Fungal Dysbiosis Correlates with the Development of Tumor-Induced Cachexia in Mice

 , , ,
, , ,
Abstract
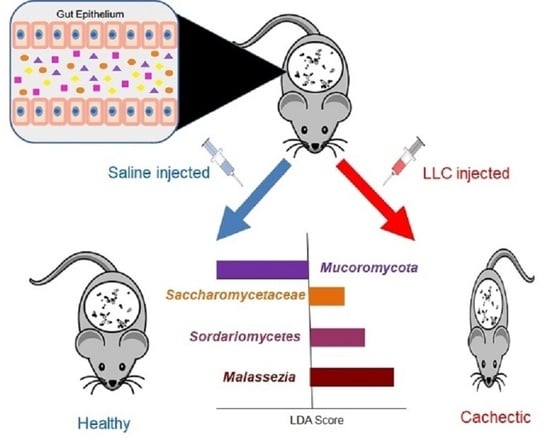
:
1. Introduction
2. Materials and Methods
2.1. Cachexia Induction in C57BL/6 Mice and Collection of Stool Samples for Characterization of the Gut Mycobiota during the Development of Cachexia
2.2. Assessment of Cachexia Progression
2.3. Sample Processing, Sequencing, and Analysis of ITS1 Amplicon Libraries
2.4. Fungal Population and Diversity Analysis of the Gut Mycobiota from SC and CC Mice
2.5. Identification of Differentially Represented Taxa in SC and CC Mice, Using Linear Discriminant Analysis Effect Size (LEfSe)
2.6. Quantitative Real-Time PCR (qPCR)
3. Results
3.1. Cachexia Induction and Characterization in Mice
3.2. Characterization and Comparison of Fungal Gut Populations (Mycobiota) in SC and CC Animals
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animals | Total of Amplicons Sequenced |
|---|---|
| SC1 | 109,236 |
| SC2 | 66,436 |
| SC3 | 120,142 |
| SC4 | 135,404 |
| SC5 | 163,377 |
| SC6 | 144,145 |
| SC7 | 133,910 |
| SC8 | 153,817 |
| CC1 | 155,586 |
| CC2 | 141,331 |
| CC3 | 121,782 |
| CC4 | 124,370 |
| CC5 | 104,937 |
| CC6 | 137,693 |
| CC7 | 130,312 |
| CC8 | 120,530 |
| Total | 1,036,541 |
References
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Bruera, E.; Hui, D. Conceptual models for integrating palliative care at cancer centers. J. Palliat. Med. 2012, 15, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Bindels, L.B.; Beck, R.; Schakman, O.; Martin, J.C.; De Backer, F.; Sohet, F.M.; Dewulf, E.M.; Pachikian, B.D.; Neyrinck, A.M.; Thissen, J.P.; et al. Restoring Specific Lactobacilli Levels Decreases Inflammation and Muscle Atrophy Markers in an Acute Leukemia Mouse Model. PLoS ONE 2012, 7, e37971. [Google Scholar] [CrossRef] [Green Version]
- Bindels, L.B.; Neyrinck, A.M.; Claus, S.P.; Le Roy, C.I.; Grangette, C.; Pot, B.; Martinez, I.; Walter, J.; Cani, P.D.; Delzenne, N.M. Synbiotic approach restores intestinal homeostasis and prolongs survival in leukaemic mice with cachexia. ISME J. 2016, 10, 1456–1470. [Google Scholar] [CrossRef] [PubMed]
- Tözün, N.; Vardareli, E. Gut Microbiome and Gastrointestinal Cancer: Les liaisons Dangereuses. J. Clin. Gastroenterol. 2016, 50 (Suppl. S2), S191–S196. [Google Scholar] [CrossRef]
- Qin, Q.; Xu, Y.; He, T.; Qin, C.; Xu, J. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 2012, 22, 90–106. [Google Scholar] [CrossRef] [Green Version]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buttó, L.F.; Haller, D. Functional relevance of microbiome signatures: The correlation era requires tools for consolidation. J. Allergy Clin. Immunol. 2017, 139, 1092–1098. [Google Scholar] [CrossRef] [Green Version]
- Pötgens, S.A.; Brossel, H.; Sboarina, M.; Catry, E.; Cani, P.D.; Neyrinck, A.M.; Delzenne, N.M.; Bindels, L.B. Klebsiella oxytoca expands in cancer cachexia and acts as a gut pathobiont contributing to intestinal dysfunction. Sci. Rep. 2018, 17, 12321. [Google Scholar] [CrossRef] [Green Version]
- Bennani-Baiti, N.; Walsh, D. Animal models of the cancer anorexia–cachexia syndrome. Support Care Cancer 2011, 19, 1451–1463. [Google Scholar] [CrossRef]
