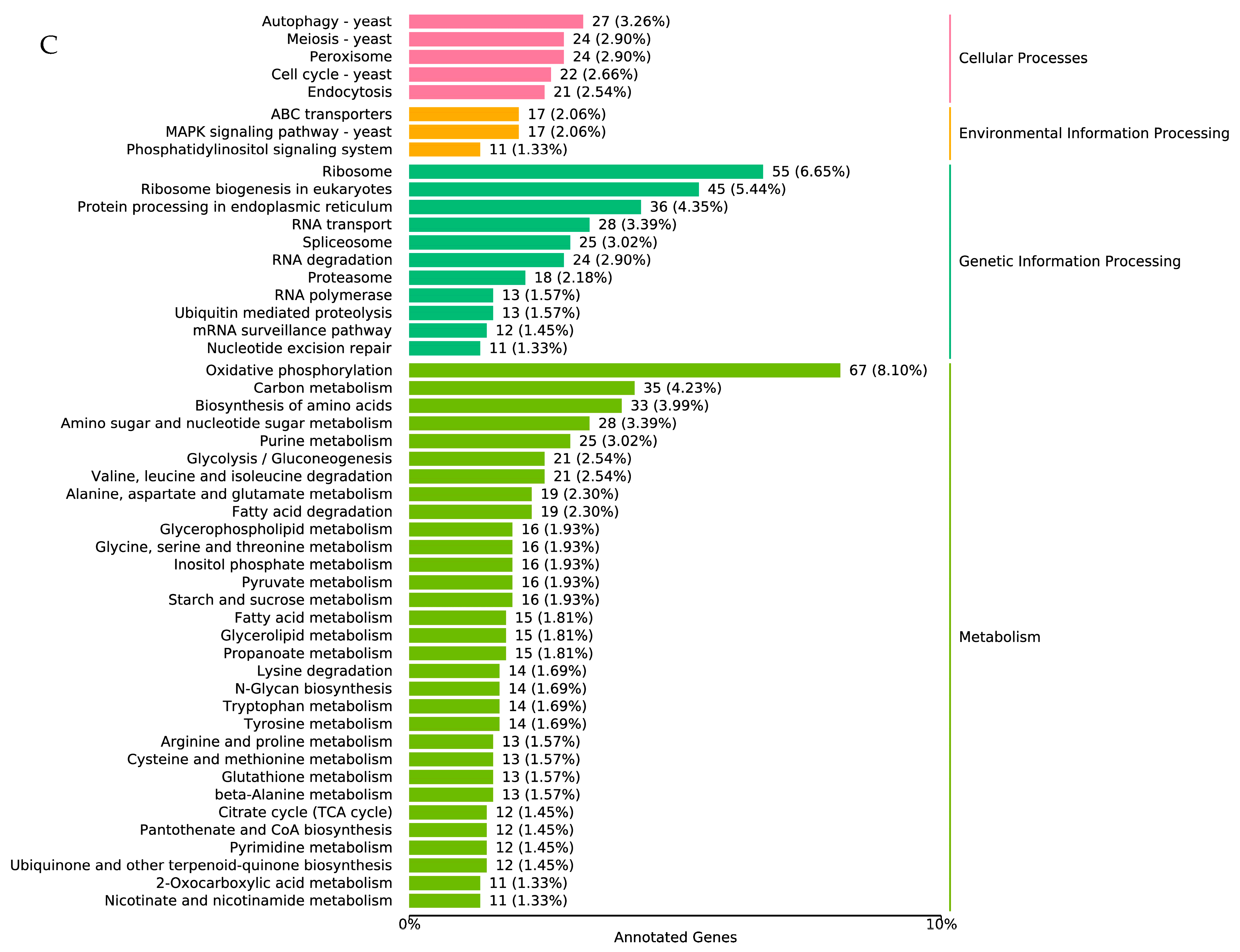
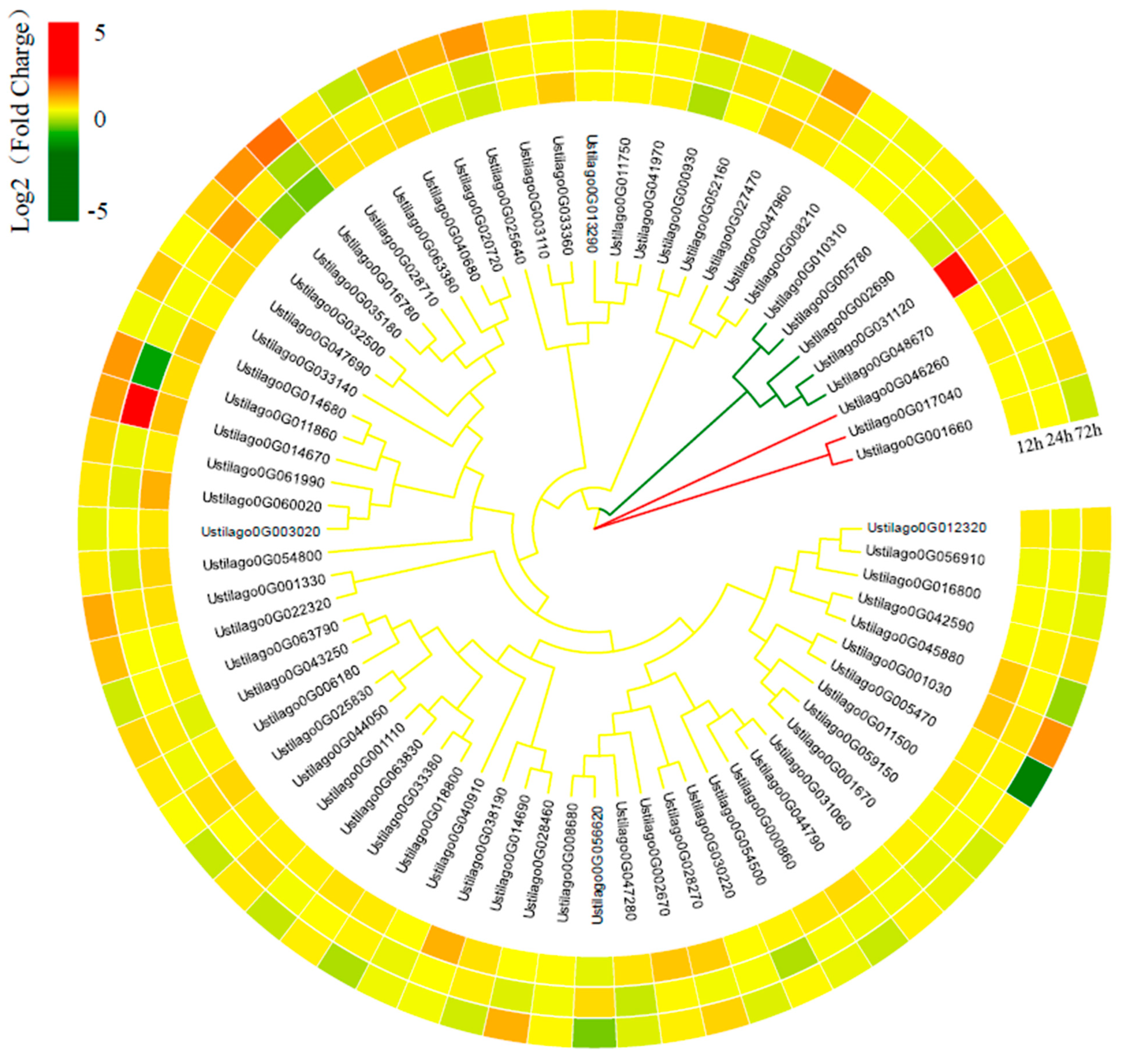
3.6. Important Genes Involved in Pathogenicity
3.7. U. crameri Candidate Effectors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kumar, B. First record of smut disease of foxtail millet caused by Ustilago crameri Korn. J. Mycol. Plant Pathol. 2011, 41, 459–461. [Google Scholar]
- Wang, C.S. Physiologic specialization and the control of millet smut. Phytopathology 1944, 34, 1050–1055. [Google Scholar]
- Lata, C.; Gupta, S.; Prasad, M. Foxtail millet: A model crop for genetic and genomic studies in bioenergy grasses. Crit. Rev. Biotechnol. 2013, 33, 328–343. [Google Scholar] [CrossRef] [PubMed]
- Muthamilarasan, M.; Prasad, M. Advances in Setaria genomics for genetic improvement of cereals and bioenergy grasses. TAG. Theoretical and applied genetics. Theor. Appl. Genet. 2015, 128, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, Z.; Jin, K. Investigation on occurrence and control measures of millet smut in 2003 and 2004 in southeastern Shanxi Province. Shaanxi Agric. Sci. 2007, 1, 94–95. [Google Scholar]
- Hao, L.; Liu, J.; Zhang, A.; Han, Y.; Yi, H. Differential gene expression in foxtail millet during interaction with the smut fungus Ustilago crameri. Physiol. Mol. Plant Pathol. 2020, 110, 101459. [Google Scholar] [CrossRef]
- Lockhart, D.J.; Winzeler, E.A. Genomics, gene expression and DNA arrays. Nature 2000, 405, 827–836. [Google Scholar] [CrossRef]
- Xue, W.; Li, J.T.; Zhu, Y.P.; Hou, G.Y.; Kong, X.F.; Kuang, Y.Y.; Sun, X.W. L_RNA_scaffolder: Scaffolding genomes with transcripts. BMC Genom. 2013, 14, 604. [Google Scholar] [CrossRef]
- Kumar, A.; Pandey, V.; Singh, M.; Pandey, D.; Saharan, M.S.; Marla, S.S. Draft genome sequence of Karnal bunt pathogen (Tilletia indica) of wheat provides insights into the pathogenic mechanisms of quarantined fungus. PLoS ONE 2017, 12, e0171323. [Google Scholar] [CrossRef]
- Gurjar, M.S.; Aggarwal, R.; Jogawat, A.; Kulshreshtha, D.; Sharma, S.; Solanke, A.U.; Dubey, H.; Jain, R.K. De novo genome sequencing and secretome analysis of Tilletia indica inciting Karnal bunt of wheat provides pathogenesis-related genes. 3 Biotech. 2019, 9, 219. [Google Scholar] [CrossRef]
