1. Introduction
Aspergillus fumigatus is a saprophytic fungus that can colonize diverse ecological niches facilitated by its great plasticity to adapt to different environments and the wide dispersal of its small, airborne spores called conidia [
1,
2]. The conidia allow
A. fumigatus to reach all chambers of the human respiratory tract, where the fungus is able to cause a wide range of infections depending on the immune status of the patient [
3,
4,
5,
6].
The biological characteristics that allow
A. fumigatus to colonize and/or infect the respiratory tract are known as virulence factors. These include the fungal cell wall and the proteins involved in its formation, the resistance of the conidia to ultraviolet radiation and dryness, the mechanisms to evade the immune response, and the production of proteases and toxins [
6]. In fact, the
A. fumigatus genome contains a great variety of biosynthetic clusters [
7,
8] that provide the fungus with complex and bioactive secondary metabolites, including the production of toxins [
9,
10,
11]. Among the variety of toxins produced by
A. fumigatus, several have been linked with enhanced virulence and include gliotoxin [
12,
13,
14,
15], fumitremorgin A and B [
16,
17], hexadehydroastechrome [
18], hemolysin, and mitogillin [
19].
The role of fumagillin in fungal virulence has not been completely elucidated yet. This toxin has been widely studied for other reasons, such as tumor control due to the inhibition of endothelial cell proliferation and its effect as an anti-tumor molecule [
20]. Furthermore, it has been used as an antibiotic agent in the treatment against different pathogens, such as microsporidia, parasites, and as a treatment of diseases such as AIDS and obesity [
21]. The mycotoxin is produced by a series of enzymes encoded in a biosynthetic cluster located on chromosome 8 of
A. fumigatus [
22]. The members of the biosynthetic cluster are overexpressed in the lungs of intranasally infected mice [
23] and include the fumagillin pathway-specific transcription factor [
24]. In fact, a non-fumagillin producer mutant strain caused significantly less cellular damage than the wild type in vitro, demonstrating a potential relevance of this toxin in virulence [
23].
Given these previous data and the need to deeply analyze the involvement of secondary metabolites in virulence [
25], the aim of the present study was to assess the role of fumagillin in the pathogenesis of
A. fumigatus. For this purpose, we studied the effect of commercial fumagillin on
A. fumigatus itself, macrophages, and epithelial cells. Additionally, a non-fumagillin producer mutant strain was used in co-incubation with cell cultures and during murine infection to determine the potential impact of the toxin on fungal virulence.
2. Materials and Methods
2.1. Aspergillus fumigatus Strains, Media, and Growth Conditions
The Δ
akuBku80 strain of
A. fumigatus was used as a wild-type (Wt) strain during this study. Furthermore, we used the previously published [
22] deletion mutant strain ∆
fmaA (a non-fumagillin producer strain) and its complemented strain ∆
fmaA::
fmaA [
23]. All the strains were grown on glucose minimal medium agar (GMM) for seven days at 37 °C, as previously described [
26]. Conidia were harvested and cleaned twice with saline-Tween solution (SS-T; 0.9% NaCl and 0.02% Tween 20). The number of conidia for each experiment was adjusted using a Bürker counting chamber.
2.2. Aspergillus fumigatus Phenotypic Characterization
The phenotypes of the mutant
A. fumigatus strains used in this work were evaluated using the spot dilution assay protocol, as previously described [
27]. Briefly, we supplemented GMM medium with 80 µg/mL congo red (CR), 40 µg/mL calcofluor white (CW) or 0.0125% sodium dodecyl sulfate (SDS) as cell wall/membrane stress agents; 1 M NaCl, 1 M KCl, or 1.2 M sorbitol as osmotic stress agents; or hydrogen peroxide (30%, 15%, 7.5%, and 3.75%
v/
v) as an oxidative stress agent.
2.3. Cell Lines
Two different cell lines obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) were used in this study: the murine macrophage cell line RAW 264.7 and the human alveolar epithelial cell line A549. Cell cultures were maintained in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), 200 mM l-glutamine, 100 U/mL penicillin, and 0.1 mg/mL streptomycin (complete RPMI), and incubated at 37 °C, 5% CO2, and 95% of humidity (cell culture atmosphere). Cell line passages were used only when the viability of the cells was higher than 90%. All culture media components were from Sigma–Aldrich (St. Louis, MO, USA).