- Deboer, M.D. Animal models of anorexia and cachexia. Expert Opin Drug Discov. 2009, 4, 1145–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macpherson, A.J.; McCoy, K. Standardised animal models of host microbial mutualism. Mucosal Immunol. 2015, 8, 476–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, C.L.; Ericsson, A.C. Microbiota and reproducibility of rodent models. Lab. animal 2017, 46, 114. [Google Scholar] [CrossRef] [PubMed]
- Henriques, F.S.; Sertié, R.A.L.R.; Franco, F.O.; Knobl, P.; Neves, R.X.; Andreotti, S.; Lima, F.B.; Guilherme, A.; Seelaender, M.; Batista, M.L., Jr. Early suppression of adipocyte lipid turnover induces immunometabolic modulation in cancer cachexia syndrome. FASEB J. 2017, 31, 1976–1986. [Google Scholar] [CrossRef] [Green Version]
- Voltarelli, F.A.; Frajacomo, F.T.; de Souza Padilha, C.; Testa, M.T.J.; Cella, P.S.; Ribeiro, D.F.; de Oliveira, D.X.; Veronez, L.C.; Bisson, G.S.; Moura, F.A.; et al. Syngeneic B16F10 Melanoma Causes Cachexia and Impaired Skeletal Muscle Strength and Locomotor Activity in Mice. Front. Physiol. 2017, 8, 715. [Google Scholar] [CrossRef] [Green Version]
- Batista, M.L., Jr.; Henriques, F.S.; Neves, R.X.; Olivan, M.R.; Matos-Neto, E.M.; Alcântara, P.S.; Maximiano, L.F.; Otoch, J.P.; Alves, M.J.; Seelaender, M. Cachexia-associated adipose tissue morphological rearrangement in gastrointestinal cancer patients. J. Cachexia Sarcopenia Muscle 2016, 7, 37–47. [Google Scholar] [CrossRef]
- Guo, K.; Mogen, J.; Struzzi, S.; Zhang, Y. Preadipocyte transplantation: An in vivo study of direct leptin signaling on adipocyte morphogenesis and cell size. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, 1339–1347. [Google Scholar] [CrossRef]
- Rose-John, S. IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef]
- Kemik, O.; Sumer, A.; Kemik, A.S.; Hasirci, I.; Purisa, S.; Dulger, A.C.; Demiriz, B.; Tuzun, S. The relationship among acute-phase response proteins, cytokines and hormones in cachectic patients with colon cancer. World J. Surg. Oncol. 2010, 8, 85. [Google Scholar] [CrossRef] [Green Version]
- Batista, M.L., Jr.; Olivan, M.; Alcantara, P.S.M.; Sandoval, R.; Peres, S.B.; Neves, R.X.; Silverio, R.; Maximiano, L.F.; Otoch, J.P.; Seelaender, M. Adipose tissue-derived factors as potential biomarkers in cachectic cancer patients. Cytokine 2013, 61, 532–539. [Google Scholar] [CrossRef] [Green Version]
- Mallick, H.; Ma, S.; Franzosa, E.A.; Vatanen, T.; Morgan, X.C.; Huttenhower, C. Experimental design and quantitative analysis of microbial community multiomics. Genome Biol. 2017, 18, 228. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 799–821. [Google Scholar] [CrossRef] [PubMed]
- Coker, O.O.; Nakatsu, G.; Dai, R.Z.; Wu, W.K.K.; Wong, S.H.; Ng, S.C.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. Enteric fungal microbiota dysbiosis and ecological alterations in colorectal cancer. Gut 2019, 68, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.J.; Lewis, K.E.; Huws, S.A.; Lin, W.; Hegarty, M.J.; Lewis, P.D.; Mur, L.A.; Pachebat, J.A. Metagenomic Sequencing of the Chronic Obstructive Pulmonary Disease Upper Bronchial Tract Microbiome Reveals Functional Changes Associated with Disease Severity. PLoS ONE 2016, 11, e0149095. [Google Scholar] [CrossRef] [Green Version]