- Wang, A.; Pang, L.; Wang, N.; Ai, P.; Yin, D.; Li, S.; Deng, Q.; Zhu, J.; Liang, Y.; Zhu, J.; et al. The pathogenic mechanisms of Tilletia horrida as revealed by comparative and functional genomics. Sci. Rep. 2018, 8, 15413. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Shu, X.; Niu, X.; Yi, X.; Zheng, A. Transcriptome analysis and whole genome re-sequencing provide insights on rice kernel smut (Tilletia horrida) pathogenicity. J. Plant Pathol. 2020, 102, 155–167. [Google Scholar] [CrossRef]
- Wang, A.; Pan, L.; Niu, X.; Shu, X.; Yi, X.; Yamamoto, N.; Li, S.; Deng, Q.; Zhu, J.; Liang, Y.; et al. Comparative secretome analysis of different smut fungi and identification of plant cell death-inducing secreted proteins from Tilletia horrida. BMC Plant Biol. 2019, 19, 360. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Yin, D.; Liang, J.; Xu, D.; Jiang, Y.; Xiang, T.; Wang, Y.; Jiao, C.; Li, P.; Zheng, A.; et al. ThSCSP_12: Novel effector in Tilletia horrida that induces cell death and defense responses in non-host plants. Int. J. Mol. Sci. 2022, 23, 14752. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Xu, D.; Jiang, Y.; Liang, J.; Xiang, T.; Wang, Y.; Zhang, W.; Han, X.; Jiao, C.; Zheng, A.; et al. Functional analyses of a small secreted cysteine-rich protein ThSCSP_14 in Tilletia horrida. Int. J. Mol. Sci. 2022, 23, 15042. [Google Scholar] [CrossRef]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Zdobnov, E.M. BUSCO: Assessing genomic data quality and beyond. Curr. Protoc. 2021, 1, e323. [Google Scholar] [CrossRef]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef]
- Stanke, M.; Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 2003, 19, ii215–ii225. [Google Scholar] [CrossRef]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef]
- Blanco, E.; Parra, G.; Guigó, R. Using geneid to identify genes. Curr. Protoc. Bioinform. 2007, 18, 4.3.1–4.3.28. [Google Scholar] [CrossRef]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Keilwagen, J.; Wenk, M.; Erickson, J.L.; Schattat, M.H.; Grau, J.; Hartung, F. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 2016, 44, e89. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.A.; Haas, B.J.; Hamilton, J.P.; Mount, S.M.; Buell, C.R. Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genom. 2006, 7, 327. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using Evidence Modeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef]
- Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2004, 25, 4.10.1–4.10.14. [Google Scholar]
