

Deciphering the Genomic Landscape and Virulence Mechanisms of the Wheat Powdery Mildew Pathogen Blumeria graminis f. sp. tritici Wtn1: Insights from Integrated Genome Assembly and Conidial Transcriptomics


 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virulence Profiling and Selection of the Isolate
2.2. Selection of the Powdery Mildew Pathogen for WGS
2.3. The Wheat Cultivar and Axenic Culturing of the Powdery Mildew Pathogen
2.4. Mass Production of Conidia and Microscopy
2.5. Genomic DNA Isolation and Strain Identification
2.6. Amplification of the Pathogenesis-Related Genes
2.7. Genomic Libraries and Whole-Genome Sequencing
2.8. Sequence Curation and De Novo Genome Assembly
2.9. Molecular Phylogeny
2.10. Gene Prediction and Annotation
2.11. Comparative Genomic Analysis
2.12. Transcriptomics of B. graminis f. sp. tritici Wtn1
2.13. Genome Analysis for Nucleotide Variation
3. Results
3.1. Sequence Read Quality, Genome Assembly and Evaluation
3.2. Identification of Repeats and Transposable Elements
3.3. Gene Prediction, Annotation Validation, and Comparative Protein Coding Genes
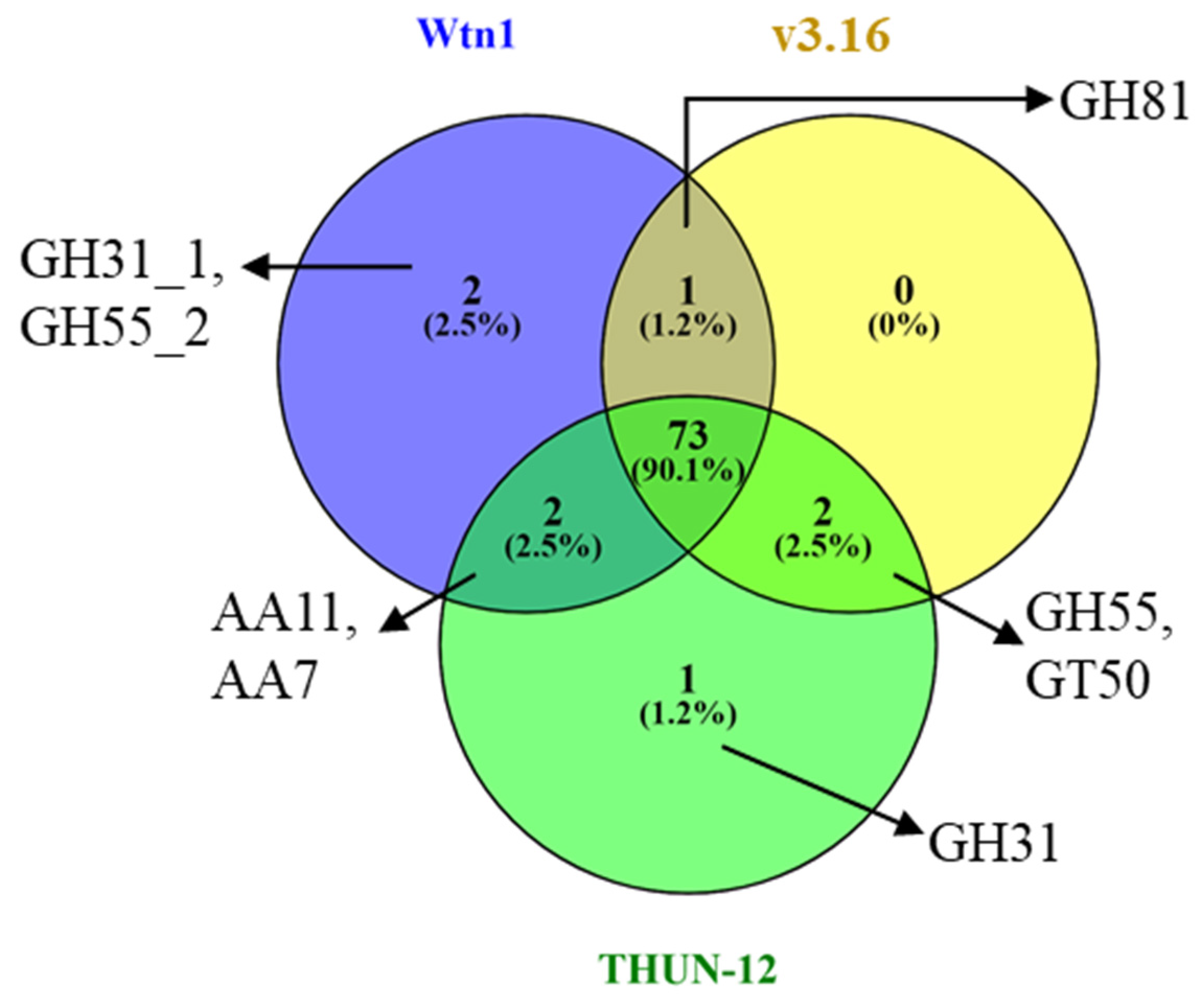
3.4. Analysis of the Orthologous Genes and CAZymes in the Assembled Genomes
3.5. Deciphering the tRNA Anticodons in the Genome
3.6. Gene Ontology Annotation
3.7. KOG Classification of the Genome
3.8. KEGG Pathways
3.9. KEGG Mapper BRITE Classification
3.10. Prediction of Pathogenicity and Virulence Effectors
3.11. Transcriptome-Driven Gene Expression and Functional Annotation
3.12. Functional Annotation of the Conidial Transcriptome
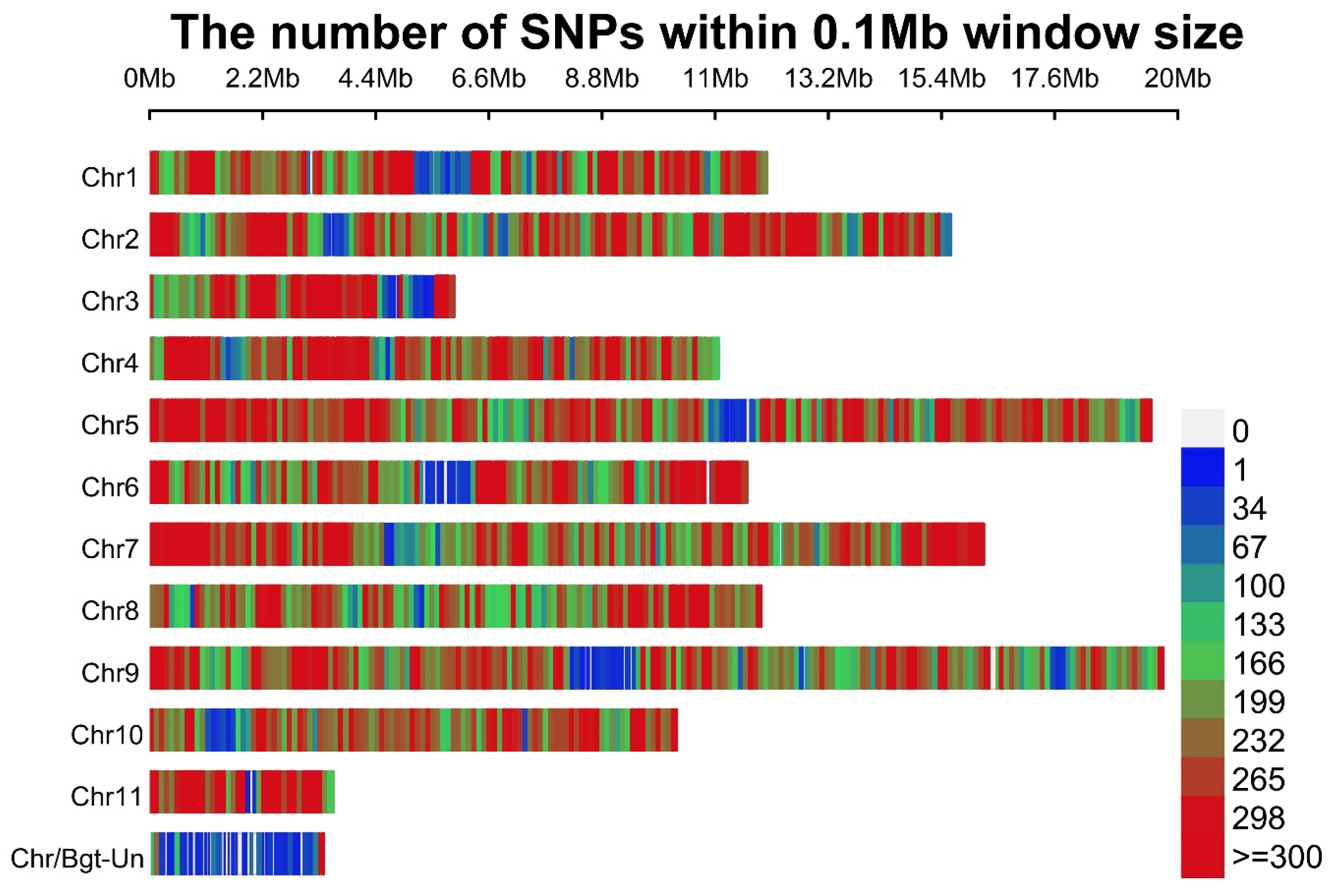
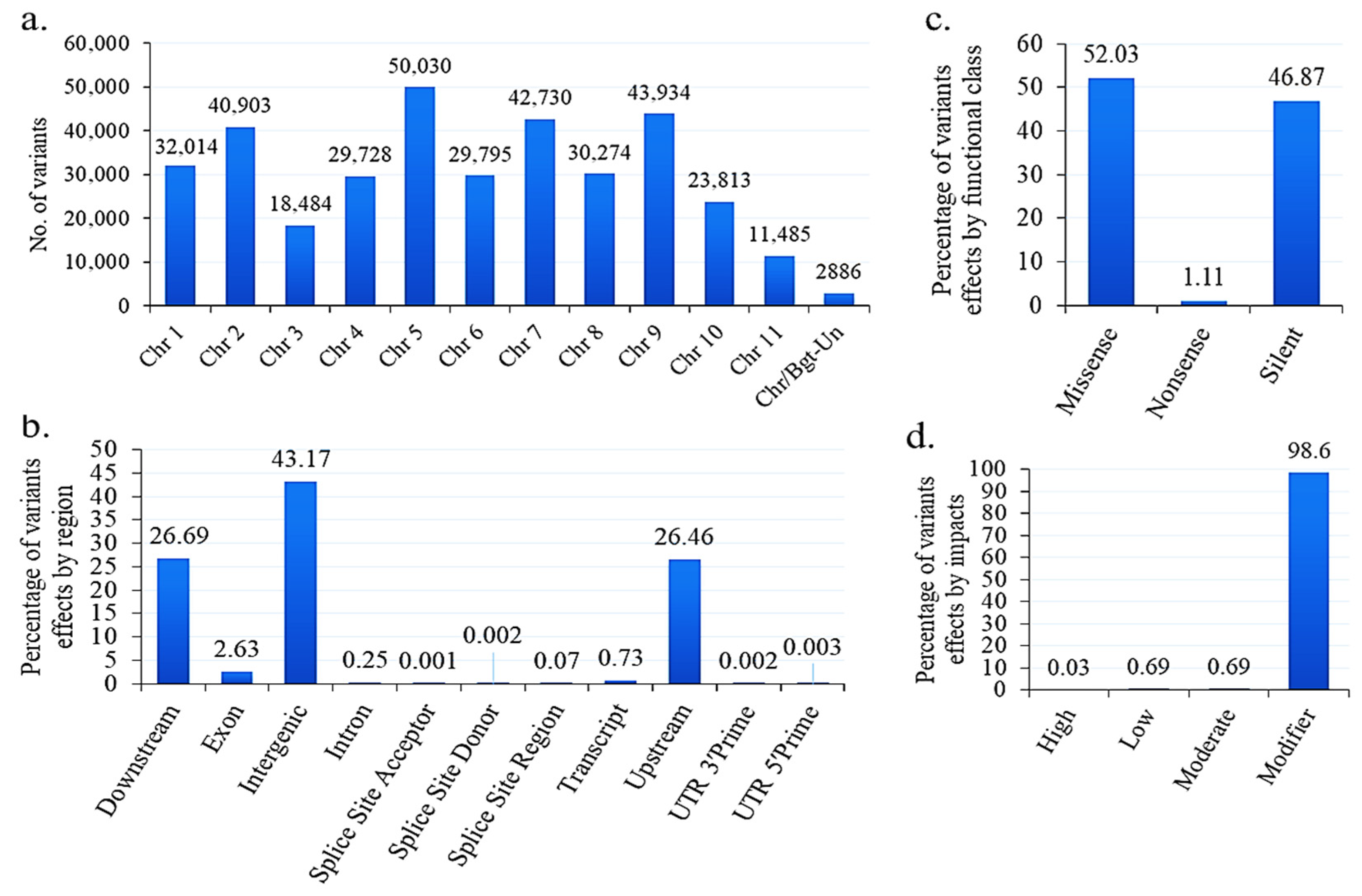
3.13. SNPs and Indels in B. graminis f. sp. tritici
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sharma, I. Diseases in Wheat Crops-an Introduction. In Disease Resistance in Wheat; Sharma, I., Ed.; CABI Digital Library: Wallingford, UK, 2012; pp. 1–17. [Google Scholar] [CrossRef]
- Dean, R.; Van Kan, J.A.L.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 Fungal Pathogens in Molecular Plant Pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Traskovetskaya, V.; Gorash, A.; Liatukas, Ž.; Saulyak, N.; Ternovyi, K.; Babayants, O.; Ruzgas, V.; Leistrumaitė, A. Virulence and Diversity of the Blumeria graminis f. sp. tritici Populations in Lithuania and Southern Ukraine. Zemdirb.-Agric. 2019, 106, 107–116. [Google Scholar] [CrossRef]
- Wolfe, M.; Schwarzbach, E. Patterns of Race Changes in Powdery Mildews. Annu. Rev. Phytopathol. 1978, 16, 159–180. [Google Scholar] [CrossRef]
- Hermansen, J.E.; Torp, U.; Prahm, L.P. Studies of Transport of Live Spores of Cereal Mildew and Rust Fungi across the North Sea. Grana 1978, 17, 41–46. [Google Scholar] [CrossRef]
- Parks, R.; Carbone, I.; Murphy, J.P.; Marshall, D.; Cowger, C. Virulence Structure of the Eastern U.S. Wheat Powdery Mildew Population. Plant Dis. 2008, 92, 1074–1082. [Google Scholar] [CrossRef]
- De Miccolis Angelini, R.M.; Pollastro, S.; Rotondo, P.R.; Laguardia, C.; Abate, D.; Rotolo, C.; Faretra, F. Transcriptome Sequence Resource for the Cucurbit Powdery Mildew Pathogen Podosphaera xanthii. Sci. Data 2019, 6, 1–7. [Google Scholar] [CrossRef]
- Ben-David, R.; Parks, R.; Dinoor, A.; Kosman, E.; Wicker, T.; Keller, B.; Cowger, C. Differentiation Among Blumeria graminis f. sp. tritici Isolates Originating from Wild Versus Domesticated Triticum Species in Israel. Phytopathology 2016, 106, 861–870. [Google Scholar] [CrossRef]
- Mackie, A.J.; Roberts, A.M.; Callow, J.A.; Green, J.R. Molecular Differentiation in Pea Powdery-Mildew Haustoria—Identification of a 62-KDa N-Linked Glycoprotein Unique to the Haustorial Plasma Membrane. Planta 1991, 183, 399–408. [Google Scholar] [PubMed]
- Panstruga, R.; Dodds, P.N. Terrific Protein Traffic: The Mystery of Effector Protein Delivery by Filamentous Plant Pathogens. Science 2009, 324, 748. [Google Scholar] [CrossRef]