- Astudillo-García, C.; Bell, J.J.; Webster, N.S.; Glasl, B.; Jompa, J.; Montoya, J.M.; Taylor, M.W. Evaluating the core microbiota in complex communities: A systematic investigation. Environ. Microbiol. 2017, 19, 1450–1462. [Google Scholar] [CrossRef]
- Flemer, B.; Gaci, N.; Borrel, G.; Sanderson, I.R.; Chaudhary, P.P.; Tottey, W.; O’Toole, P.W.; Brugère, J.F. Fecal microbiota variation across the lifespan of the healthy laboratory rat. Gut Microbes 2017, 8, 428–439. [Google Scholar] [CrossRef]
- Björk, J.R.; O’Hara, R.B.; Ribes, M.; Coma, R.; Montoya, J.M. The dynamic core microbiome: Structure, dynamics and stability. bioRxiv 2018, 137885. [Google Scholar] [CrossRef]
- Toju, H.; Peay, K.G.; Yamamichi, M.; Narisawa, K.; Hiruma, K.; Naito, K.; Fukuda, S.; Ushio, M.; Nakaoka, S.; Onoda, Y.; et al. Core microbiomes for sustainable agroecosystems. Nat. Plants 2018, 4, 247–257. [Google Scholar] [CrossRef]
- Russel, J.; Roesch, L.; Atkinson, M.A.; Schatz, D.; Triplett, E.W.; Ludvigsson, J. Genetic Risk for Type 1 Diabetes Profoundly Influences the Core Gut Microbiome in Children. Diabetes 2018, 67, 209. [Google Scholar] [CrossRef]
- Piampiano, E.; Pini, F.; Biondi, N.; Pastorelli, R.; Giovannetti, L.; Viti, C. Analysis of microbiota in cultures of the green microalga Tetraselmis suecica. Eur. J. Phycol. 2019, 54, 497–508. [Google Scholar] [CrossRef]
- Suenami, S.; Konishi Nobu, M.; Miyazaki, R. Community analysis of gut microbiota in hornets, the largest eusocial wasps, Vespa mandarinia and V. simillima. Sci. Rep. 2019, 9, 9830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, R.J.; Sasson, G.; Garnsworthy, P.C.; Tapio, I.; Gregson, E.; Bani, P.; Huhtanen, P.; Bayat, A.R.; Strozzi, F.; Biscarini, F. A heritable subset of the core rumen microbiome dictates dairy cow productivity and emissions. Sci. Adv. 2019, 5, eaav8391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clos-Garcia, M.; Andrés-Marin, N.; Fernández-Eulate, G.; Abecia, L.; Lavín, J.L.; van Liempd, S.; Cabrera, D.; Royo, F.; Valero, A.; Errazquin, N.; et al. Gut microbiome and serum metabolome analyses identify molecular biomarkers and altered glutamate metabolism in fibromyalgia. EBioMedicine 2019, 46, 499–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugita, T.; Takashima, M.; Kodama, M.; Tsuboi, R.; Nishikawa, A. Description of a New Yeast Species, Malassezia japonica, and Its Detection in Patients with Atopic Dermatitis and Healthy Subjects. J. Clin. Microbiol. 2003, 41, 4695–4699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagao, K.; Ota, T.; Tanikawa, A.; Takae, Y.; Mori, T.; Udagawa, S.; Nishikawa, T. Genetic identification and detection of human pathogenic Rhizopus species, a major mucormycosis agent, by multiplex PCR based on internal transcribed spacer region of rRNA gene. J. Dermatol. Sci. 2005, 39, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.W.; Nam, Y.D.; Sung, Y.; Kim, K.H.; Roh, S.W.; Yoon, J.H.; An, K.G.; Bae, J.W. Quantitative real time PCR assays for the enumeration of Saccharomyces cerevisiae and the Saccharomyces sensu stricto complex in human feces. J. Microbiol. Methods 2007, 71, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Hierro, N.; Esteve-Zarzoso, B.; Mas, A.; Guillamón, J.M. Monitoring of Saccharomyces and Hanseniaspora populations during alcoholic fermentation by real-time quantitative PCR. FEMS Yeast Res. 2007, 7, 1340–1349. [Google Scholar] [CrossRef] [Green Version]