- Lowe, T.M.; Eddy, S.R. tRNA scan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Burge, S.W.; Bateman, A.; Daub, J.; Eberhardt, R.Y.; Eddy, S.R.; Floden, E.W.; Gardner, P.P.; Jones, T.A.; Tate, J.; et al. Rfam 12.0: Updates to the RNA families database. Nucleic Acids Res. 2015, 43, D130–D137. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32 (Suppl. S1), D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P.; Chen, Y.W.; He, F.C. Integrated nr database in protein annotation system and its localization. Comput. Eng. 2006, 32, 71–74. [Google Scholar]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I.; et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Winnenburg, R.; Baldwin, T.K.; Urban, M.; Rawlings, C.; Köhler, J.; Hammond-Kosack, K.E. PHI-base: A new database for pathogen host interactions. Nucleic Acids Res. 2006, 34, D459–D464. [Google Scholar] [CrossRef]
- Lu, T.; Yao, B.; Zhang, C. DFVF: Database of fungal virulence factors. Database 2012, 2012, bas032. [Google Scholar] [CrossRef]
- Fischer, M.; Knoll, M.; Sirim, D.; Wagner, F.; Funke, S.; Pleiss, J. The Cytochrome P450 Engineering Database: A navigation and prediction tool for the cytochrome P450 protein family. Bioinformatics 2007, 23, 2015–2017. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Kriventseva, E.V.; Fleischmann, W.; Zdobnov, E.M.; Apweiler, R. CluSTr: A database of clusters of SWISS-PROT+TrEMBL proteins. Nucleic Acids Res. 2001, 29, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for trna genes in genomic sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar] [PubMed]
- Griffiths-Jones, S.; Moxon, S.; Marshall, M.; Khanna, A.; Eddy, S.R.; Bateman, A. Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005, 33, D121–D124. [Google Scholar] [CrossRef] [PubMed]
- Zheng, A.; Lin, R.; Zhang, D.; Qin, P.; Xu, L.; Ai, P.; Ding, L.; Wang, Y.; Chen, Y.; Liu, Y.; et al. The evolution and pathogenic mechanisms of the rice sheath blight pathogen. Nat. Commun. 2013, 4, 1424. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.I.H. Functions and mechanisms: Polygalacturonases from plant pathogenic fungi as pathogenicity and virulence factors. J. Gen. Plant Pathol. 2019, 85, 243–250. [Google Scholar] [CrossRef]
- Cord-Landwehr, S.; Melcher, R.L.; Kolkenbrock, S.; Moerschbacher, B.M. A chitin deacetylase from the endophytic fungus Pestalotiopsis sp. efficiently inactivates the elicitor activity of chitin oligomers in rice cells. Sci. Rep. 2016, 6, 38018. [Google Scholar] [CrossRef]