- Stergiopoulos, I.; De Wit, P.J.G.M. Fungal Effector Proteins. Annu. Rev. Phytopathol. 2009, 47, 233–263. [Google Scholar] [CrossRef]
- Voegele, R.T.; Mendgen, K. Rust Haustoria: Nutrient Uptake and Beyond. N. Phytol. 2003, 159, 93–100. [Google Scholar] [CrossRef]
- Lo Presti, L.; Lanver, D.; Schweizer, G.; Tanaka, S.; Liang, L.; Tollot, M.; Zuccaro, A.; Reissmann, S.; Kahmann, R. Fungal Effectors and Plant Susceptibility. Annu. Rev. Plant Biol. 2015, 66, 513–545. [Google Scholar] [CrossRef]
- Liu, N.; Lewis Liu, Z.; Gong, G.; Zhang, M.; Wang, X.; Zhou, Y.; Qi, X.; Chen, H.; Yang, J.; Luo, P. Virulence Structure of Blumeria graminis f. sp. tritici and Its Genetic Diversity by ISSR and SRAP Profiling Analyses. PLoS ONE 2015, 10, e0130881. [Google Scholar] [CrossRef]
- Yuan, H.; Jin, C.; Pei, H.; Zhao, L.; Li, X.; Li, J.; Huang, W.; Fan, R.; Liu, W.; Shen, Q.H. The Powdery Mildew Effector CSEP0027 Interacts With Barley Catalase to Regulate Host Immunity. Front. Plant Sci. 2021, 12, 733237. [Google Scholar] [CrossRef]
- Wyand, R.A.; Brown, J.K.M. Genetic and Forma Specialis Diversity in Blumeria graminis of Cereals and Its Implications for Host-Pathogen Co-Evolution. Mol. Plant Pathol. 2003, 4, 187–198. [Google Scholar] [CrossRef]
- Troch, V.; Audenaert, K.; Bekaert, B.; Höfte, M.; Haesaert, G. Phylogeography and Virulence Structure of the Powdery Mildew Population on Its “new” Host Triticale. BMC Evol. Biol. 2012, 12, 1–12. [Google Scholar] [CrossRef]
- Spanu, P.D.; Abbott, J.C.; Amselem, J.; Burgis, T.A.; Soanes, D.M.; Stüber, K.; Van Themaat, E.V.L.; Brown, J.K.M.; Butcher, S.A.; Gurr, S.J.; et al. Genome Expansion and Gene Loss in Powdery Mildew Fungi Reveal Tradeoffs in Extreme Parasitism. Science 2010, 330, 1543–1546. [Google Scholar] [CrossRef]
- Wicker, T.; Oberhaensli, S.; Parlange, F.; Buchmann, J.P.; Shatalina, M.; Roffler, S.; Ben-David, R.; Doležel, J.; Šimková, H.; Schulze-Lefert, P. The Wheat Powdery Mildew Genome Shows the Unique Evolution of an Obligate Biotroph. Nat. Genet. 2013, 45, 1092–1096. [Google Scholar] [CrossRef]
- Hacquard, S.; Kracher, B.; Maekawa, T.; Vernaldi, S.; Schulze-Lefert, P.; Van Themaat, E.V.L. Mosaic Genome Structure of the Barley Powdery Mildew Pathogen and Conservation of Transcriptional Programs in Divergent Hosts. Proc. Natl. Acad. Sci. USA 2013, 110, E2219–E2228. [Google Scholar] [CrossRef]
- Menardo, F.; Praz, C.R.; Wyder, S.; Ben-David, R.; Bourras, S.; Matsumae, H.; McNally, K.E.; Parlange, F.; Riba, A.; Roffler, S.; et al. Hybridization of Powdery Mildew Strains Gives Rise to Pathogens on Novel Agricultural Crop Species. Nat. Genet. 2016, 48, 201–205. [Google Scholar] [CrossRef]
- Müller, M.C.; Praz, C.R.; Sotiropoulos, A.G.; Menardo, F.; Kunz, L.; Schudel, S.; Oberhänsli, S.; Poretti, M.; Wehrli, A.; Bourras, S.; et al. A Chromosome-Scale Genome Assembly Reveals a Highly Dynamic Effector Repertoire of Wheat Powdery Mildew. N. Phytol. 2019, 221, 2176–2189. [Google Scholar] [CrossRef]
- Kim, S.; Subramaniyam, S.; Jung, M.; Oh, E.A.; Kim, T.H.; Kim, J.G. Genome Resource of Podosphaera xanthii, the Host-Specific Fungal Pathogen That Causes Cucurbit Powdery Mildew. Mol. Plant-Microbe Interact. MPMI 2021, 34, 457–459. [Google Scholar] [CrossRef]
- Kusch, S.; Vaghefi, N.; Takamatsu, S.; Liu, S.Y.; Németh, M.Z.; Seress, D.; Frantzeskakis, L.; Chiu, P.E.; Panstruga, R.; Kiss, L. First Draft Genome Assemblies of Pleochaeta shiraiana and Phyllactinia moricola, Two Tree-Parasitic Powdery Mildew Fungi with Hemiendophytic Mycelia. Phytopathology 2022, 112, 961–967. [Google Scholar] [CrossRef]
