
Analysis of the Putative Nucleoporin POM33 in the Filamentous Fungus Sordaria macrospora



Abstract
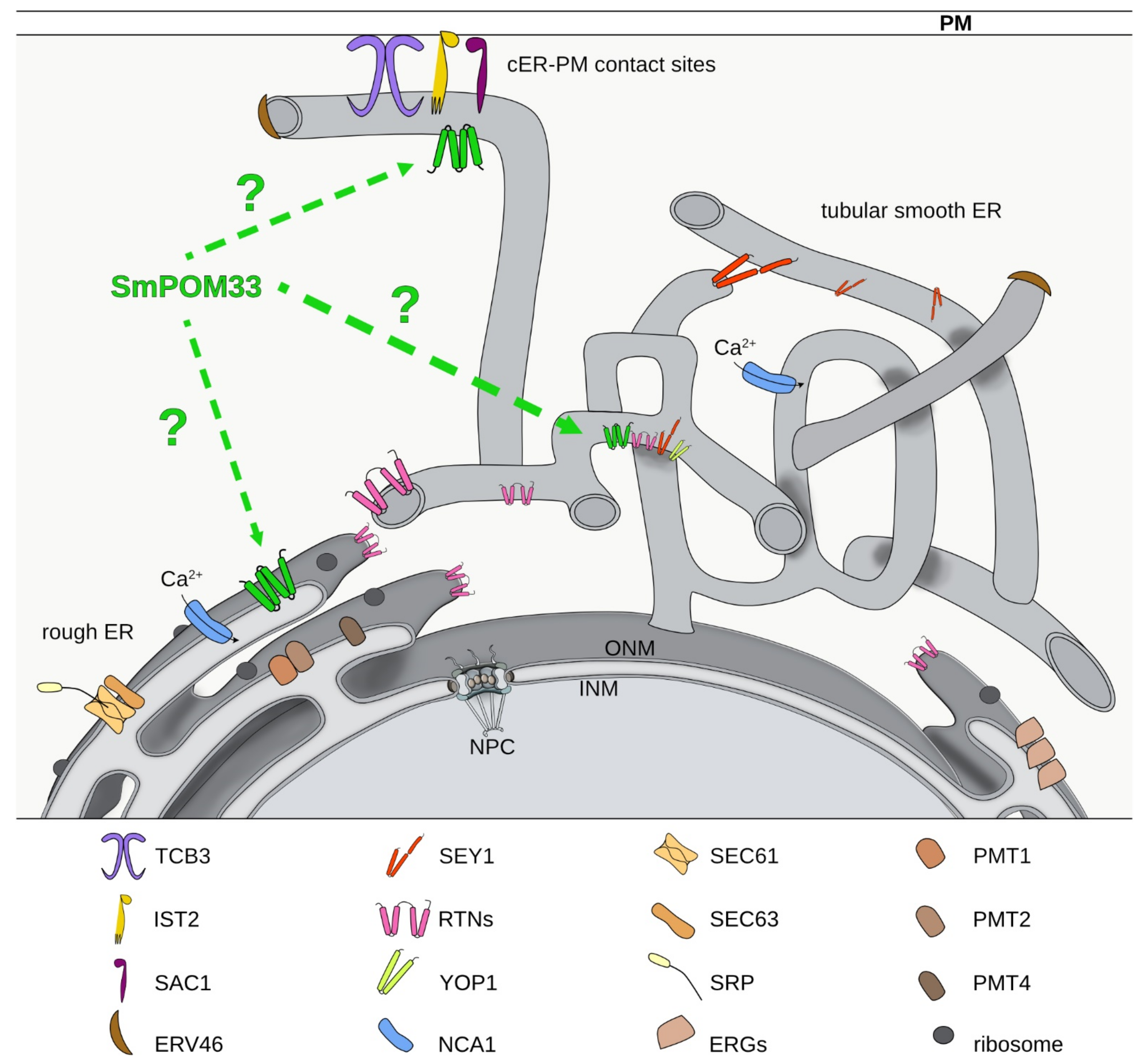
:1. Introduction
2. Materials and Methods
2.1. Strains, Media and Growth Conditions
2.2. Generation of Plasmids
2.3. Generation of the Knockout Strains Δku80 and Δpom33
2.4. Light and Fluorescence Microscopy
2.5. Protein Sample Preparation and Western Blot Hybridization
2.6. Pulldown Experiments, LC/MS and Data Analysis
2.6.1. Pulldown Experiments
2.6.2. Trypsin In-Gel Digest of Proteins and C18 Stage Tip Purification
2.6.3. Liquid Chromatography—Mass Spectrometry (LC/MS) Analysis
2.7. Data Acquisition and Analysis
2.8. Protein Domain Analysis
3. Results
3.1. S. macrospora POM33 Belongs to the Pom33/ TMEM33 Protein Family
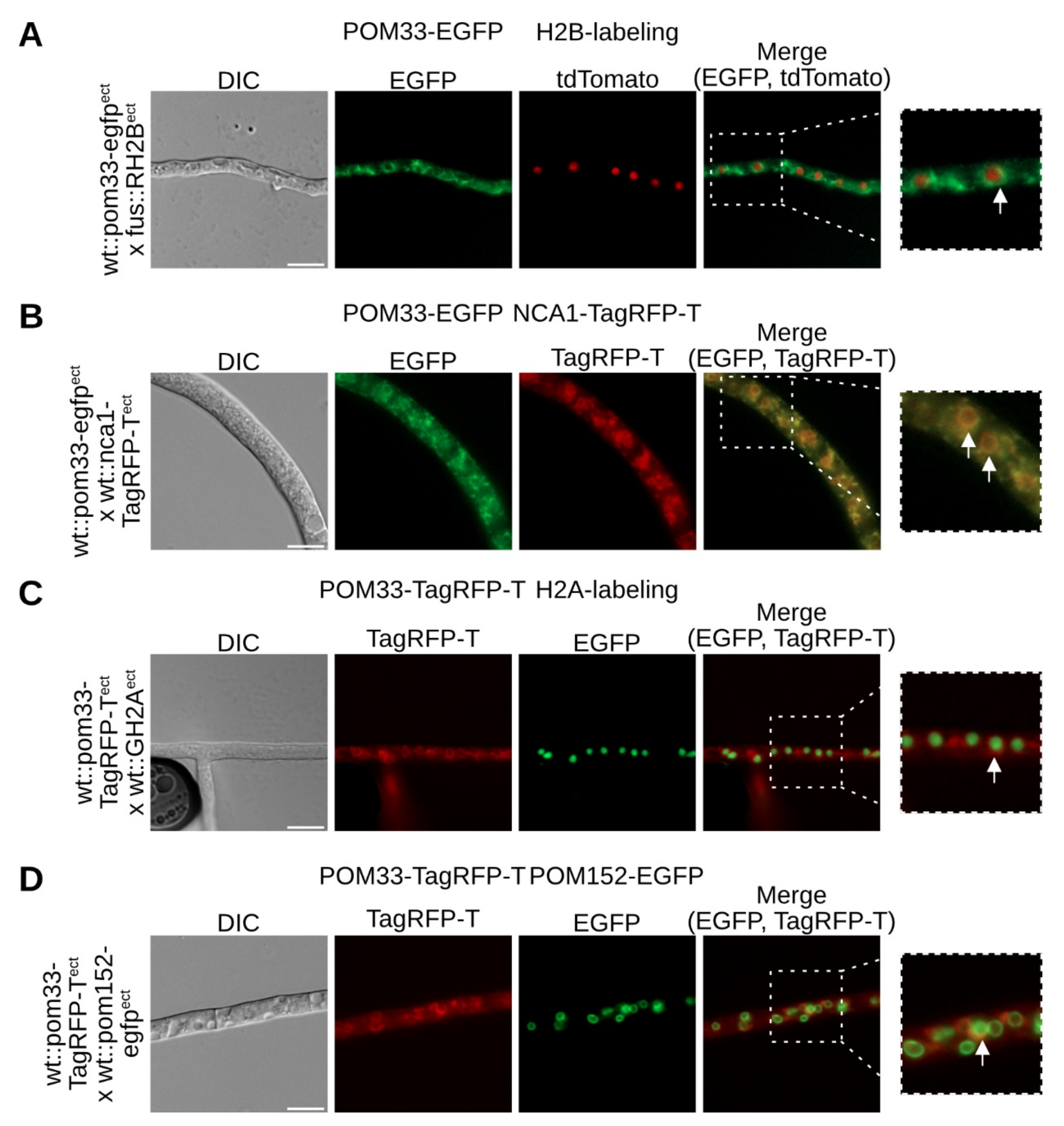
3.2. SmPOM33 Localizes at the Nuclear Envelope and the ER
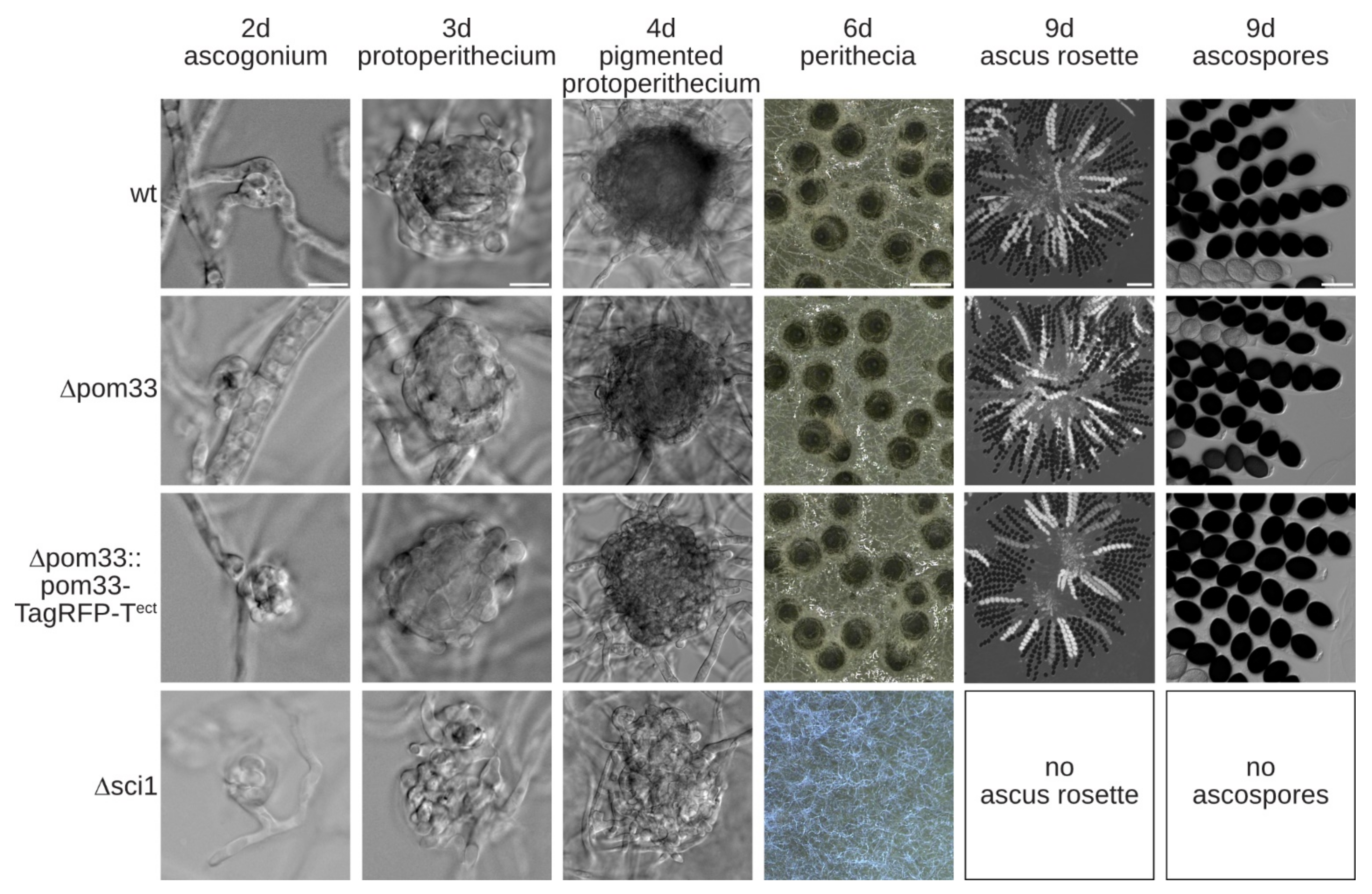
3.3. Deletion of pom33 Displays no Distinct Phenotype Compared to the Wildtype
3.4. The Mutant Δpom33 Exhibits no Sensitivity against a Series of Stress Conditions
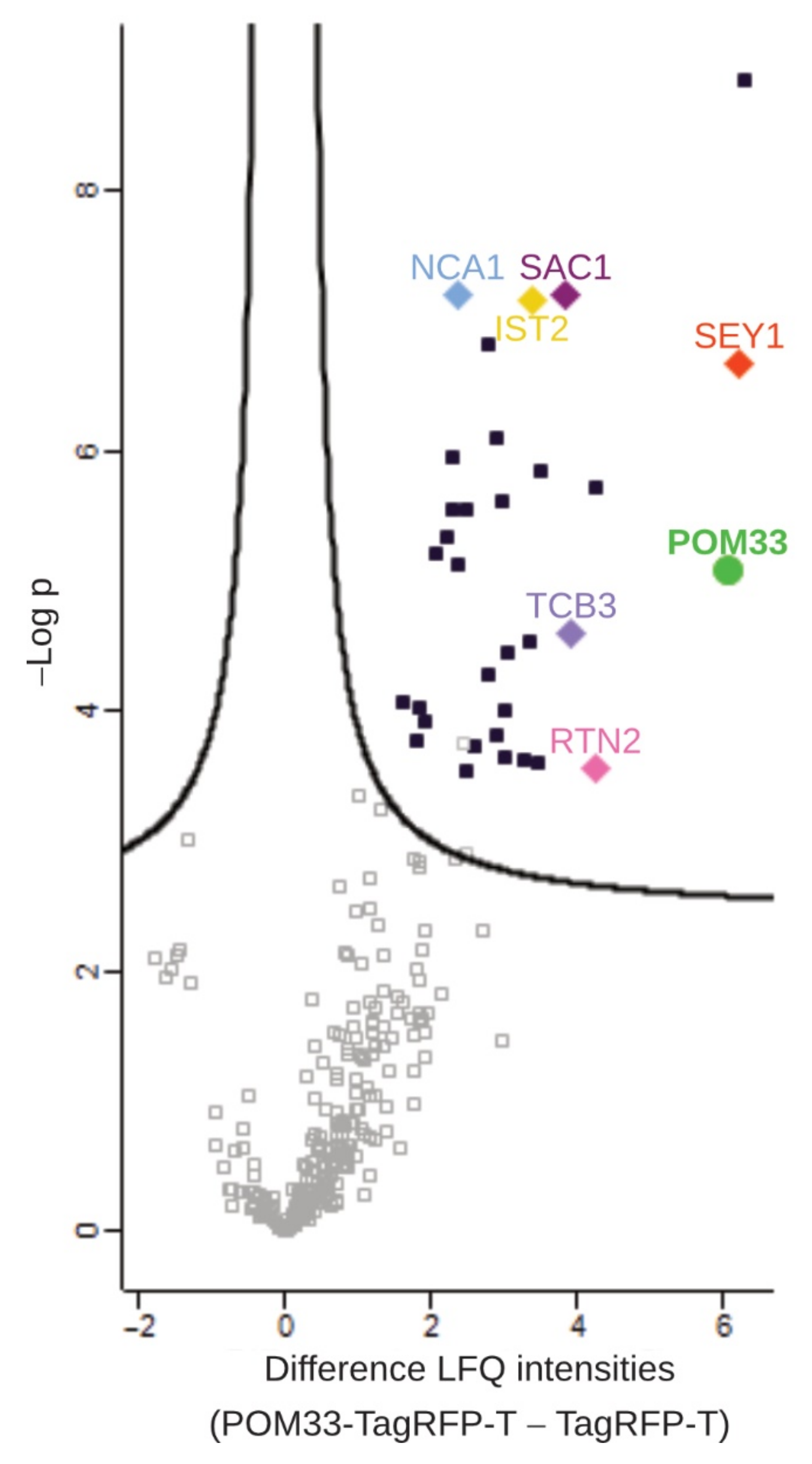
3.5. Proteins of the ER Are Putative Interaction Partners of POM33
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hwang, J.; Pallas, D.C. STRIPAK complexes: Structure, biological function, and involvement in human diseases. Int. J. Biochem. Cell Biol. 2014, 47, 118–148. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.; Jiao, S.; Zhou, Z. STRIPAK complexes in cell signaling and cancer. Oncogene 2016, 35, 4549–4557. [Google Scholar] [CrossRef]
- Benoist, M.; Gaillard, S.; Castets, F. The striatin family: A new signaling platform in dendritic spines. J. Physiol. Paris 2006, 99, 146–153. [Google Scholar] [CrossRef]
- Teichert, I.; Nowrousian, M.; Pöggeler, S.; Kück, U. The filamentous fungus Sordaria macrospora as a genetic model to study fruiting body development. Adv. Genet. 2014, 87, 199–244. [Google Scholar]
- Teichert, I.; Pöggeler, S.; Nowrousian, M. Sordaria macrospora: 25 years as a model organism for studying the molecular mechanisms of fruiting body development. Appl. Microbiol. Biotechnol. 2020, 104, 3691–3704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloemendal, S.; Bernhards, Y.; Bartho, K.; Dettmann, A.; Voigt, O.; Teichert, I.; Seiler, S.; Wolters, D.A.; Pöggeler, S.; Kück, U. A homologue of the human STRIPAK complex controls sexual development in fungi. Mol. Microbiol. 2012, 84, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Pöggeler, S.; Kück, U. A WD40 repeat protein regulates fungal cell differentiation and can be replaced functionally by the mammalian homologue striatin. Eukaryot. Cell 2004, 3, 232–240. [Google Scholar] [CrossRef] [Green Version]