- Vuran, E.; Karaarslan, A.; Karasartova, D.; Turegun, B.; Sahin, F. Identification of Malassezia species from pityriasis versicolor lesions with a new multiplex PCR method. Mycopathologia 2014, 177, 41–49. [Google Scholar] [CrossRef]
- Xie, Z.; Ran, Y.; Zhang, H.; Zhang, M.; Wan, H.; Li, C. An analysis of the Malassezia species distribution in the skin of patients with pityriasis versicolor in Chengdu, China. Sci. World J. 2014, 2014, 182596. [Google Scholar] [CrossRef]
- Urubschurov, V.; Busing, K.; Janczyk, P.; Souffrant, W.B.; Zeyner, A. Development and Evaluation of qPCR Assay for Quantitation of Kazachstania slooffiae and Total Yeasts Occurring in the Porcine Gut. Curr. Microbiol. 2015, 71, 373–381. [Google Scholar] [CrossRef]
- McKernan, K.; Spangler, J.; Zhang, L.; Tadigotla, V.; Helbert, Y.; Foss, T.; Smith, D. Cannabis microbiome sequencing reveals several mycotoxic fungi native to dispensary grade Cannabis flowers. F1000Research 2015, 4, 1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolatabadi, S.; de Hoog, G.S.; Meis, J.F.; Walther, G. Species boundaries and nomenclature of Rhizopus arrhizus (syn. R. oryzae). Mycoses 2014, 57, 108–127. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Liu, E.J.; Kheradman, R.; Fagan, A.; Heuman, D.M.; White, M.; Gavis, E.A.; Hylemon, P.; Sikaroodi, M.; Gillevet, P.M. Fungal dysbiosis in cirrhosis. Gut 2018, 67, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Mar Rodríguez, M.; Pérez, D.; Javier Chaves, F.; Esteve, E.; Marin-Garcia, P.; Xifra, G.; Vendrell, J.; Jové, M.; Pamplona, R.; Ricart, W.; et al. Obesity changes the human gut mycobiome. Sci. Rep. 2015, 5, 14600. [Google Scholar] [CrossRef]
- Walsh, A.M.; Sweeney, T.; Bahar, B.; O’Doherty, J.V. Multi-functional roles of chitosan as a potential protective agent against obesity. PLoS ONE 2013, 8, e53828. [Google Scholar] [CrossRef]
- Tasar, O.C.; Erdal, S.; Taskin, M. Chitosan production by psychrotolerant Rhizopus oryzae in non-sterile openfermentation conditions. Int. J. Biol. Macromol. 2016, 89, 428–433. [Google Scholar] [CrossRef]
- Londoño-Hernández, L.; Ramírez-Toro, C.; Ruiz, H.A.; Ascacio-Valdés, J.A.; Aguilar-Gonzalez, M.A.; Rodríguez-Herrera, R.; Aguilar, C.N. Rhizopus oryzae—Ancient microbial resource with importance in modern food industry. Int. J. Food Microbiol. 2017, 257, 110–127. [Google Scholar] [CrossRef]
- Leow, S.S.; Sekaran, S.D.; Sundram, K.; Tan, Y.; Sambanthamurthi, R. Gene expression changes in spleens and livers of tumour-bearing mice suggest delayed inflammation and attenuated cachexia in response to oil palm phenolics. J. Nutrigenet. Nutr. 2013, 6, 305–326. [Google Scholar] [CrossRef] [Green Version]
- Dludla, P.V.; Nkambule, B.B.; Jack, B.; Mkandla, Z.; Mutize, T.; Silvestri, S.; Orlando, P.; Tiano, L.; Louw, J.; Mazibuko-Mbeje, S.E. Inflammation and Oxidative Stress in an Obese State and the Protective Effects of Gallic Acid. Nutrients 2019, 11, 23. [Google Scholar] [CrossRef] [Green Version]