- Szunics, L.; Szunics, L.; Vida, G.; Bedö, Z.; Svec, M. Dynamics of Changes in the Races and Virulence of Wheat Powdery Mildew in Hungary between 1971 and 1999. Euphytica 2001, 119, 143–147. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Parks, R.; Carbone, I.; Murphy, J.P.; Cowger, C. Population Genetic Analysis of an Eastern U.S. Wheat Powdery Mildew Population Reveals Geographic Subdivision and Recent Common Ancestry with U.K. and Israeli Populations. Phytopathology 2009, 99, 840–849. [Google Scholar] [CrossRef]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid Adapter Trimming, Identification, and Read Merging. BMC Res. Notes 2016, 9, 1–7. [Google Scholar] [CrossRef]
- Zimin, A.V.; Marçais, G.; Puiu, D.; Roberts, M.; Salzberg, S.L.; Yorke, J.A. The MaSuRCA Genome Assembler. Bioinformatics 2013, 29, 2669–2677. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A Web Server for Gene Prediction in Eukaryotes That Allows User-Defined Constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef] [PubMed]
- Aramaki, T.; Blanc-Mathieu, R.; Endo, H.; Ohkubo, K.; Kanehisa, M.; Goto, S.; Ogata, H. KofamKOALA: KEGG Ortholog Assignment Based on Profile HMM and Adaptive Score Threshold. Bioinformatics 2020, 36, 2251–2252. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. EggNOG 5.0: A Hierarchical, Functionally and Phylogenetically Annotated Orthology Resource Based on 5090 Organisms and 2502 Viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Urban, M.; Cuzick, A.; Seager, J.; Wood, V.; Rutherford, K.; Venkatesh, S.Y.; Sahu, J.; Vijaylakshmi Iyer, S.; Khamari, L.; De Silva, N.; et al. PHI-Base in 2022: A Multi-Species Phenotype Database for Pathogen–Host Interactions. Nucleic Acids Res. 2022, 50, D837–D847. [Google Scholar] [CrossRef]
- Buchfink, B.; Reuter, K.; Drost, H.G. Sensitive Protein Alignments at Tree-of-Life Scale Using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein Domains Identifier. Nucleic Acids Res. 2005, 33, W116. [Google Scholar] [CrossRef]
- Sperschneider, J.; Dodds, P.N. EffectorP 3.0: Prediction of Apoplastic and Cytoplasmic Effectors in Fungi and Oomycetes. Mol. Plant-Microbe Interact. 2022, 35, 146–156. [Google Scholar] [CrossRef]
- Afgan, E.; Nekrutenko, A.; Grüning, B.A.; Blankenberg, D.; Goecks, J.; Schatz, M.C.; Ostrovsky, A.E.; Mahmoud, A.; Lonie, A.J.; Syme, A.; et al. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2022 Update. Nucleic Acids Res. 2022, 50, W345–W351. [Google Scholar] [CrossRef]
- Kameshwar, A.K.S.; Ramos, L.P.; Qin, W. CAZymes-Based Ranking of Fungi (CBRF): An Interactive Web Database for Identifying Fungi with Extrinsic Plant Biomass Degrading Abilities. Bioresour. Bioprocess. 2019, 6, 51. [Google Scholar] [CrossRef]
- Sun, J.; Lu, F.; Luo, Y.; Bie, L.; Xu, L.; Wang, Y. OrthoVenn3: An Integrated Platform for Exploring and Visualizing Orthologous Data across Genomes. Nucleic Acids Res. 2023, 51, W397–W403. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential Gene and Transcript Expression Analysis of RNA-Seq Experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, 1–11. [Google Scholar] [CrossRef]
- Javed, M.; Reddy, B.; Sheoran, N.; Ganesan, P.; Kumar, A. Unraveling the Transcriptional Network Regulated by MiRNAs in Blast-Resistant and Blast-Susceptible Rice Genotypes during Magnaporthe oryzae Interaction. Gene 2023, 886, 147718. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M. Twelve Years of SAMtools and BCFtools. GigaScience 2021, 10, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, M.; Huang, X.; Zhang, G.; Zeng, L.; Zhang, G.; Wu, S.; Wang, Y. SRplot: A Free Online Platform for Data Visualization and Graphing. PLoS ONE 2023, 18, e0294236. [Google Scholar] [CrossRef] [PubMed]
- Glawe, D.A. The Powdery Mildews: A Review of the World’s Most Familiar (yet Poorly Known) Plant Pathogens. Annu. Rev. Phytopathol. 2008, 46, 27–51. [Google Scholar] [CrossRef]