- Lee, J.; Min, K.R.; Kim, Y.C.; Kim, C.K.; Lim, J.Y.; Yoon, H.; Min, K.H.; Lee, K.S.; Kim, Y. Cloning of salicylate hydroxylase gene and catechol 2,3-dioxygenase gene and sequencing of an intergenic sequence between the two genes of Pseudomonas putida KF715. Biochem. Biophys. Res. Commun. 1995, 211, 382–388. [Google Scholar] [CrossRef]
- Jennings, D.B. The Role of Mannitol and Mannitol Dehydrogenase in Plant-Pathogen Interactions. Ph.D. Thesis, North Carolina State University, Raleigh, NC, USA, 2023. [Google Scholar]
- Nelson, D.R. Cytochrome P450 and the individuality of species. Arch. Biochem. Biophys. 1999, 369, 1–10. [Google Scholar] [CrossRef]
- Que, Y.; Xu, L.; Wu, Q.; Liu, Y.; Ling, H.; Liu, Y.; Zhang, Y.; Guo, J.; Su, Y.; Chen, J.; et al. Genome sequencing of Sporisorium scitamineum provides insights into the pathogenic mechanisms of sugarcane smut. BMC Genom. 2014, 15, 996. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Hatabayashi, H.; Arai, H.; Kitamoto, H.K.; Yabe, K. Function of the cypX and moxY genes in aflatoxin biosynthesis in Aspergillus parasiticus. Appl. Environ. Microbiol. 2005, 71, 3192–3198. [Google Scholar] [CrossRef] [PubMed]
- Mueller, O.; Kahmann, R.; Aguilar, G.; Trejo-Aguilar, B.; Wu, A.; de Vries, R.P. The secretome of the maize pathogen Ustilago maydis. Fungal Genet. Biol. 2008, 45, S63–S70. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Lei, L.; Liu, C.; Zhang, Y.; Xu, Q.; Zhu, J.; Guo, Z.; Wang, Y.; Li, Q.; Li, Y.; et al. Major Facilitator Superfamily Transporter Gene FgMFS1 Is Essential for Fusarium graminearum to Deal with Salicylic Acid Stress and for Its Pathogenicity towards Wheat. Int. J. Mol. Sci. 2021, 22, 8497. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.; Thorborg, K.; Nielsen, M.B.; Budtz-Jørgensen, E.; Hölmich, P. Preventive effect of eccentric training on acute hamstring injuries in men’s soccer: A cluster-randomized controlled trial. Am. J. Sports Med. 2011, 39, 2296–2303. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Laurie, J.D.; Linning, R.; Cervantes-Chávez, J.A.; Gaudet, D.; Bakkeren, G. An immunity-triggering effector from the Barley smut fungus Ustilago hordei resides in an Ustilaginaceae-specific cluster bearing signs of transposable element-assisted evolution. PLoS Pathog. 2014, 10, e1004223. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hao, Z.; Sun, H.; Liu, J.; Shen, S.; Zhou, C.; Li, Z. Genome Sequence Resource of Ustilago crameri, a Fungal Pathogen Causing Millet Smut Disease of Foxtail Millet. Plant Dis. 2023, 107, 546–548. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, K.; Fang, A.; Han, Y.; Yang, J.; Xue, M.; Bao, J.; Hu, D.; Zhou, B.; Sun, X.; et al. Specific adaptation of Ustilaginoidea virens in occupying host florets revealed by comparative and functional genomics. Nat. Commun. 2014, 5, 3849. [Google Scholar] [CrossRef]