2.4. Isolation and Culture of Mouse Primary Macrophages
Mouse bone marrow-derived macrophages (BMMs) were generated following the method previously published using 8–12 week-old C57Bl/6 mice [
28]. Briefly, bone marrow material was obtained by flushing mouse femurs and tibias in complete RPMI. The cell suspensions were filtered through a 70 µm-nylon mesh (ThermoFisher, Waltham, MA, USA) and then centrifuged at 400 rpm for 5 min. Then, ammonium–chloride–potassium (ACK) lysis buffer was used to remove red blood cells. The remaining cells were incubated in untreated 100 mm × 15 mm Petri dishes for 7 days in the presence of complete RPMI supplemented with 30 ng/mL of M-CSF (Miltenyi Biotec, Bergisch Gladbach, Germany). Fresh medium was added after 3 days of culture.
2.5. Quantification of Fumagillin Secretion by Aspergillus fumigatus
To study the ability of each fungal strain to produce fumagillin, we seeded 5 × 106 conidia/mL of each strain in 2 mL of complete RPMI using 6-well plates (ThermoFisher). The plates were incubated at 37 °C, 5% CO2, and 95% humidity. After 48 h of incubation, we centrifuged an aliquot of 1 mL at 14,000 rpm for 5 min, and the supernatant was transferred to a light-safe microtube and kept on ice until its measurement by UHPLC. Each assay was done using four independent conidia batches of each strain harvested just before the assay start point.
2.6. Fungal Response to Increasing Concentrations of Fumagillin
To study if fumagillin affects fungal strains, we performed an 8 h germination study in the presence of 0.5, 1, and 2 µg/mL of fumagillin. We seeded 2 × 106 conidia/well using 24 cell culture well plates. In each case, we calculated fungal germination and hyphal branching using a Nikon Eclipse TE2000-U inverted microscope.
Growth curves of the fungal strains were assessed for 70 h in 96-well plates (ThermoFisher, Waltham, MA, USA). We seeded 5 × 105 fresh conidia per well in 150 µL of MM supplemented with 0.5, 1, and 2 µg/mL of fumagillin. The plate was incubated in the microplate reader SynergyTM HT (BioTek, Winooski, VT, USA) at 37 °C, and automatic absorbance measures at 600 nm were done each 30 min for 70 h.
2.7. Fumagillin Absorption Ability by Cell Lines
To study the absorption of fumagillin by mammalian cells, we seeded 1 × 106 cells/mL of the RAW 264.7 and A549 cell lines in a final volume of 2 mL of complete RPMI using 6-well plates. After 1 h of incubation to allow cell attachment, we replaced the medium with 2 mL of complete RPMI supplemented with 1 µg/mL of fumagillin (Sigma–Aldrich). Aliquots of 500 µL were collected after 8, 20, and 24 h of cell exposition in light-safe microtubes and processed as previously indicated until its measurement by UHPLC (Waters, Milford, CT, USA).
In order to study whether fumagillin binds to the cell line instead of being degraded, we grew RAW 264.7 and A549 cells in the presence of 2 µg/mL of toxin. After 24 h of exposition, we discarded the supernatants and, after a saline solution wash, we lysed the cells using 1 mL of RIPA buffer. Finally, we centrifuged the samples at 14,000 rpm for 5 min, and the supernatants were measured using the UHPLC method. A toxin degradation study was done by exposition of a 1 µg/mL fumagillin solution to different environmental conditions as 80 °C or exposition to light for 96 h and exposition to pH 1.
2.8. Quantification of Fumagillin by UHPLC (Ultra High-Performance Liquid Chromatography) in RPMI Samples
Quantitative analysis of fumagillin was carried out using an Acquity Ultra-High-Performance Liquid Chromatography (UHPLC) system (Waters, Milford, CT, USA) coupled to a photodiode array (PDA) detector, following a methodology previously published [
29]. The chromatographic separation was performed on an Acquity BEH C18 column (2.1 mm × 50 mm, 1.7 μm) from Waters. The mobile phases used were a 10 mM ammonia/ammonium buffer (pH 10) as the aqueous mobile phase (A) and acetonitrile as the organic modifier (B). A flow rate of 0.40 mL/min was used with an elution gradient as follows: 0–0.5 min, 20% B; 0.5–5.5 min, linear change from 20% to 95% B; 5.5–6.5 min, 95% B; 6.5–7.0 min, from 95% to 20%. During the chromatographic analysis, the column was maintained at 35 °C with a thermostat, and the samples were kept at 4 °C in the autosampler. Wavelengths of 336 and 280 nm were employed for fumagillin and diclofenac (internal standard, IS), respectively. System control, data collection, and data processing were accomplished using Empower 2 software.