- Zulak, K.G.; Cox, B.A.; Tucker, M.A.; Oliver, R.P.; Lopez-Ruiz, F.J. Improved Detection and Monitoring of Fungicide Resistance in Blumeria graminis f. sp. hordei with High-Throughput Genotype Quantification by Digital PCR. Front. Microbiol. 2018, 9, 706. [Google Scholar] [CrossRef]
- Meyers, E.; Arellano, C.; Cowger, C. Sensitivity of the U.S. Blumeria graminis f. sp. tritici Population to Demethylation Inhibitor Fungicides. Plant Dis. 2019, 103, 3108–3116. [Google Scholar] [CrossRef] [PubMed]
- Aylward, J.; Steenkamp, E.T.; Dreyer, L.L.; Roets, F.; Wingfield, B.D.; Wingfield, M.J. A Plant Pathology Perspective of Fungal Genome Sequencing. IMA Fungus 2017, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.; Kumar, A.; Mehta, S.; Sheoran, N.; Chinnusamy, V.; Prakash, G. Hybrid de Novo Genome-Reassembly Reveals New Insights on Pathways and Pathogenicity Determinants in Rice Blast Pathogen Magnaporthe oryzae RMg_Dl. Sci. Rep. 2021, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Cruz, J.; Romero, D.; De La Torre, F.N.; Fernández-Ortun, D.; Torés, J.A.; De Vicente, A.; Pérez-García, A. The Functional Characterization of Podosphaera xanthii Candidate Effector Genes Reveals Novel Target Functions for Fungal Pathogenicity. Mol. Plant-Microbe Interact. 2018, 31, 914–931. [Google Scholar] [CrossRef] [PubMed]
- Menardo, F.; Praz, C.R.; Wicker, T.; Keller, B. Rapid Turnover of Effectors in Grass Powdery Mildew (Blumeria graminis). BMC Evol. Biol. 2017, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.L.; Sawbridge, T.; Mann, R.; Kaur, J.; May, T.W.; Edwards, J. Rediscovering an Old Foe: Optimised Molecular Methods for DNA Extraction and Sequencing Applications for Fungarium Specimens of Powdery Mildew (Erysiphales). PLoS ONE 2020, 15, e0232535. [Google Scholar] [CrossRef]
- Brown, J.K.M.; Wolfe, M.S. Structure and Evolution of a Population of Erysiphe graminis f.sp. hordei. Plant Pathol. 1990, 39, 376–390. [Google Scholar] [CrossRef]
- Stewart, C.N.; Via, L.E. A Rapid CTAB DNA Isolation Technique Useful for RAPD Fingerprinting and Other PCR Applications. Biotechniques 1993, 14, 748–750. [Google Scholar] [PubMed]
- Inuma, T.; Khodaparast, S.A.; Takamatsu, S. Multilocus Phylogenetic Analyses within Blumeria graminis, a Powdery Mildew Fungus of Cereals. Mol. Phylogenetics Evol. 2007, 44, 741–751. [Google Scholar] [CrossRef]
- Weiß, C.L.; Schuenemann, V.J.; Devos, J.; Shirsekar, G.; Reiter, E.; Gould, B.A.; Stinchcombe, J.R.; Krause, J.; Burbano, H.A. Temporal Patterns of Damage and Decay Kinetics of Dna Retrieved from Plant Herbarium Specimens. R. Soc. Open Sci. 2016, 3, 160239. [Google Scholar] [CrossRef]
- JiLiang, C.; XiaoLong, H.; AnDi, W.; ShuangQing, Z.; DongYi, H.; JinPing, L. An Efficient Extraction Method of Pathogenic Fungus DNA for PCR. Mycosystema 2011, 30, 147–149. [Google Scholar]
- Feehan, J.M.; Scheibel, K.E.; Bourras, S.; Underwood, W.; Keller, B.; Somerville, S.C. Purification of High Molecular Weight Genomic DNA from Powdery Mildew for Long-Read Sequencing. J. Vis. Exp. JoVE 2017, 2017, 55463. [Google Scholar] [CrossRef]
- Frantzeskakis, L.; Németh, M.Z.; Barsoum, M.; Kusch, S.; Kiss, L.; Takamatsu, S.; Panstrugaa, R. The Parauncinula polyspora Draft Genome Provides Insights into Patterns of Gene Erosion and Genome Expansion in Powdery Mildew Fungi. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Kusch, S.; Németh, M.Z.; Vaghefi, N.; Ibrahim, H.M.M.; Panstruga, R.; Kiss, L. A Short-Read Genome Assembly Resource for Leveillula taurica Causing Powdery Mildew Disease of Sweet Pepper (Capsicum Annuum). Mol. Plant-Microbe Interact. MPMI 2020, 33, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Basenko, E.Y.; Pulman, J.A.; Shanmugasundram, A.; Harb, O.S.; Crouch, K.; Starns, D.; Warrenfeltz, S.; Aurrecoechea, C.; Stoeckert, C.J.; Kissinger, J.C.; et al. FungiDB: An Integrated Bioinformatic Resource for Fungi and Oomycetes. J. Fungi 2018, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Ciufo, S.; Kannan, S.; Sharma, S.; Badretdin, A.; Clark, K.; Turner, S.; Brover, S.; Schoch, C.L.; Kimchi, A.; DiCuccio, M. Using Average Nucleotide Identity to Improve Taxonomic Assignments in Prokaryotic Genomes at the NCBI. Int. J. Syst. Evol. Microbiol. 