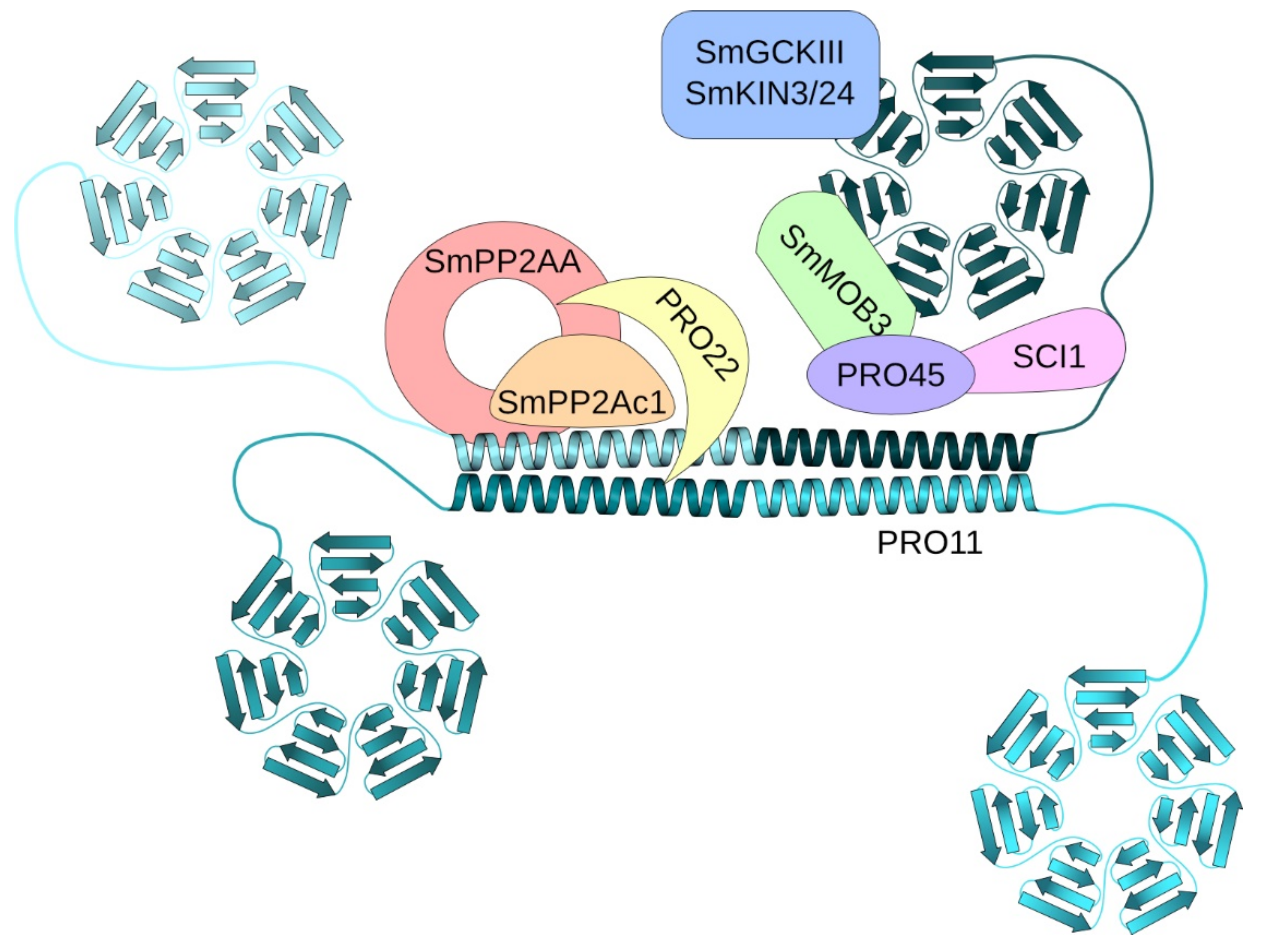
- Kück, U.; Beier, A.M.; Teichert, I. The composition and function of the striatin-interacting phosphatases and kinases (STRIPAK) complex in fungi. Fungal Genet. Biol. 2016, 90, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Nordzieke, S.; Zobel, T.; Franzel, B.; Wolters, D.A.; Kück, U.; Teichert, I. A fungal sarcolemmal membrane-associated protein (SLMAP) homolog plays a fundamental role in development and localizes to the nuclear envelope, endoplasmic reticulum, and mitochondria. Eukaryot. Cell 2015, 14, 345–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beier, A.; Teichert, I.; Krisp, C.; Wolters, D.A.; Kück, U. Catalytic Subunit 1 of Protein Phosphatase 2A Is a Subunit of the STRIPAK Complex and Governs Fungal Sexual Development. mBio 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, S.; Reschka, E.J.; Pöggeler, S. Germinal Center Kinases SmKIN3 and SmKIN24 Are Associated with the Sordaria macrospora Striatin-Interacting Phosphatase and Kinase (STRIPAK) Complex. PLoS ONE 2015, 10, e0139163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radchenko, D.; Teichert, I.; Pöggeler, S.; Kück, U. A Hippo pathway-related GCK controls both sexual and vegetative developmental processes in the fungus Sordaria macrospora. Genetics 2018, 210, 137–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernhards, Y.; Pöggeler, S. The phocein homologue SmMOB3 is essential for vegetative cell fusion and sexual development in the filamentous ascomycete Sordaria macrospora. Curr Genet. 2011, 57, 133–149. [Google Scholar] [CrossRef] [Green Version]
- Reschka, E.J.; Nordzieke, S.; Valerius, O.; Braus, G.H.; Pöggeler, S. A novel STRIPAK complex component mediates hyphal fusion and fruiting-body development in filamentous fungi. Mol. Microbiol. 2018, 110, 513–532. [Google Scholar] [CrossRef]
- Jeong, B.C.; Bae, S.J.; Ni, L.; Zhang, X.; Bai, X.C.; Luo, X. Cryo-EM structure of the Hippo signaling integrator human STRIPAK. Nat. Struct. Mol. Biol. 2021, 28, 290–299. [Google Scholar] [CrossRef]
- De Magistris, P.; Antonin, W. The dynamic nature of the nuclear envelope. Curr. Biol. 2018, 28, R487–R497. [Google Scholar] [CrossRef] [Green Version]