Prior to the chromatographic analysis, a solid-phase extraction (SPE) procedure was applied to the RPMI samples. Each 500 μL RPMI sample was spiked with 25 μL of 20 mg/mL IS solution in methanol and 475 μL of phosphate buffer (100 mM, pH 12). After vortex mixing, the solution was transferred to Oasis MAX cartridges (30 mg, 1 cm3) from Waters. The cartridges had been previously activated with 1 mL methanol and conditioned with 1 mL phosphate buffer (100 mM, pH 12). After sample loading, the cartridges were washed with 1 mL phosphate buffer (100 mM, pH 12):methanol (55:45) followed by 5 min drying at high vacuum, and then 1 mL of 3.5% formic acid solution in methanol was used for eluting the analyte. Subsequently, 500 μL of aqueous mobile phase was added to 500 μL of the eluate, and after centrifugation, the solution was transferred to autosampler vials and injected into the UHPLC system for analysis. For the quantification of fumagillin, a calibration curve in RPMI was built with a linear range between 25 and 1500 μg/L.
2.9. Cellular Electron Transport Chain Activity
The electron transport chain (ETC) activity of cells in contact with different concentrations of fumagillin was measured using an MTT assay. Briefly, we seeded 8 × 104 cells per well (RAW 264.7 or A549) in 96-well plates (ThermoFisher) with 150 µL of complete RPMI. After 2 h of incubation to allow cell attachment, we replaced the complete RPMI with 150 µL of RPMI supplemented with different concentrations of fumagillin (0, 0.5, 1, and 2 µg/mL), and the cells were incubated using the abovementioned conditions of cell culture atmosphere. After 24 h of incubation, we eliminated the medium, and we added 150 µL/well of complete RPMI supplemented with 0.5 mg/mL of MTT (Sigma–Aldrich) and the plate was incubated for 4 h to allow the reduction of the tetrazolium dye MTT to formazan crystals. Finally, we replaced MTT solution with 150 µL of DMSO in order to dissolve formazan crystals, and after 15 min, the absorbance was measured at 560 nm using a microplate reader SynergyTM HT (BioTek, Winooski, VT, USA). Results were expressed as a percentage of metabolic activity.
2.10. Wound Healing Assay
In order to evaluate the growth inhibitory capacity of fumagillin over A549 epithelial cells, we performed wound healing assays. Briefly, 1.5 × 105 cells per well were seeded in 24-well plates and, after an incubation of 24 h, confluent monolayers were obtained. Using a 1000 µL pipette tip, a vertical and a horizontal scratch was made and then washed twice with PBS to remove detached cells. Then, fresh medium alone or containing 0.5, 1, or 2 µg/mL of fumagillin was added to the cultures. Images were taken from one field of view down between the intersection of the two scratches at 24 and 48 h after fumagillin addition, using a Nikon Eclipse TE2000-U (Nikon, Minato, Tokyo, Japan) inverted microscope. Finally, the percentage of scratch reduction was calculated and normalized to the untreated control.
2.11. Analysis of Cell Proliferation and Viability by Flow Cytometry
Flow cytometry-based assays were carried out in the general services from the University of the Basque Country (SGIker, UPV/EHU) using a Beckman Coulter Gallios cytometer (Beckman Coulter, Brea, CA, USA).
Changes in cell proliferation of A549 and RAW 264.7 cells in the presence of fumagillin at different concentrations were determined using the fluorescent dye CFSE (Invitrogen, Carlsbad, CA, USA). Cells were stained using 2 µM of CFSE in PBS for 30 min at 37 °C and 5% CO2 and then washed twice in RPMI supplemented with 1% of FBS. Finally, we seeded 1 × 105 cells/well in 6-well plates. At time 0 h, the just stained cells were analyzed in the flow cytometer (517 nm) alongside a set of unstained cells. Then, cells were exposed to 0.5, 1, or 2 µg/mL of fumagillin, or left untreated. The study was extended for 72 h, and daily samples were analyzed by flow cytometry. For that, cells were detached from each well using a cell scraper and resuspended in RPMI. The flow cytometer was set up to analyze 10,000 events from each sample, exciting the sample at 492 nm and measuring the fluorescence at 517 nm. In order to calculate the statistical differences between conditions, the average fluorescence of the cells without treatment was compared with the average fluorescence of the cells treated with the abovementioned fumagillin concentrations.