2018, 68, 2386. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Raffaele, S.; Kamoun, S. The Two-Speed Genomes of Filamentous Pathogens: Waltz with Plants. Curr. Opin. Genet. Dev. 2015, 35, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Möller, M.; Stukenbrock, E.H. Evolution and Genome Architecture in Fungal Plant Pathogens. Nat. Rev. Microbiol. 2017, 15, 756–771. [Google Scholar] [CrossRef]
- Dutech, C.; Feau, N.; Lesur, I.; Ehrenmann, F.; Letellier, T.; Li, B.; Mouden, C.; Guichoux, E.; Desprez-Loustau, M.L.; Gross, A. An Easy and Robust Method for Isolation and Validation of Single-Nucleotide Polymorphic Markers from a First Erysiphe aAlphitoides Draft Genome. Mycol. Prog. 2020, 19, 615–628. [Google Scholar] [CrossRef]
- Spanu, P.D. Messages from Powdery Mildew DNA: How the Interplay with a Host Moulds Pathogen Genomes. J. Integr. Agric. 2014, 13, 233–236. [Google Scholar] [CrossRef]
- Ma, L.J.; Van Der Does, H.C.; Borkovich, K.A.; Coleman, J.J.; Daboussi, M.J.; Di Pietro, A.; Dufresne, M.; Freitag, M.; Grabherr, M.; Henrissat, B.; et al. Comparative Genomics Reveals Mobile Pathogenicity Chromosomes in Fusarium. Nature 2010, 464, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Castanera, R.; López-Varas, L.; Borgognone, A.; LaButti, K.; Lapidus, A.; Schmutz, J.; Grimwood, J.; Pérez, G.; Pisabarro, A.G.; Grigoriev, I.V.; et al. Transposable Elements versus the Fungal Genome: Impact on Whole-Genome Architecture and Transcriptional Profiles. PLoS Genet. 2016, 12, e1006108. [Google Scholar] [CrossRef] [PubMed]
- Bindschedler, L.V.; Burgis, T.A.; Mills, D.J.S.; Ho, J.T.C.; Cramer, R.; Spanu, P.D. In Planta Proteomics and Proteogenomics of the Biotrophic Barley Fungal Pathogen Blumeria graminis f. sp. hordei. Mol. Cell. Proteomics MCP 2009, 8, 2368–2381. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Lefert, P.; Panstruga, R. A Molecular Evolutionary Concept Connecting Nonhost Resistance, Pathogen Host Range, and Pathogen Speciation. Trends Plant Sci. 2011, 16, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Praz, C.R.; Menardo, F.; Robinson, M.D.; Müller, M.C.; Wicker, T.; Bourras, S.; Keller, B. Non-Parent of Origin Expression of Numerous Effector Genes Indicates a Role of Gene Regulation in Host Adaption of the Hybrid Triticale Powdery Mildew Pathogen. Front. Plant Sci. 2018, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Nowara, D.; Schweizer, P.; Gay, A.; Lacomme, C.; Shaw, J.; Ridout, C.; Douchkov, D.; Hensel, G.; Kumlehn, J. HIGS: Host-Induced Gene Silencing in the Obligate Biotrophic Fungal Pathogen Blumeria graminis. Plant Cell 2010, 22, 3130–3141. [Google Scholar] [CrossRef] [PubMed]
- Ridout, C.J.; Skamnioti, P.; Porritt, O.; Sacristan, S.; Jones, J.D.G.; Brown, J.K.M. Multiple Avirulence Paralogues in Cereal Powdery Mildew Fungi May Contribute to Parasite Fitness and Defeat of Plant Resistance. Plant Cell 2006, 18, 2402–2414. [Google Scholar] [CrossRef] [PubMed]
- Sacristán, S.; Vigouroux, M.; Pedersen, C.; Skamnioti, P.; Thordal-Christensen, H.; Micali, C.; Brown, J.K.M.; Ridout, C.J. Coevolution between a Family of Parasite Virulence Effectors and a Class of LINE-1 Retrotransposons. PLoS ONE 2009, 4, e7463. [Google Scholar] [CrossRef]
- Shen, Q.H.; Saijo, Y.; Mauch, S.; Biskup, C.; Bieri, S.; Keller, B.; Seki, H.; Ülker, B.; Somssich, I.E.; Schulze-Lefert, P. Nuclear Activity of MLA Immune Receptors Links Isolate-Specific and Basal Disease-Resistance Responses. Science 2007, 315, 1098–1103. [Google Scholar] [CrossRef]
