
ITS2 Sequencing and Targeted Meta-Proteomics of Infant Gut Mycobiome Reveal the Functional Role of Rhodotorula sp. during Atopic Dermatitis Manifestation

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Microbial DNA Extraction of Stool Samples
2.3. Amplification and Sequencing of the Internal Transcribed Spacer 2 (ITS2) Region
2.4. Mycobiome Determination from ITS2 Region Sequencing
2.5. Quantitative Real-Time PCR (qPCR) Analysis of Five Distinct Genera
2.5.1. Primer Design
2.5.2. Plasmid Construction and Standard Curve Creation
2.5.3. Quantification of Targeted Genera in Each Sample by qPCR
2.6. Meta-Proteomics Analysis of Five Distinct Genera
2.6.1. Sample Preparation
2.6.2. Liquid Chromatography–Tandem Mass Spectrophotometry (LC–MS/MS)
2.6.3. Quantification of Detected Protein Using Bioinformatics Tools
2.6.4. Determination of Meta-Proteomics Activity of Five Distinct Genera
2.7. Statistical Analysis
3. Results
3.1. Relation of Infant Characteristics
3.2. Diversity of Infant Gut Mycobiome
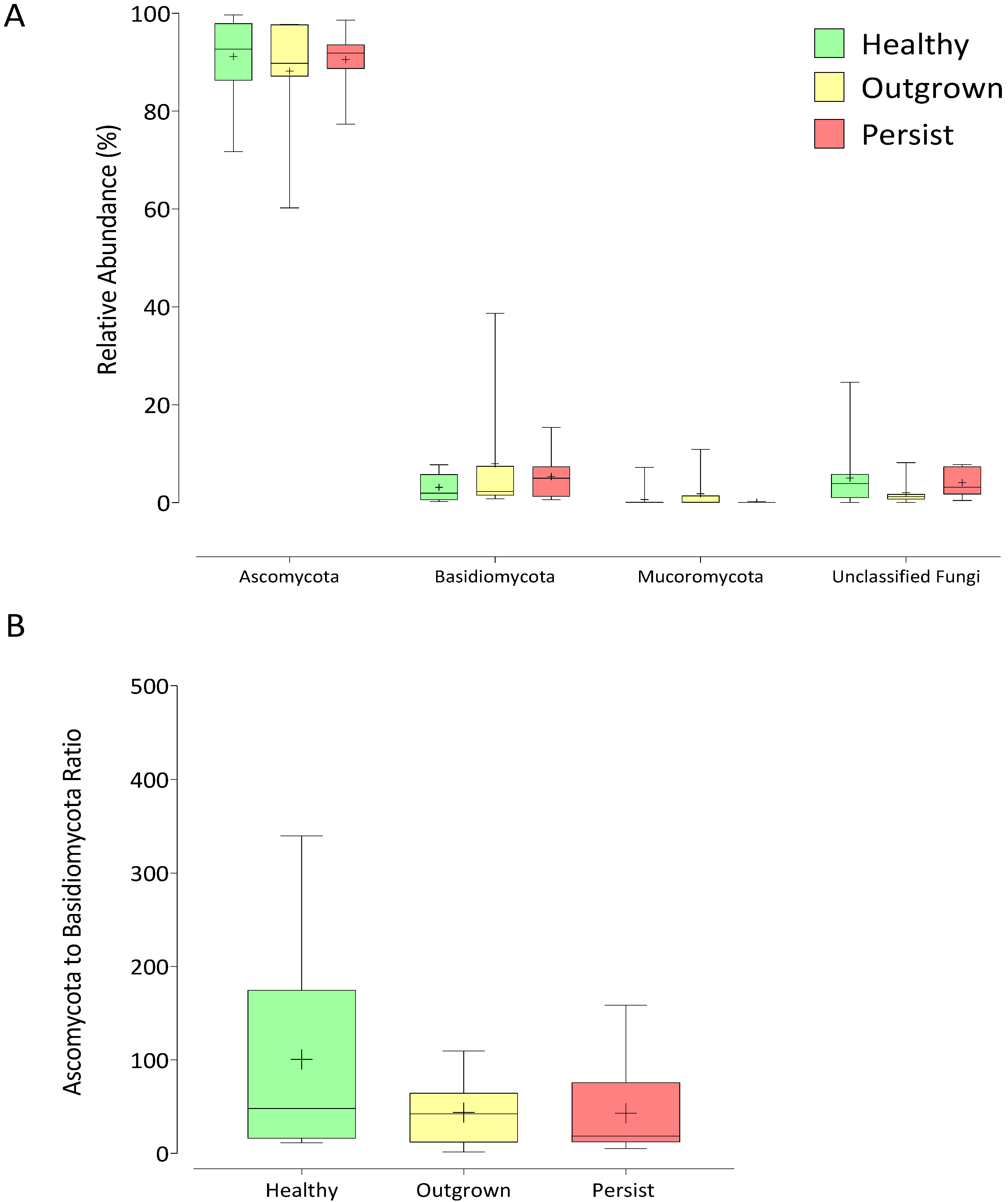
3.3. Distinctive Gut Mycobiome Profile between Healthy, Outgrown, and Persisted Infant Groups