- Baumann, O.; Walz, B. Endoplasmic reticulum of animal cells and its organization into structural and functional domains. Int. Rev. Cytol. 2001, 205, 149–214. [Google Scholar]
- Wang, N.; Rapoport, T.A. Reconstituting the reticular ER network–mechanistic implications and open questions. J. Cell Sci. 2019, 132, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Shibata, Y.; Voeltz, G.K.; Rapoport, T.A. Rough sheets and smooth tubules. Cell 2006, 126, 435–439. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Chen, L.B. Dynamic behavior of endoplasmic reticulum in living cells. Cell 1988, 54, 37–46. [Google Scholar] [CrossRef]
- Du, Y.; Ferro-Novick, S.; Novick, P. Dynamics and inheritance of the endoplasmic reticulum. J. Cell Sci. 2004, 117, 2871–2878. [Google Scholar] [CrossRef] [Green Version]
- Hampoelz, B.; Andres-Pons, A.; Kastritis, P.; Beck, M. Structure and assembly of the nuclear pore complex. Annu. Rev. Biophys. 2019, 48, 515–536. [Google Scholar] [CrossRef] [PubMed]
- Hetzer, M.W.; Walther, T.C.; Mattaj, I.W. Pushing the envelope: Structure, function, and dynamics of the nuclear periphery. Annu. Rev. Cell Dev. Biol. 2005, 21, 347–380. [Google Scholar] [CrossRef]
- Antonin, W.; Ellenberg, J.; Dultz, E. Nuclear pore complex assembly through the cell cycle: Regulation and membrane organization. FEBS Lett. 2008, 582, 2004–2016. [Google Scholar] [CrossRef] [Green Version]
- Doucet, C.M.; Hetzer, M.W. Nuclear pore biogenesis into an intact nuclear envelope. Chromosoma 2010, 119, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Doucet, C.M.; Talamas, J.A.; Hetzer, M.W. Cell cycle-dependent differences in nuclear pore complex assembly in metazoa. Cell 2010, 141, 1030–1041. [Google Scholar] [CrossRef] [Green Version]
- Chial, H.J.; Rout, M.P.; Giddings Jr, T.H.; Winey, M. Saccharomyces cerevisiae Ndc1p is a shared component of nuclear pore complexes and spindle pole bodies. J. Cell Biol. 1998, 143, 1789–1800. [Google Scholar] [CrossRef] [Green Version]
- Mansfeld, J.; Güttinger, S.; Hawryluk-Gara, L.A.; Panté, N.; Mall, M.; Galy, V.; Haselmann, U.; Mühlhäusser, P.; Wozniak, R.W.; Mattaj, I.W.; et al. The conserved transmembrane nucleoporin NDC1 is required for nuclear pore complex assembly in vertebrate cells. Mol. Cell 2006, 22, 93–103. [Google Scholar] [CrossRef]
- Stavru, F.; Hülsmann, B.B.; Spang, A.; Hartmann, E.; Cordes, V.C.; Görlich, D. NDC1: A crucial membrane-integral nucleoporin of metazoan nuclear pore complexes. J. Cell Biol. 2006, 173, 509–519. [Google Scholar] [CrossRef] [Green Version]