On the other hand, cell viability assays were carried out using Propidium Iodide Ready Flow™ Reagent (Invitrogen, Carlsbad, CA, USA) for 15 min following the manufacturer’s instructions. Cell density, detachment protocol, and the number of cells analyzed in each run were the same as in the proliferation assays. After cell staining, samples were excited at 535 nm, following the manufacturer’s instructions, and the emission was at 617 nm. Results were expressed as a percentage of cell viability.
2.12. Phagocytosis Assay and Fungal Growth
To analyze the interactions of the fungal strains with RAW 264.7 macrophages and BMMs, we seeded 2 × 105 cells/mL in 500 µL of complete RPMI using 24-well plates (ThermoFisher) which contained 12 mm-diameter coverslips (ThermoFisher). After overnight incubation, immune cells were co-cultured with A. fumigatus conidia of each strain (Wt, ∆fmaA, ∆fmaA::fmaA) at a multiplicity of infection of 10 (ten conidia per immune cell). In parallel, we seeded the same number of each fungal strain conidia but in this case without immune cells to analyze fungal growth in the absence of murine cells. At each incubation time (2, 4, 6, and 8 h), we removed the coverslip to a new plate in order to calculate the percentage of phagocytosis, fungal germination, and hyphal branching. For that purpose, a minimum of 500 fungal cells was counted using a Nikon Eclipse TE2000-U inverted microscope.
Furthermore, we performed the same phagocytosis assay under two additional conditions. The first one, using RAW 264.7 macrophages pre-treated for 24 h with 1 µg/mL of fumagillin (before the co-incubation) to determine the effect of the toxin itself, and the second one using heat-inactivated conidia (121 °C for 30 min) of each A. fumigatus strain to ensure that the genetic manipulation of the strains did not produce structural changes of the conidia.
On the other hand, to study changes in the hyphal length of strains when growing alone or combined with either of the macrophage models, we performed CW staining on coverslips after 8 h of incubation. For that, samples were washed with PBS and stained with 100 µL of CW solution for 20 min in the dark and at room temperature. Finally, we washed the coverslips again with PBS, and they were mounted for fluorescence microscopy. Ten micrographs per coverslip were taken randomly using an Eclipse Ni microscope fluorescence microscope and a Nikon Ds-Fi2 camera (Nikon). Image analysis was carried out using ImageJ software.
2.13. Mouse Model of Pulmonary Aspergillosis
Mice used in this study were kept in the General Animal Unit Service of the University of the Basque Country (SGIker, UPV/EHU), with water and food ad libitum, handled in biological safety cabinets, and kept in sterilized cages with negative-pressure ventilation and filters.
SWISS female mice weighing 25–35 g were used in this study. All mice were immunosuppressed by the daily administration of 100 mg/kg cyclophosphamide (Sigma–Aldrich), starting four days before infection. Five animals per group were infected intranasally with 20 µL SS-T solution containing 1 × 107 resting conidia of one A. fumigatus strain, either Wt, ∆fmaA or ∆fmaA::fmaA. Two independent experiments were performed.
Mice were monitored daily to analyze changes in weight and clinical scores due to the infection process. Clinical scoring was performed using a scale of 0–10, in which we evaluated different aspects of the disease (
Table S1) following the recommendations of experts from our institution animal facility and the ethical committee members. Those mice that reached humane endpoints (10 points) were euthanized to minimize mice suffering, and their cause of death was considered as fungal infection.
To study the fungal burden in mice lungs, organs were extracted and were homogenized in 1 mL of SS-T by vortexing in a tube in the presence of a sterile glass stick. Finally, an aliquot of 0.1 mL was inoculated in potato dextrose agar plates supplemented with 10 μg/mL chloramphenicol and 25 μg/mL gentamicin (both from Sigma–Aldrich). Plates were incubated at 37 °C, and the number of Colony-Forming Units (CFUs) was determined after 3 days.