- Kemen, E.; Gardiner, A.; Schultz-Larsen, T.; Kemen, A.C.; Balmuth, A.L.; Robert-Seilaniantz, A.; Bailey, K.; Holub, E.; Studholme, D.J.; Maclean, D.; et al. Gene gain and loss during evolution of obligate parasitism in the white rust pathogen of Arabidopsis thaliana. PLoS Biol. 2011, 9, e1001094. [Google Scholar] [CrossRef]
- Kämper, J.; Kahmann, R.; Bölker, M.; Ma, L.J.; Brefort, T.; Saville, B.J.; Banuett, F.; Kronstad, J.W.; Gold, S.E.; Müller, O.; et al. Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 2006, 444, 97–101. [Google Scholar] [CrossRef]
- Gao, Q.; Jin, K.; Ying, S.H.; Zhang, Y.; Xiao, G.; Shang, Y.; Duan, Z.; Hu, X.; Xie, X.Q.; Zhou, G.; et al. Genome sequencing and comparative transcriptomics of the model entomopathogenic fungi Metarhizium anisopliae and M. acridum. PLoS Genet. 2011, 7, e1001264. [Google Scholar] [CrossRef] [PubMed]
- Alexander, N.J.; McCormick, S.P.; Hohn, T.M. TRI12, a trichothecene efflux pump from Fusarium sporotrichioides: Gene isolation and expression in yeast. Mol. Gen. Genet. 1999, 261, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Callahan, T.M.; Rose, M.S.; Meade, M.J.; Ehrenshaft, M.; Upchurch, R.G. CFP, the putative cercosporin transporter of Cercospora kikuchii, is required for wild type cercosporin production, resistance, and virulence on soybean. Mol. Plant Microbe Interact. 1999, 12, 901–940. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, M.C.; Valent, B. Filamentous plant pathogen effectors in action. Nat. Rev. Microbiol. 2013, 11, 800–814. [Google Scholar] [CrossRef] [PubMed]
- Mueller, A.N.; Ziemann, S.; Treitschke, S.; Aßmann, D.; Doehlemann, G. Compatibility in the Ustilago maydis-maize interaction requires inhibition of host cysteine proteases by the fungal effector Pit2. PLoS Pathog. 2013, 9, e1003177. [Google Scholar] [CrossRef] [PubMed]
- Redkar, A.; Villajuana-Bonequi, M.; Doehlemann, G. Conservation of the Ustilago maydis effector See1 in related smuts. Plant Signal. Behav. 2015, 10, e1086855. [Google Scholar] [CrossRef]