- Chadrin, A.; Hess, B.; San Roman, M.; Gatti, X.; Lombard, B.; Loew, D.; Barral, Y.; Palancade, B.; Doye, V. Pom33, a novel transmembrane nucleoporin required for proper nuclear pore complex distribution. J. Cell Biol. 2010, 189, 795–811. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Oliferenko, S. Tts1, the fission yeast homologue of the TMEM33 family, functions in NE remodeling during mitosis. Mol. Biol. Cell 2014, 25, 2970–2983. [Google Scholar] [CrossRef]
- Urade, T.; Yamamoto, Y.; Zhang, X.; Ku, Y.; Sakisaka, T. Identification and characterization of TMEM33 as a reticulon-binding protein. Kobe J. Med. Sci. 2014, 60, E57–E65. [Google Scholar]
- Sambrook, J.; Fritsch, E.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Colot, H.V.; Park, G.; Turner, G.E.; Ringelberg, C.; Crew, C.M.; Litvinkova, L.; Weiss, R.L.; Borkovich, K.A.; Dunlap, J.C. A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc. Natl. Acad. Sci. USA 2006, 103, 10352–10357. [Google Scholar] [CrossRef] [Green Version]
- James, P.; Halladay, J.; Craig, E.A. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 1996, 144, 1425–1436. [Google Scholar] [CrossRef]
- Walz, M.; Kuck, U. Transformation of Sordaria macrospora to hygromycin B resistance: Characterization of transformants by electrophoretic karyotyping and tetrad analysis. Curr. Genet. 1995, 29, 88–95. [Google Scholar] [CrossRef]
- Kück, U.; Hoff, B. Application of the nourseothricin acetyltransferase gene (nat1) as dominant marker for the transformation of filamentous fungi. Fungal Genet. Newsl. 2006, 53, 9–11. [Google Scholar] [CrossRef] [Green Version]
- Elleuche, S.; Pöggeler, S. Visualization of peroxisomes via SKL-tagged DsRed protein in Sordaria macrospora. Fungal Genet. Rep. 2008, 55, 9–12. [Google Scholar] [CrossRef] [Green Version]
- Esser, K. Cryptogams: Cyanobacteria, Algae, Fungi, Lichens; Cambridge University Press: Cambridge, UK, 1982. [Google Scholar]
- Nowrousian, M.; Ringelberg, C.; Dunlap, J.C.; Loros, J.J.; Kück, U. Cross-species microarray hybridization to identify developmentally regulated genes in the filamentous fungus Sordaria macrospora. Mol. Genet. Genom. 2005, 273, 137–149. [Google Scholar] [CrossRef]