2.14. Statistics
All the assays were done at least by triplicate in three independent days. All statistical analyses of this study were performed using GraphPad Prism 7 software (GraphPad Software Inc., San Diego, CA, USA). At least three biological replicates were performed to measure each parameter in each experimental condition; any statistically significant differences were analyzed as required. All data present in this study followed a normal distribution. t-test or ANOVA was used to study differences between conditions depending on if we compared punctual data or multiple comparisons, respectively.
4. Discussion
The importance of gliotoxin for
A. fumigatus virulence has been widely described [
12,
13,
14,
15,
30,
31,
32,
33]. However, the relevance of other toxins produced by
A. fumigatus, such as fumagillin, has not been studied in depth.
Fumagillin is a macrolide that targets methionine aminopeptidase 2 (MetAP2) by covalent binding to the His231, inhibiting the proper functioning of the protein. Recent publications have pointed out that fumagillin could induce lung epithelial cell damage, both acting alone [
23] and in synergy with gliotoxin [
34]. Moreover, this mycotoxin is able to inhibit cell proliferation and angiogenesis in endothelial cells [
20]. However, little is known about its effect on the fungus or other cell types or its role in fungal virulence in vivo during the development of pulmonary aspergillosis.
To study this, we used a deletion mutant strain of the
fmaA gene and its complement strain where
fmaA encodes a terpene cyclase required for fumagillin synthesis [
22]. First, an UHPLC method for the detection of fumagillin in samples of RPMI obtained after in vitro assays was standardized. Using this method, we were able to detect as low as 0.1 µg/mL of fumagillin. Furthermore, we confirmed that the ∆
fmaA strain was not able to secrete fumagillin, corroborating the previously published data [
22].
Then, we performed a macroscopic phenotypical and growth study that showed that the mutant strain ∆
fmaA did not present any difference compared to the Wt strain when they grew in the presence of structural, osmotic, and oxidative stresses (
Figures S1 and S3). We also observed that
A. fumigatus was not susceptible to exogenous fumagillin (
Figure S2), but that fumagillin may be a germination-stimulating factor (
Figure 1). These results could support our hypothesis that the gene Afu8g00410, encoding a methionine aminopeptidase located inside the fumagillin cluster, confers resistance to the fungus. This gene would be co-expressed with the rest of the cluster, providing a resistant MetAP2 to the fungus and may explain the results obtained in
Figure 1. Continuing with fungal strains’ germination ability, it is notable that the ∆