- Pedersen, C.; van Themaat, E.V.L.; McGuffin, L.J.; Abbott, J.C.; Burgis, T.A.; Barton, G.; Bindschedler, L.V.; Lu, X.; Maekawa, T.; Weßling, R.; et al. Structure and Evolution of Barley Powdery Mildew Effector Candidates. BMC Genom. 2012, 13, 1–21. [Google Scholar] [CrossRef]
- Urban, M.; Pant, R.; Raghunath, A.; Irvine, A.G.; Pedro, H.; Hammond-Kosack, K.E. The Pathogen-Host Interactions Database (PHI-Base): Additions and Future Developments. Nucleic Acids Res. 2015, 43, D645. [Google Scholar] [CrossRef]
- Wright, D.P.; Baldwin, B.C.; Shephard, M.C.; Scholes, J.D. Source-Sink Relationships in Wheat Leaves Infected with Powdery Mildew. I. Alterations in Carbohydrate Metabolism. Physiol. Mol. Plant Pathol. 1995, 47, 237–253. [Google Scholar] [CrossRef]
- Huang, Q.; Li, X.; Chen, W.Q.; Xiang, Z.P.; Zhong, S.F.; Chang, Z.J.; Zhang, M.; Zhang, H.Y.; Tan, F.Q.; Ren, Z.L. Genetic Mapping of a Putative Thinopyrum intermedium-Derived Stripe Rust Resistance Gene on Wheat Chromosome 1B. TAG. Theor. Appl. Genet. Theor. Und Angew. Genet. 2014, 127, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Yike, I. Fungal Proteases and Their Pathophysiological Effects. Mycopathologia 2011, 171, 299–323. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y. Proteases in Pathogenesis and Plant Defence. Cell. Microbiol. 2004, 6, 905–913. [Google Scholar] [CrossRef]
- Reddy, B.; Mehta, S.; Prakash, G.; Sheoran, N.; Kumar, A. Structured Framework and Genome Analysis of Magnaporthe grisea Inciting Pearl Millet Blast Disease Reveals Versatile Metabolic Pathways, Protein Families, and Virulence Factors. J. Fungi 2022, 8, 614. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Zhang, S.; Zhou, X.; Wang, C.; Xiang, P.; Zheng, Q.; Xu, J.R. The FgHOG1 Pathway Regulates Hyphal Growth, Stress Responses, and Plant Infection in Fusarium graminearum. PLoS ONE 2012, 7, e49495. [Google Scholar] [CrossRef]
- Both, M.; Csukai, M.; Stumpf, M.P.H.; Spanu, P.D. Gene Expression Profiles of Blumeria graminis Indicate Dynamic Changes to Primary Metabolism during Development of an Obligate Biotrophic Pathogen. Plant Cell 2005, 17, 2107. [Google Scholar] [CrossRef]
- Li, Y.; Yan, X.; Wang, H.; Liang, S.; Ma, W.B.; Fang, M.Y.; Talbot, N.J.; Wang, Z.Y. MoRic8 Is a Novel Component of G-Protein Signaling during Plant Infection by the Rice Blast Fungus Magnaporthe oryzae. Mol. Plant-Microbe Interact. MPMI 2010, 23, 317–331. [Google Scholar] [CrossRef]
- Lee, Y.-H.; Dean’, R.A. CAMP Regulates Infection Structure Formation in the Plant Pathogenic Fungus Magnaporthe grisea. Plant Cell 1993, 5, 693–700. [Google Scholar] [CrossRef]
- Adachi, K.; Hamer, J.E. Divergent CAMP Signaling Pathways Regulate Growth and Pathogenesis in the Rice Blast Fungus Magnaporthe grisea. Plant Cell 1998, 10, 1361–1373. [Google Scholar] [CrossRef] [PubMed]
- Waugh, M.S.; Nichols, C.B.; DeCesare, C.M.; Cox, G.M.; Heitman, J.; Alspaugh, J.A. Ras1 and Ras2 Contribute Shared and Unique Roles in Physiology and Virulence of Cryptococcus neoformans. Microbiology 2002, 148, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Pentland, D.R.; Piper-Brown, E.; Mühlschlegel, F.A.; Gourlay, C.W. Ras Signalling in Pathogenic Yeasts. Microb. Cell 2018, 5, 63. [Google Scholar] [CrossRef]
- Wolanin, P.M.; Thomason, P.A.; Stock, J.B. Histidine Protein Kinases: Key Signal Transducers Outside the Animal Kingdom. Genome Biol. 2002, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.G.; Lee, D.; Studer, R.A.; Dawson, N.L.; Sillitoe, I.; Das, S.; Yeats, C.; Dessailly, B.H.; Rentzsch, R.; Orengo, C.A. Gene3D: Multi-Domain Annotations for Protein Sequence and Comparative Genome Analysis. Nucleic Acids Res. 2014, 42, D240–D245. [Google Scholar] [CrossRef] [PubMed]