- Maresca, M.; Fantini, J. Some food-associated mycotoxins as potential risk factors in humans predisposed to chronic intestinal inflammatory diseases. Toxicon 2010, 56, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Liguori, G.; Lamas, B.; Richard, M.L.; Brandi, G.; da Costa, G.; Hoffmann, T.W.; Di Simone, M.P.; Calabrese, C.; Poggioli, G.; Langella, P.; et al. Fungal Dysbiosis in Mucosa-associated Microbiota of Crohn’s Disease Patients. J. Crohns Colitis 2016, 10, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandarano, A.H.; Giloteaux, L.; Keller, B.A.; Levine, S.M.; Hanson, M.R. Eukaryotes in the gut microbiota in myalgic encephalomyelitis/chronic fatigue syndrome. PeerJ 2018, 6, e4282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luan, C.; Xie, L.; Yang, X.; Miao, H.; Lv, N.; Zhang, R.; Xiao, X.; Hu, Y.; Liu, Y.; Wu, N.; et al. Dysbiosis of fungal microbiota in the intestinal mucosa of patients with colorectal adenomas. Sci. Rep. 2015, 5, 7980. [Google Scholar] [CrossRef] [PubMed]
- Antonissen, G.; Martel, A.; Pasmans, F.; Ducatelle, R.; Verbrugghe, E.; Vandenbroucke, V.; Li, S.; Haesebrouck, F.; Van Immerseel, F.; Croubels, S. The impact of Fusarium mycotoxins on human and animal host susceptibility to infectious diseases. Toxins 2014, 6, 430–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chikina, A.S.; Nadalin, F.; Maurin, M.; San-Roman, M.; Thomas-Bonafos, T.; Li, X.V.; Lameiras, S.; Baulande, S.; Henri, S.; Malissen, B.; et al. Macrophages Maintain Epithelium Integrity by Limiting Fungal Product Absorption. Cell 2020, 183, 411–428. [Google Scholar] [CrossRef]
- Klein, G.L.; Petschow, B.W.; Shaw, A.L.; Weaver, E. Gut barrier dysfunction and microbial translocation in cancer cachexia: A new therapeutic target. Curr. Opin. Support Palliat Care 2013, 7, 361–367. [Google Scholar] [CrossRef] [Green Version]
- Kurtzman, C.P.; Robnett, C.J.; Ward, J.M.; Brayton, C.; Gorelick, P.; Walsh, T.J. Multigene phylogenetic analysis of pathogenic candida species in the Kazachstania (Arxiozyma) telluris complex and description of their ascosporic states as Kazachstania bovina sp. nov., K. heterogenica sp. nov., K. pintolopesii sp. nov., and K. slooffiae sp. nov. J. Clin. Microbiol. 2005, 43, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Arfken, A.M.; Frey, J.F.; Ramsay, T.G.; Summers, K.L. Yeasts of Burden: Exploring the Mycobiome-Bacteriome of the Piglet GI Tract. Front. Microbiol. 2019, 10, 2286. [Google Scholar] [CrossRef]
- Jawhara, S.; Habib, K.; Maggiotto, F.G.; Pignede, G.; Vandekerckove, P.; Maes, E.; Dubuquoy, L.; Fontaine, T.; Guerardel, Y.; Poulain, D. Modulation of intestinal inflammation by yeasts and cell wall extracts: Strain dependence and unexpected anti-inflammatory role of glucan fractions. PLoS ONE 2012, 7, e40648. [Google Scholar] [CrossRef] [PubMed]
- Chiaro, T.R.; Soto, R.; Zac Stephens, W.; Kubinak, J.L.; Petersen, C.; Gogokhia, L.; Bell, R.; Delgado, J.C.; Cox, J.; Voth, W.; et al. A member of the gut mycobiota modulates host purine metabolism exacerbating colitis in mice. Sci. Transl. Med. 2017, 9, 380. [Google Scholar] [CrossRef] [PubMed]
- Lamprinaki, D.; Beasy, G.; Zhekova, A.; Wittmann, A.; James, S.; Dicks, J.; Iwakura, Y.; Saijo, S.; Wang, X.; Chow, C.W.; et al. LC3-Associated Phagocytosis Is Required for Dendritic Cell Inflammatory Cytokine Response to Gut Commensal Yeast Saccharomyces cerevisiae. Front. Immunol. 2017, 8, 1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsuyama, K.; Niwa, M.; Takedatsu, H.; Yamasaki, H.; Kuwaki, K.; Yoshioka, S.; Yamauchi, R.; Fukunaga, S.; Torimura, T. Antibody markers in the diagnosis of inflammatory bowel disease. World J. Gastroenterol. 2016, 22, 1304–1310. [Google Scholar] [CrossRef] [PubMed]