- Nowrousian, M.; Teichert, I.; Masloff, S.; Kück, U. Whole-Genome Sequencing of Sordaria macrospora Mutants Identifies Developmental Genes. G3 2012, 2, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Pöggeler, S.; Kück, U. Highly efficient generation of signal transduction knockout mutants using a fungal strain deficient in the mammalian ku70 ortholog. Gene 2006, 378, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Voigt, O.; Pöggeler, S. Autophagy genes Smatg8 and Smatg4 are required for fruiting-body development, vegetative growth and ascospore germination in the filamentous ascomycete Sordaria macrospora. Autophagy 2013, 9, 33–49. [Google Scholar] [CrossRef] [Green Version]
- Werner, A.; Otte, K.; Stahlhut, G.; Hanke, L.M.; Pöggeler, S. The Glyoxysomal Protease LON2 Is Involved in Fruiting-Body Development, Ascosporogenesis and Stress Resistance in Sordaria macrospora. J. Fungi 2021, 7, 82. [Google Scholar] [CrossRef]
- Dahlmann, T.A.; Terfehr, D.; Becker, K.; Teichert, I. Golden Gate vectors for efficient gene fusion and gene deletion in diverse filamentous fungi. Curr. Genet. 2021, 67, 317–330. [Google Scholar] [CrossRef]
- Klix, V.; Nowrousian, M.; Ringelberg, C.; Loros, J.J.; Dunlap, J.C.; Pöggeler, S. Functional characterization of MAT1-1-specific mating-type genes in the homothallic ascomycete Sordaria macrospora provides new insights into essential and nonessential sexual regulators. Eukaryot. Cell 2010, 9, 894–905. [Google Scholar] [CrossRef] [Green Version]
- Christianson, T.W.; Sikorski, R.S.; Dante, M.; Shero, J.H.; Hieter, P. Multifunctional yeast high-copy-number shuttle vectors. Gene 1992, 110, 119–122. [Google Scholar] [CrossRef]
- Carroll, A.M.; Sweigard, J.A.; Valent, B. Improved vectors for selecting resistance to hygromycin. Fungal Genet. Rep. 1994, 41, 22. [Google Scholar] [CrossRef] [Green Version]
- Towbin, H.; Staehelin, T.; Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 1979, 76, 4350–4354. [Google Scholar] [CrossRef] [Green Version]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef]
- Rappsilber, J.; Ishihama, Y.; Mann, M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 2003, 75, 663–670. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Blank-Landeshammer, B.; Teichert, I.; Märker, R.; Nowrousian, M.; Kück, U.; Sickmann, A. Combination of Proteogenomics with peptide de novo sequencing identifies new genes and hidden posttranscriptional modifications. MBio 2019, 10, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Oliveros, J.C. VENNY. An Interactive Tool for Comparing Lists with Venn Diagrams (2007–2015). Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 12 April 2021).