- Bornberg-Bauer, E.; Albà, M.M. Dynamics and Adaptive Benefits of Modular Protein Evolution. Curr. Opin. Struct. Biol. 2013, 23, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Stergiopoulos, I.; Zwiers, L.H.; De Waard, M.A. Secretion of Natural and Synthetic Toxic Compounds from Filamentous Fungi by Membrane Transporters of the ATP-Binding Cassette and Major Facilitator Superfamily. Eur. J. Plant Pathol. 2002, 108, 719–734. [Google Scholar] [CrossRef]
- Lee, J.; Iwata, Y.; Suzuki, Y.; Suzuki, I. Rapid Phosphate Uptake via an ABC Transporter Induced by Sulfate Deficiency in Synechocystis Sp. PCC 6803. Algal Res. 2021, 60, 102530. [Google Scholar] [CrossRef]
- Komínková, E.; Dreiseitl, A.; Maleèková, E.; Doležel, J.; Valárik, M. Genetic Diversity of Blumeria graminis f. sp. hordei in Central Europe and Its Comparison with Australian Population. PLoS ONE 2016, 11, e0167099. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Attributes | B. graminis f. sp. tritici Wtn1 |
|---|---|
| Illumina HiSeq 2500 Raw (PE) | 92,496,414 (PE reads), 13.97 Gb |
| HiSeq 2500 Clean (PE) | 9,24,13,253 (PE reads), 13.34 Gb |
| Illumina Nanopore PromethION | 32,25,945 (Single end), 4.94 Gb |
| Scaffolds | 93 |
| Largest contig | 19,534,821 |
| Total length | 140,604,965 |
| GC (%) | 43.72 |
| N50 | 16,029,079 |
| N75 | 9,486,802 |
| L50 | 47 |
| L75 | 101 |
| No. of genes | 8480 |
| Proteins | 7707 |
| rRNA | 190 |
| 18S | 61 |
| 28S | 61 |
| 5S | 6 |
| 5.8S | 62 |
| tRNA | 583 |
| CAZymes | 156 |
| Repeat element bases | 102.14 Mb (73.8%) |
| #N’sper100 kbp | 0.07 |
| Complete BUSCOs (C) | 723 (95.4%) |
| Complete and single-copy BUSCOs (S) | 714 (94.2%) |
| Complete and duplicated BUSCOs (D) | 9 (1.2%) |
| Fragmented BUSCOs (F) | 12 (1.6%) |
| Missing BUSCOs (M) | 23 (3.0%) |
| Total BUSCOs | 758 (100%) |
| Repeat Elements | Number of Elements | Length Occupied (bp) | Percentage of Sequence |
|---|---|---|---|
| Retro-elements | 95,626 | 86,147,131 | 62.24 |
| SINEs: | 1118 | 106,274 | 0.08 |
| Penelope | 7 | 317 | 0.00 |
| LINEs | 34,624 | 42,918,846 | 31.01 |
| CRE/SLACS | 2 | 398 | 0.00 |
| L2/CR1/Rex | 0 | 0 | 0.00 |
| R1/LOA/Jockey | 9 | 685 | 0.00 |
| R2/R4/NeSL | 0 | 0 | 0.00 |
| RTE/Bov-B | 0 | 0 | 0.00 |
| L1/CIN4 | 0 | 0 | 0.00 |
| LTR elements: | 59,884 | 43,122,011 | 31.16 |
| BEL/Pao | 0 | 0 | 0.00 |
| Ty1/Copia | 26,614 | 19,968,861 | 14.43 |
| Gypsy/DIRS1 | 26,883 | 21,158,915 | 15.29 |
| Retroviral | 0 | 0 | 0.00 |
| DNA transposons | 2358 | 1,136,913 | 0.82 |
| hobo-Activator | 0 | 0 | 0.00 |
| Tc1-IS630-Pogo | 2334 | 1,135,200 | 0.82 |
| En-Spm | 0 | 0 | 0.00 |
| MuDR-IS905 | 0 | 0 | 0.00 |
| PiggyBac | 0 | 0 | 0.00 |
| Tourist/Harbinger | 7 | 414 | 0.00 |
| Other (Mirage) | 2 | 51 | 0.00 |
| P-element, transit | |||
| Rolling-circle | 0 | 0 | 0.00 |
| Unclassified | 29,224 | 14,859,654 bp | 10.74 |
| Small RNA | 150 | 305,896 | 0.22 |
| Satellites | 284 | 70,281 | 0.05 |
| Simple repeats | 1183 | 27,3126 | 0.20 |
| Low complexity | 80 | 12,044 | 0.01 |
| Total interspersed repeats: 102,143,698 bp | 73.80 | ||
| Category | B graminis f. sp. tritici 96224 v3.16 | B graminis f. sp. triticale THUN-12 | B graminis f. sp. tritici Wtn1 |
|---|---|---|---|
| AA | 9 | 13 | 13 |
| CBM | 4 | 4 | 4 |
| CE | 11 | 10 | 11 |
| GH | 63 | 72 | 70 |
| GT | 56 | 58 | 58 |
| Total | 143 | 157 | 156 |
| Carbohydrate Metabolism Pathways | Gene Count |
|---|---|
| 00010 Glycolysis/Gluconeogenesis | 21 |
| 00020 Citrate cycle (TCA cycle) | 19 |
| 00030 Pentose phosphate pathway | 15 |
| 00040 Pentose and glucuronate interconversions | 8 |
| 00051 Fructose and mannose metabolism | 13 |
| 00052 Galactose metabolism | 8 |
| 00053 Ascorbate and aldarate metabolism | 1 |
| 00500 Starch and sucrose metabolism | 18 |
| 00520 Amino sugar and nucleotide sugar metabolism | 18 |
| 00620 Pyruvate metabolism | 22 |
| 00630 Glyoxylate and dicarboxylate metabolism | 17 |
| 00640 Propanoate metabolism | 16 |
| 00650 Butanoate metabolism | 13 |