fmaA strain reached higher germination and branching rates than the other strains growing without cells and in the presence of fumagillin. The increased branching rates observed in the ∆
fmaA strain may indicate that fumagillin could be involved in a polarization process, or in contrast, the increased branching observed could be a consequence of the adaptive process to the fumagillin presence. In any case, both hypotheses should be studied in depth.
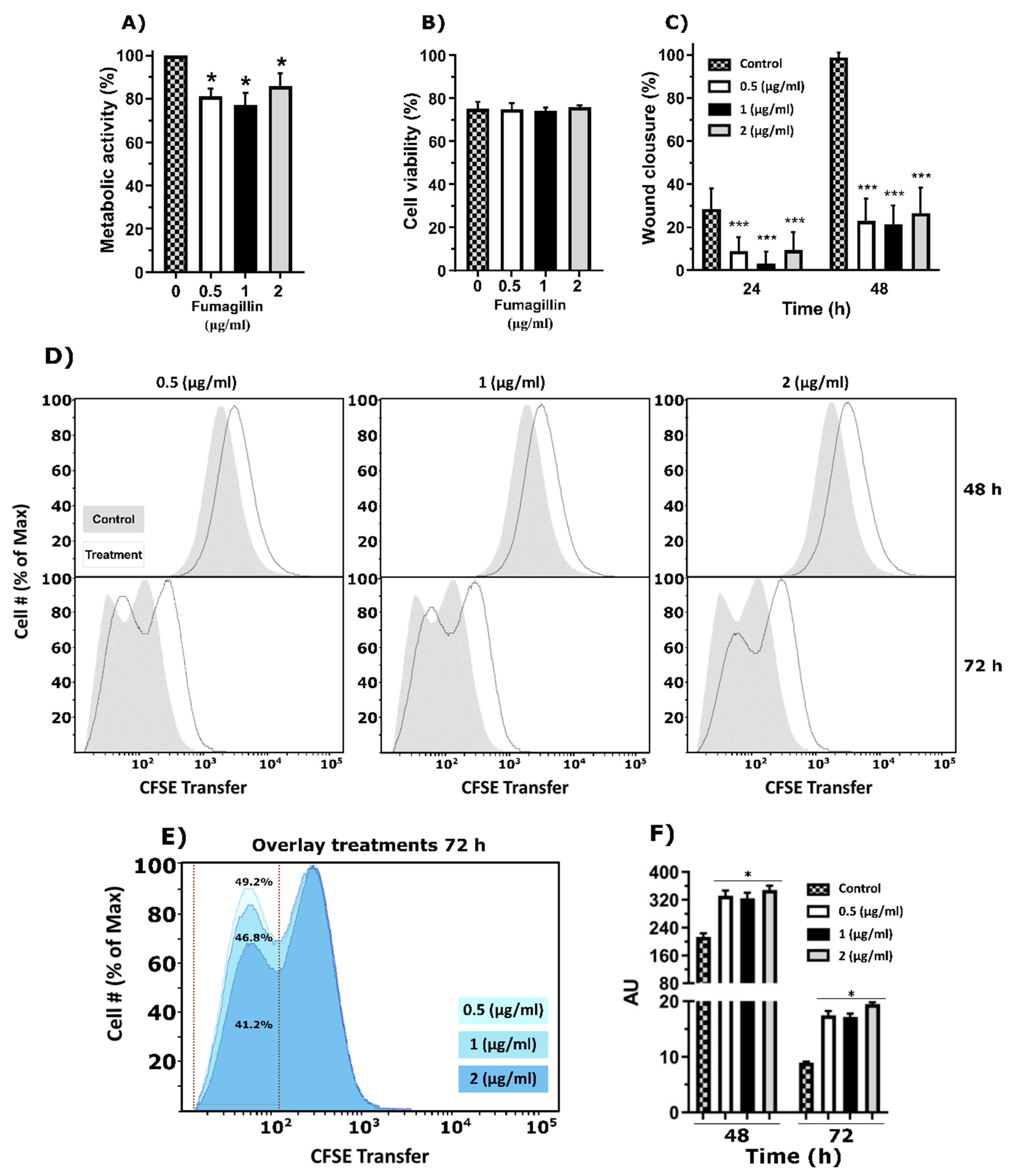
Analysis of the effect of synthetic fumagillin on both A549 pneumocytes and RAW 264.7 macrophages, which are common cell lines to perform toxin/molecule/drug response analysis [
23,
34,
35], demonstrated that each cell type uptakes the mycotoxin in different ways (
Figure 2). Fumagillin was clearly depleted from the supernatant (93.5%) of pneumocytes but to a much lesser degree (23.5%) macrophages culture. Furthermore, the lysis of both cell types using RIPA buffer and subsequent detection of the toxin by UHPLC suggested that the reduction in fumagillin from the media was due to its uptake by the cell. One explanation could be attributed to the different expression levels of the MetAP2 enzyme between both cell types (
https://www.proteinatlas.org; accessed 15 August 2021). Moreover, we cannot forget that macrophages can express different efflux systems that could expel the toxin from the cytosol. Finally, we cannot totally discard that cells could also degrade fumagillin, but no peaks similar to the ones observed after the fumagillin degradation assays (light, temperature, and acid environment) were detected either in the supernatants or the pellets (
Figure S2). Therefore, it seems that fumagillin could passively cross the cell membrane reaching the cell cytosol [
36] and bind to MetAP2, thus leading to loss of the toxin in the supernatant.
The MTT results shown in this study are in accordance with the ones previously published [
34,
37,
38], but our interpretation of the MTT results varies with the other studies. In this study, we used the MTT assay as a technique suitable to measure the ETC [
39], while other authors used it to measure cell viability. On the other hand, it is important to note that our highest fumagillin concentration used (2 µg/mL) was the lowest of all these above-cited studies. In contrast, it is important to note that during the infection process in vivo or during the contact between the fungus and host cells, the toxin concentration reached could be higher. For instance, the production of candidalys in by
Candida albicans in the invasion pocket produced an extremely high concentration of the toxin at the contact site between the fungus and the cell [
40]. This hypothesis could explain why a previous study performed with this same ∆
fmaA strain demonstrated that it was less toxic than the Wt to the A549 cell line as assessed by
51Cr release assays [
23].