- Velegraki, A.; Cafarchia, C.; Gaitanis, G.; Iatta, R.; Boekhout, T. Malassezia Infections in Humans and Animals: Pathophysiology, Detection, and Treatment. PLoS Pathog. 2015, 11, e1004523. [Google Scholar] [CrossRef] [Green Version]
- Limon, J.J.; Tang, J.; Li, D.; Wolf, A.J.; Michelsen, K.S.; Funari, V.; Gargus, M.; Nguyen, C.; Sharma, P.; Maymi, V.; et al. Malassezia Is Associated with Crohn’s Disease and Exacerbates Colitis in Mouse Models. Cell Host Microbe 2019, 25, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Aykut, B.; Pushalkar, S.; Chen, R.; Li, Q.; Abengozar, R.; Kim, J.I.; Shadaloey, S.A.; Wu, D.; Preiss, P.; Verma, N.; et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 2019, 574, 264–267. [Google Scholar] [CrossRef]
- Van Asbeck, E.C.; Hoepelman, A.I.; Scharringa, J.; Herpers, B.L.; Verhoef, J. Mannose binding lectin plays a crucial role in innate immunity against yeast by enhanced complement activation and enhanced uptake of polymorphonuclear cells. BMC Microbiol. 2008, 8, 229. [Google Scholar] [CrossRef] [Green Version]
- Honkanen, J.; Vuorela, A.; Muthas, D.; Orivuori, L.; Luopajärvi, K.; Tejesvi, M.; Lavrinienko, A.; Pirttilä, A.M.; Fogarty, C.L.; Härkönen, T.; et al. Fungal Dysbiosis and Intestinal Inflammation in Children with Beta-Cell Autoimmunity. Front. Immunol. 2020, 11, 468. [Google Scholar] [CrossRef] [Green Version]
- Kostovcikova, K.; Coufal, S.; Galanova, N.; Fajstova, A.; Hudcovic, T.; Kostovcik, M.; Prochazkova, P.; Jiraskova Zakostelska, Z.; Cermakova, M.; Sediva, B.; et al. Diet Rich in Animal Protein Promotes Pro-inflammatory Macrophage Response and Exacerbates Colitis in Mice. Front. Immunol. 2019, 10, 919. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jabes, D.L.; de Maria, Y.N.L.F.; Aciole Barbosa, D.; Santos, K.B.N.H.; Carvalho, L.M.; Humberto, A.C.; Alencar, V.C.; Costa de Oliveira, R.; Batista, M.L., Jr.; Menegidio, F.B.; et al. Fungal Dysbiosis Correlates with the Development of Tumor-Induced Cachexia in Mice. J. Fungi 2020, 6, 364. https://doi.org/10.3390/jof6040364
Jabes DL, de Maria YNLF, Aciole Barbosa D, Santos KBNH, Carvalho LM, Humberto AC, Alencar VC, Costa de Oliveira R, Batista ML Jr., Menegidio FB, et al. Fungal Dysbiosis Correlates with the Development of Tumor-Induced Cachexia in Mice. Journal of Fungi. 2020; 6(4):364. https://doi.org/10.3390/jof6040364
Chicago/Turabian StyleJabes, Daniela L., Yara N. L. F. de Maria, David Aciole Barbosa, Kaltinaitis B. N. H. Santos, Lucas M. Carvalho, Ana Carolina Humberto, Valquíria C. Alencar, Regina Costa de Oliveira, Miguel L. Batista, Jr., Fabiano B. Menegidio, and et al. 2020. "Fungal Dysbiosis Correlates with the Development of Tumor-Induced Cachexia in Mice" Journal of Fungi 6, no. 4: 364. https://doi.org/10.3390/jof6040364
APA StyleJabes, D. L., de Maria, Y. N. L. F., Aciole Barbosa, D., Santos, K. B. N. H., Carvalho, L. M., Humberto, A. C., Alencar, V. C., Costa de Oliveira, R., Batista, M. L., Jr., Menegidio, F. B., & Nunes, L. R. (2020). Fungal Dysbiosis Correlates with the Development of Tumor-Induced Cachexia in Mice. Journal of Fungi, 6(4), 364. https://doi.org/10.3390/jof6040364