- Cherry, J.M.; Adler, C.; Ball, C.; Chervitz, S.A.; Dwight, S.S.; Hester, E.T.; Jia, Y.; Juvik, G.; Roe, T.; Schroeder, M.; et al. SGD: Saccharomyces Genome Database. Nucleic Acids Res. 1998, 26, 73–79. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Blum, M.; Chang, H.Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef] [PubMed]
- Combet, C.; Blanchet, C.; Geourjon, C.; Deleage, G. NPS@: Network protein sequence analysis. Trends Biochem. Sci. 2000, 25, 147–150. [Google Scholar] [CrossRef]
- Klausen, M.S.; Jespersen, M.C.; Nielsen, H.; Jensen, K.K.; Jurtz, V.I.; Sonderby, C.K.; Sommer, M.O.A.; Winther, O.; Nielsen, M.; Petersen, B.; et al. NetSurfP-2.0: Improved prediction of protein structural features by integrated deep learning. Proteins 2019, 87, 520–527. [Google Scholar] [CrossRef] [Green Version]
- Asakawa, H.; Yang, H.J.; Yamamoto, T.G.; Ohtsuki, C.; Chikashige, Y.; Sakata-Sogawa, K.; Tokunaga, M.; Iwamoto, M.; Hiraoka, Y.; Haraguchi, T. Characterization of nuclear pore complex components in fission yeast Schizosaccharomyces pombe. Nucleus 2014, 5, 149–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pöggeler, S.; Masloff, S.; Hoff, B.; Mayrhofer, S.; Kück, U. Versatile EGFP reporter plasmids for cellular localization of recombinant gene products in filamentous fungi. Curr. Genet. 2003, 43, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Fell, V.L.; Schild-Poulter, C. The Ku heterodimer: Function in DNA repair and beyond. Mutat. Res. Rev. Mutat. Res. 2015, 763, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Ferreira, M.E.; Kress, M.R.; Savoldi, M.; Goldman, M.H.S.; Härtl, A.; Heinekamp, T.; Brakhage, A.A.; Goldman, G.H. The akuBKU80 mutant deficient for nonhomologous end joining is a powerful tool for analyzing pathogenicity in Aspergillus fumigatus. Eukaryot. Cell 2006, 5, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Ninomiya, Y.; Suzuki, K.; Ishii, C.; Inoue, H. Highly efficient gene replacements in Neurospora strains deficient for nonhomologous end-joining. Proc. Natl. Acad. Sci. USA 2004, 101, 12248–12253. [Google Scholar] [CrossRef] [Green Version]
- Monnerjahn, C.; Techel, D.; Meyer, U.; Rensing, L. The grp78 promoter of Neurospora crassa: Constitutive, stress and differentiation-dependent protein-binding patterns. Curr. Genet. 2001, 39, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Orlean, P.; Albright, C.; Robbins, P.W. Cloning and sequencing of the yeast gene for dolichol phosphate mannose synthase, an essential protein. J. Biol. Chem. 1988, 263, 17499–17507. [Google Scholar] [CrossRef]
- Bowman, B.J.; Draskovic, M.; Freitag, M.; Bowman, E.J. Structure and distribution of organelles and cellular location of calcium transporters in Neurospora crassa. Eukaryot. Cell 2009, 8, 1845–1855. [Google Scholar] [CrossRef] [Green Version]
- Floch, A.G.; Tareste, D.; Fuchs, P.F.; Chadrin, A.; Naciri, I.; Leger, T.; Schlenstedt, G.; Palancade, B.; Doye, V. Nuclear pore targeting of the yeast Pom33 nucleoporin depends on karyopherin and lipid binding. J. Cell Sci. 2015, 128, 305–316. [Google Scholar] [CrossRef] [Green Version]
- Sakabe, I.; Hu, R.; Jin, L.; Clarke, R.; Kasid, U.N. TMEM33: A new stress-inducible endoplasmic reticulum transmembrane protein and modulator of the unfolded protein response signaling. Breast Cancer Res. Treat. 2015, 153, 285–297. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Vjestica, A.; Oliferenko, S. The cortical ER network limits the permissive zone for actomyosin ring assembly. Curr. Biol. 2010, 20, 1029–1034. [Google Scholar] [CrossRef] [Green Version]
- Doye, V.; Hurt, E. From nucleoporins to nuclear pore complexes. Curr. Opin. Cell Biol. 1997, 9, 401–411. [Google Scholar] [CrossRef]
- Dawson, T.R.; Lazarus, M.D.; Hetzer, M.W.; Wente, S.R. ER membrane-bending proteins are necessary for de novo nuclear pore formation. J. Cell Biol. 2009, 184, 659–675. [Google Scholar] [CrossRef] [Green Version]
- Oertle, T.; Klinger, M.; Stuermer, C.A.; Schwab, M.E. A reticular rhapsody: Phylogenic evolution and nomenclature of the RTN/Nogo gene family. FASEB J. 2003, 17, 1238–1247. [Google Scholar] [CrossRef] [Green Version]
- Voeltz, G.K.; Prinz, W.A.; Shibata, Y.; Rist, J.M.; Rapoport, T.A. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell 2006, 124, 573–586. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Shibata, Y.; Voss, C.; Shemesh, T.; Li, Z.; Coughlin, M.; Kozlov, M.M.; Rapoport, T.A.; Prinz, W.A. Membrane proteins of the endoplasmic reticulum induce high-curvature tubules. Science 2008, 319, 1247–1250. [Google Scholar] [CrossRef] [PubMed]
- Shibata, Y.; Voss, C.; Rist, J.M.; Hu, J.; Rapoport, T.A.; Prinz, W.A.; Voeltz, G.K. The reticulon and DP1/Yop1p proteins form immobile oligomers in the tubular endoplasmic reticulum. J. Biol. Chem. 2008, 283, 18892–18904. [Google Scholar] [CrossRef] [Green Version]