- Hemetsberger, C.; Herrberger, C.; Zechmann, B.; Hillmer, M.; Doehlemann, G. The Ustilago maydis effector Pep1 suppresses plant immunity by inhibition of host peroxidase activity. inhibition of host cysteine proteases by the fungal effector Pit2. PLoS Pathog. 2012, 8, e1002684. [Google Scholar] [CrossRef]
- Djamei, A.; Schipper, K.; Rabe, F.; Ghosh, A.; Vincon, V.; Kahnt, J.; Osorio, S.; Tohge, T.; Fernie, A.R.; Feussner, I.; et al. Metabolic priming by a secreted fungal effector. Nature 2011, 478, 395–398. [Google Scholar] [CrossRef]
- Tanaka, S.; Brefort, T.; Neidig, N.; Djamei, A.; Kahnt, J.; Vermerris, W.; Koenig, S.; Feussner, K.; Feussner, I.; Kahmann, R. A secreted Ustilago maydis effector promotes virulence by targeting anthocyanin biosynthesis in maize. eLife 2014, 3, e01355. [Google Scholar] [CrossRef]
- Stergiopoulos, I.; de Wit, P.J. Fungal effector proteins. Annu. Rev. Phytopathol. 2009, 47, 233–263. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genomic Features | Numbers |
|---|---|
| Genome size (Mb) | 19.55 |
| Coverage | 151.06× |
| Number of contigs | 73 |
| N50 (bp) | 840,209 |
| N90 (bp) | 396,038 |
| GC content (%) | 54.09 |
| Repeat rate (%) | 3.72 |
| Predicted protein-coding genes | 6576 |
| Average gene length (bp) | 2308.48 |
| Exons number | 10,145 |
| Average exon length (bp) | 1438.95 |
| Introns number | 3569 |
| Average intron length (bp) | 163.19 |
| tRNA | 357 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, J.; Yin, D.; Shu, X.; Xiang, T.; Zhang, C.; Li, H.; Wang, A. Integrated Genome Sequencing and Transcriptome Analysis Identifies Candidate Pathogenicity Genes from Ustilago crameri. J. Fungi 2024, 10, 82. https://doi.org/10.3390/jof10010082
Liang J, Yin D, Shu X, Xiang T, Zhang C, Li H, Wang A. Integrated Genome Sequencing and Transcriptome Analysis Identifies Candidate Pathogenicity Genes from Ustilago crameri. Journal of Fungi. 2024; 10(1):82. https://doi.org/10.3390/jof10010082
Chicago/Turabian StyleLiang, Juan, Desuo Yin, Xinyue Shu, Ting Xiang, Chao Zhang, Honglian Li, and Aijun Wang. 2024. "Integrated Genome Sequencing and Transcriptome Analysis Identifies Candidate Pathogenicity Genes from Ustilago crameri" Journal of Fungi 10, no. 1: 82. https://doi.org/10.3390/jof10010082
APA StyleLiang, J., Yin, D., Shu, X., Xiang, T., Zhang, C., Li, H., & Wang, A. (2024). Integrated Genome Sequencing and Transcriptome Analysis Identifies Candidate Pathogenicity Genes from Ustilago crameri. Journal of Fungi, 10(1), 82. https://doi.org/10.3390/jof10010082