Our results support the affectation of the ETC activity, as both the wound healing assay and the CFSE analysis performed by flow cytometry demonstrated that the proliferation of epithelial cells was affected after fumagillin exposure but not its cellular viability. Specifically, the covalent binding of fumagillin to its molecular target, MetAP2, results in an inhibition of all functions that MetAP2 orchestrates inside the cell [
21]. They include the membrane signaling pathway involving proteins, such as Gi/o, Giβγ, PI3K, PLC, DAG, or IP3 that govern cell migration and proliferation [
41,
42,
43].
The results obtained with the RAW 264.7 cells were slightly different from those obtained from the pneumocytes since although they suffered a drop in the ETC activity and a delay in the cell proliferation after exposure to fumagillin, the effect was less marked than observed in pneumocytes and was not dependent on concentration. These results are apparently in contradiction with previous studies of other authors on rat alveolar macrophages using trypan blue to measure cell viability, which is a less sensitive method [
44]. Currently, it is unknown why macrophages, but not epithelial cells die at such a high rate; the different effects of the fumagillin depending on the cell type might be explained because the MetAP2 concentration could vary among tissues and cell types (
https://www.proteinatlas.org; accessed 15 August 2021).
In addition, fumagillin seems to protect, at least in early hours, the fungus from phagocytosis as assessed from increased phagocytosis of ∆fmaA conidia. In fact, when RAW 264.7 phagocytic cells were pre-treated with fumagillin, the phagocytic ability of these cells was significantly decreased after 6 and 8 h of co-incubation in comparison with the phagocytosis rates at this same time points of the cells untreated. Furthermore, the phagocytosis of the cells pre-treated was still significantly higher in contact with ∆fmaA strain at all times. These results, alongside the evidence that phagocytosis of the heat-inactivated conidia is similar in mutants and Wt strains, points out that the inhibition of the phagocytosis observed is because of the toxin effect over the macrophages and not because of an external structure defect of the conidia cell wall produced as a consequence of the genetic manipulation of the fungal strains. Moreover, it is important to note that the fumagillin produced by the Wt and complemented strains could be modulating the macrophages’ ETC and viability influencing the cell response, from their phagocytosis ability to TNF production. Indeed, TNF induction by BMMs was also overall lower in response to the ∆fmaA strain than to the Wt and complemented strains, although it was not statistically significant except at 4 h.
Regarding in vivo assays, Liu and coworkers evaluated the virulence ability of the mutant
A. fumigatus non-fumagillin producer strain Δ
fumR [
24]. For that, they used a corticosteroid immunosuppressed mice model, and they did not describe any virulence difference between the mutant and the Wt strain. Therefore, and due to the differences in the phagocytosis assays observed (
Figure 6), we wanted to assess a neutropenic murine aspergillosis model to look at the role of the
fmaA gene in virulence. Unexpectedly, the ∆
fmaA::
fmaA strain was significantly more lethal than the Wt and ∆
fmaA strains. In contrast, the lowest mortality rate (although not significantly different to the Wt) was observed in those mice infected with the ∆
fmaA strain.
On the other hand, the CFUs analysis showed that most of the surviving mice infected with ∆fmaA strain did not contain viable fungus in their lungs, whereas a high fungal burden was detected in mice infected with the other strains at the end of the experiment. These results may indicate that the mortality caused by ∆fmaA, only observed during the first four days of infection, could be the result of the ability of this mutant to germinate faster than the others. Furthermore, the possible production of other virulence factors, such as proteases or other toxins, could be compensating for the lack of fumagillin, but this hypothesis has not been analyzed in this study.
To our knowledge, this is not the first fumagillin research, but it is the first in-depth study of the role of fumagillin on A. fumigatus virulence, showing that this molecule can affect host cell homeostasis and that A. fumigatus shows an intrinsic resistance to its own toxin. Finally, in our murine model, whereas we found that mice infected with ∆fmaA survived with lower fungal burden than those infected with the wild type or the complemented strain, there was little overall difference in virulence dependent on this toxin.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