- Anderson, D.J.; Hetzer, M.W. Reshaping of the endoplasmic reticulum limits the rate for nuclear envelope formation. J. Cell Biol. 2008, 182, 911–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiseleva, E.; Morozova, K.N.; Voeltz, G.K.; Allen, T.D.; Goldberg, M.W. Reticulon 4a/NogoA locates to regions of high membrane curvature and may have a role in nuclear envelope growth. J. Struct. Biol. 2007, 160, 224–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Shibata, Y.; Zhu, P.P.; Voss, C.; Rismanchi, N.; Prinz, W.A.; Rapoport, T.A.; Blackstone, C. A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell 2009, 138, 549–561. [Google Scholar] [CrossRef] [Green Version]
- Orso, G.; Pendin, D.; Liu, S.; Tosetto, J.; Moss, T.J.; Faust, J.E.; Micaroni, M.; Egorova, A.; Martinuzzi, A.; McNew, J.A. Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature 2009, 460, 978–983. [Google Scholar] [CrossRef]
- Liu, T.Y.; Bian, X.; Sun, S.; Hu, X.; Klemm, R.W.; Prinz, W.A.; Rapoport, T.A.; Hu, J. Lipid interaction of the C terminus and association of the transmembrane segments facilitate atlastin-mediated homotypic endoplasmic reticulum fusion. Proc. Natl. Acad. Sci. USA 2012, 109, E2146–E2154. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Novick, P.; Ferro-Novick, S. ER structure and function. Curr. Opin. Cell Biol. 2013, 25, 428–433. [Google Scholar] [CrossRef] [Green Version]
- Antebi, A.; Fink, G.R. The yeast Ca (2+)-ATPase homologue, PMR1, is required for normal Golgi function and localizes in a novel Golgi-like distribution. Mol. Biol. Cell 1992, 3, 633–654. [Google Scholar] [CrossRef] [Green Version]
- Dürr, G.; Strayle, J.; Plemper, R.; Elbs, S.; Klee, S.K.; Catty, P.; Wolf, D.H.; Rudolph, H.K. The medial-Golgi ion pump Pmr1 supplies the yeast secretory pathway with Ca2+ and Mn2+ required for glycosylation, sorting, and endoplasmic reticulum-associated protein degradation. Mol. Biol. Cell 1998, 9, 1149–1162. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, H.K.; Antebi, A.; Fink, G.R.; Buckley, C.M.; Dorman, T.E.; LeVitre, J.; Davidow, L.S.; Mao, J.; Moir, D.T. The yeast secretory pathway is perturbed by mutations in PMR1, a member of a Ca2+ ATPase family. Cell 1989, 58, 133–145. [Google Scholar] [CrossRef]
- Manford, A.G.; Stefan, C.J.; Yuan, H.L.; Macgurn, J.A.; Emr, S.D. ER-to-plasma membrane tethering proteins regulate cell signaling and ER morphology. Dev. Cell 2012, 23, 1129–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, P.C.; Bharat, T.A.M.; Wozny, M.R.; Boulanger, J.; Miller, E.A.; Kukulski, W. Tricalbins Contribute to Cellular Lipid Flux and Form Curved ER-PM Contacts that Are Bridged by Rod-Shaped Structures. Dev. Cell 2019, 51, 488–502.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collado, J.; Kalemanov, M.; Campelo, F.; Bourgoint, C.; Thomas, F.; Loewith, R.; Martinez-Sanchez, A.; Baumeister, W.; Stefan, C.J.; Fernandez-Busnadiego, R. Tricalbin-Mediated Contact Sites Control ER Curvature to Maintain Plasma Membrane Integrity. Dev. Cell 2019, 51, 476.e477–487.e477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, V.; Blank-Landeshammer, B.; Muntjes, K.; Marker, R.; Teichert, I.; Feldbrugge, M.; Sickmann, A.; Kück, U. The STRIPAK signaling complex regulates dephosphorylation of GUL1, an RNA-binding protein that shuttles on endosomes. PLoS Genet. 2020, 16, 1–32. [Google Scholar] [CrossRef]
- Costanzo, M.; VanderSluis, B.; Koch, E.N.; Baryshnikova, A.; Pons, C.; Tan, G.; Wang, W.; Usaj, M.; Hanchard, J.; Lee, S.D.; et al. A global genetic interaction network maps a wiring diagram of cellular function. Science 2016, 353, 1–34. [Google Scholar] [CrossRef]
- Deshaies, R.J.; Schekman, R. A yeast mutant defective at an early stage in import of secretory protein precursors into the endoplasmic reticulum. J. Cell Biol. 1987, 105, 633–645. [Google Scholar] [CrossRef]
- Plath, K.; Wilkinson, B.M.; Stirling, C.J.; Rapoport, T.A. Interactions between Sec complex and prepro-alpha-factor during posttranslational protein transport into the endoplasmic reticulum. Mol. Biol. Cell 2004, 15, 1–10. [Google Scholar] [CrossRef]
- Strahl-Bolsinger, S.; Immervoll, T.; Deutzmann, R.; Tanner, W. PMT1, the gene for a key enzyme of protein O-glycosylation in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1993, 90, 8164–8168. [Google Scholar] [CrossRef] [Green Version]
- Gentzsch, M.; Tanner, W. The PMT gene family: Protein O-glycosylation in Saccharomyces cerevisiae is vital. EMBO J. 1996, 15, 5752–5759. [Google Scholar] [CrossRef]
- Gentzsch, M.; Immervoll, T.; Tanner, W. Protein O-glycosylation in Saccharomyces cerevisiae: The protein O-mannosyltransferases Pmt1p and Pmt2p function as heterodimer. FEBS Lett. 1995, 377, 128–130. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, R.; Riezman, H. Differential ER exit in yeast and mammalian cells. Curr. Opin. Cell Biol. 2004, 16, 350–355. [Google Scholar] [CrossRef]