| 00660 C5-Branched dibasic acid metabolism | 6 |
| 00562 Inositol phosphate metabolism | 18 |
| Terpenoid and Polyketide Metabolism Pathways | Gene Count |
|---|---|
| 00900 Terpenoid backbone biosynthesis | 16 |
| 00909 Sesquiterpenoid and triterpenoid biosynthesis | 2 |
| 00906 Carotenoid biosynthesis | 1 |
| 00981 Insect hormone biosynthesis | 1 |
| 00908 Zeatin biosynthesis | 1 |
| 00903 Limonene degradation | 2 |
| 00907 Pinene, camphor, and geraniol degradation | 2 |
| 01051 Biosynthesis of ansamycins | 1 |
| 00523 Polyketide sugar unit biosynthesis | 1 |
| 01055 Biosynthesis of vancomycin group antibiotics | 1 |
| Signal Transduction Pathways | Gene Count |
|---|---|
| 02020 Two-component system | 15 |
| 04010 MAPK signaling pathway | 14 |
| 04013 MAPK signaling pathway—fly | 11 |
| 04016 MAPK signaling pathway—plant | 4 |
| 04011 MAPK signaling pathway—yeast | 53 |
| 04012 ErbB signaling pathway | 4 |
| 04014 Ras signaling pathway | 15 |
| 04015 Rap1 signaling pathway | 8 |
| 04310 Wnt signaling pathway | 12 |
| 04330 Notch signaling pathway | 3 |
| 04340 Hedgehog signaling pathway | 4 |
| 04341 Hedgehog signaling pathway—fly | 6 |
| 04350 TGF-beta signaling pathway | 9 |
| 04390 Hippo signaling pathway | 8 |
| 04391 Hippo signaling pathway—fly | 6 |
| 04392 Hippo signaling pathway—multiple species | 4 |
| 04370 VEGF signaling pathway | 9 |
| 04371 Apelin signaling pathway | 13 |
| 04630 JAK-STAT signaling pathway | 3 |
| 04064 NF-kappa B signaling pathway | 4 |
| 04668 TNF signaling pathway | 3 |
| 04066 HIF-1 signaling pathway | 13 |
| 04068 FoxO signaling pathway | 16 |
| 04020 Calcium signaling pathway | 9 |
| 04070 Phosphatidylinositol signaling system | 16 |
| 04072 Phospholipase D signaling pathway | 14 |
| 04071 Sphingolipid signaling pathway | 17 |
| 04024 cAMP signaling pathway | 9 |
| 04022 cGMP-PKG signaling pathway | 9 |
| 04151 PI3K-Akt signaling pathway | 23 |
| 04152 AMPK signaling pathway | 21 |
| 04150 mTOR signaling pathway | 38 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nallathambi, P.; Umamaheswari, C.; Reddy, B.; Aarthy, B.; Javed, M.; Ravikumar, P.; Watpade, S.; Kashyap, P.L.; Boopalakrishnan, G.; Kumar, S.; et al. Deciphering the Genomic Landscape and Virulence Mechanisms of the Wheat Powdery Mildew Pathogen Blumeria graminis f. sp. tritici Wtn1: Insights from Integrated Genome Assembly and Conidial Transcriptomics. J. Fungi 2024, 10, 267. https://doi.org/10.3390/jof10040267
Nallathambi P, Umamaheswari C, Reddy B, Aarthy B, Javed M, Ravikumar P, Watpade S, Kashyap PL, Boopalakrishnan G, Kumar S, et al. Deciphering the Genomic Landscape and Virulence Mechanisms of the Wheat Powdery Mildew Pathogen Blumeria graminis f. sp. tritici Wtn1: Insights from Integrated Genome Assembly and Conidial Transcriptomics. Journal of Fungi. 2024; 10(4):267. https://doi.org/10.3390/jof10040267
Chicago/Turabian StyleNallathambi, Perumal, Chandrasekaran Umamaheswari, Bhaskar Reddy, Balakrishnan Aarthy, Mohammed Javed, Priya Ravikumar, Santosh Watpade, Prem Lal Kashyap, Govindaraju Boopalakrishnan, Sudheer Kumar, and et al. 2024. "Deciphering the Genomic Landscape and Virulence Mechanisms of the Wheat Powdery Mildew Pathogen Blumeria graminis f. sp. tritici Wtn1: Insights from Integrated Genome Assembly and Conidial Transcriptomics" Journal of Fungi 10, no. 4: 267. https://doi.org/10.3390/jof10040267
APA StyleNallathambi, P., Umamaheswari, C., Reddy, B., Aarthy, B., Javed, M., Ravikumar, P., Watpade, S., Kashyap, P. L., Boopalakrishnan, G., Kumar, S., Sharma, A., & Kumar, A. (2024). Deciphering the Genomic Landscape and Virulence Mechanisms of the Wheat Powdery Mildew Pathogen Blumeria graminis f. sp. tritici Wtn1: Insights from Integrated Genome Assembly and Conidial Transcriptomics. Journal of Fungi, 10(4), 267. https://doi.org/10.3390/jof10040267