- Weichert, M.; Lichius, A.; Priegnitz, B.E.; Brandt, U.; Gottschalk, J.; Nawrath, T.; Groenhagen, U.; Read, N.D.; Schulz, S.; Fleissner, A. Accumulation of specific sterol precursors targets a MAP kinase cascade mediating cell-cell recognition and fusion. Proc. Natl. Acad. Sci. USA 2016, 113, 11877–11882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichert, M.; Herzog, S.; Robson, S.A.; Brandt, R.; Priegnitz, B.E.; Brandt, U.; Schulz, S.; Fleissner, A. Plasma Membrane Fusion Is Specifically Impacted by the Molecular Structure of Membrane Sterols During Vegetative Development of Neurospora crassa. Genetics 2020, 216, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, 442–450. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Genotype | References |
|---|---|---|
| Escherichia coli | ||
| MACH1 | ΔrecA1398, endA1, tonA, Φ80ΔlacM15, ΔlacX74, hsdR, (rK-mK+) | Invitrogen |
| Saccharomyces cerevisiae | ||
| PJ69-4A | MATa, trp1-901, leu2-3, 112, ura3-52, his3-200, gal4Δ, gal80Δ, LYS2::GAL1-HIS3, GAL2-ADE2, met2::GAL7-lacZ | [35] |
| Sordaria macrospora | ||
| DSM997 | wild type (wt) | DSMZ |
| S23442 | mutation in fus1-1 gene, brownish ascospores, fertile | [41] |
| Δku70 | Δku70::natR, fertile | [42] |
| Δsci1 | Δsci1::hygR, ssi, sterile | [14] |
| Δsci1::nca1-TagRFP-Tect | ectopic integration of p5′nca1-TagRFP-T _nat into Δsci1; hygR, natR pt, sterile, Pnca1::nca1::TagRFP-T::TtrpC | Reschka and Pöggeler (unpublished) |
| wt::egfpect | ectopic integration of p1783-1 into DSM997; hygR, ssi, fertile, Pgpd::egfp::TtrpC | [43] |
| wt::TagRFP-Tect | ectopic integration of pTagRFP-T_nat into DSM997; natR, ssi, fertile, Pccg1::TagRFP-T::TtrpC | [44] |
| wt::GH2Aect | ectopic integration of pGH2A into DSM997; hygR, pt, fertile, Pgpd::hh2a::egfp::TtrpC | Reschka and Pöggeler (unpublished) |
| fus::RH2Bect | ectopic integration of pRH2B in S23442; hygR, pt, fertile, Pgpd::hh2b::tdTomato::TtrpC | Reschka and Pöggeler (unpublished) |
| wt::nca1-TagRFP-Tect | ectopic integration of p5′nca1-TagRFP-T_hyg into DSM997; hygR, pt, fertile, Pnca1::nca1::TagRFP-T::TtrpC | Werner and Pöggeler (unpublished) |
| Δku80 | Δku80::hygR, ssi, fertile | This study |
| Δpom33 | Δpom33::natR, ssi, fertile | This study |
| wt::pom33-TagRFP-Tect | ectopic integration of p5′pom33-TagRFP-T_nat into DSM997; natR, ssi, fertile, Ppom33::pom33:: TagRFP-T::TtrpC | This study |
| wt::pom33-TagRFP-Tect | ectopic integration of p5′pom33-TagRFP-T_hyg into DSM997; hygR, pt, fertile, Ppom33::pom33:: TagRFP-T::TtrpC | This study |
| wt::pom33-egfpect | ectopic integration of p5′pom33-egfp into DSM997; natR, ssi, fertile, Ppom33::pom33::egfpt::TtrpC | This study |
| wt::pom33-TagRFP-Tect + pom152-egfpect | ectopic integration of p5′pom33-TagRFP-T_hyg and pSmPOM152GFP into DSM997; hygR, natR, pt, fertile, Ppom33::pom33:: TagRFP-T::TtrpC; Pccg1::pom152::egfp::TtrpC | This study |
| wt::pom33-TagRFP-Tect + sci1-egfpect | ectopic integration of p5′pom33-TagRFP-T_hyg and p5′sci1gfp_nat into DSM997; hygR, natR, pt, fertile, Ppom33::pom33:: TagRFP-T::TtrpC; Psci1::sci1::egfp::TtrpC | This study |
| Δpom33::pom33-TagRFP-Tect | ectopic integration of p5′pom33-TagRFP-T_hyg into Δpom33; hygR, natR ssi, fertile, Ppom33::pom33:: TagRFP-T::TtrpC | This study |
| Δpom33::RH2Bect | ectopic integration of pRH2B into Δpom33; hygR, natR pt, fertile, Pgpd::hh2b::tdTomato::TtrpC | This study |
| Δpom33:: nca1-TagRFP-Tect | ectopic integration of p5′nca1-TagRFP-T_hyg into Δpom33; hygR, natR pt, fertile, Pnca1::nca1::TagRFP-T::TtrpC | This study |
| Δpom33::sci1-egfpect | ectopic integration of p5′sci1-egfp_hyg into Δpom33; hygR, natR pt, fertile, Psci1::sci1::egfp::TtrpC | This study |
| Δpom33::pro11-egfpect | ectopic integration of pPRO11-GFP_hyg into Δpom33; hygR, natR pt, fertile, Pccg1::HA::pro11::egfp::TtrpC | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Groth, A.; Schmitt, K.; Valerius, O.; Herzog, B.; Pöggeler, S. Analysis of the Putative Nucleoporin POM33 in the Filamentous Fungus Sordaria macrospora. J. Fungi 2021, 7, 682. https://doi.org/10.3390/jof7090682
Groth A, Schmitt K, Valerius O, Herzog B, Pöggeler S. Analysis of the Putative Nucleoporin POM33 in the Filamentous Fungus Sordaria macrospora. Journal of Fungi. 2021; 7(9):682. https://doi.org/10.3390/jof7090682
Chicago/Turabian StyleGroth, Anika, Kerstin Schmitt, Oliver Valerius, Britta Herzog, and Stefanie Pöggeler. 2021. "Analysis of the Putative Nucleoporin POM33 in the Filamentous Fungus Sordaria macrospora" Journal of Fungi 7, no. 9: 682. https://doi.org/10.3390/jof7090682
APA StyleGroth, A., Schmitt, K., Valerius, O., Herzog, B., & Pöggeler, S. (2021). Analysis of the Putative Nucleoporin POM33 in the Filamentous Fungus Sordaria macrospora. Journal of Fungi, 7(9), 682. https://doi.org/10.3390/jof7090682