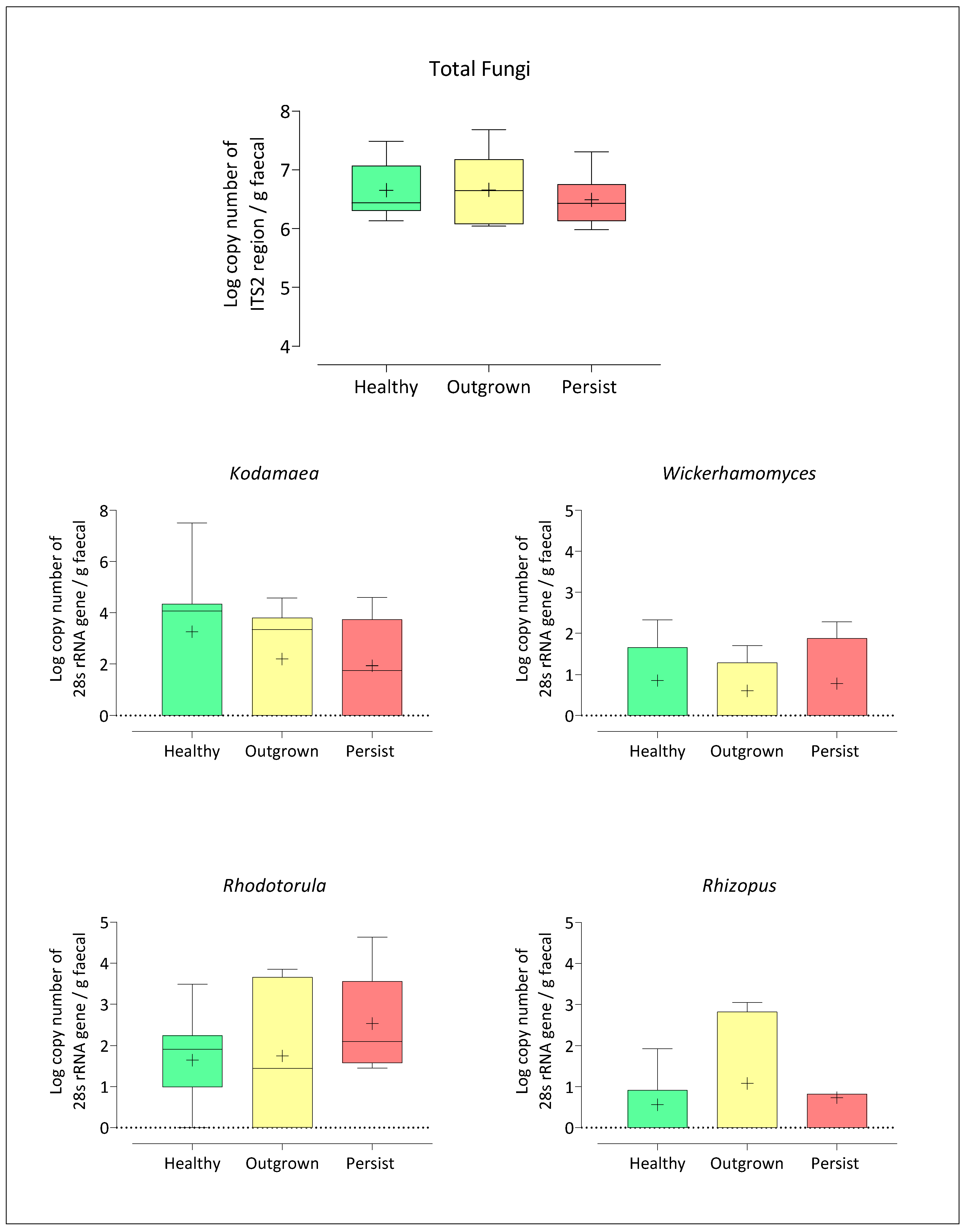
3.4. Quantification of Five Target Fungi from Healthy, Outgrown, and Persisted Infant Groups
3.5. Targeted Meta-Proteomics of Significantly Different Genera
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pearce, N.; Aït-Khaled, N.; Beasley, R.; Mallol, J.; Keil, U.; Mitchell, E.; Robertson, C. Worldwide trends in the prevalence of asthma symptoms: Phase III of the International Study of Asthma and Allergies in Childhood (ISAAC). Thorax 2007, 62, 758–766. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, D.; Sjöberg, O.; Foucard, T. Development of allergies and asthma in infants and young children with atopic dermatitis—A prospective follow-up to 7 years of age. Allergy 2000, 55, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Tsakok, T.; Marrs, T.; Mohsin, M.; Baron, S.; du Toit, G.; Till, S.; Flohr, C. Does atopic dermatitis cause food allergy? A systematic review. J. Allergy Clin. Immunol. 2016, 137, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maliyar, K.; Sibbald, C.; Pope, E.; Gary Sibbald, R. Diagnosis and Management of Atopic Dermatitis: A Review. Adv. Ski. Wound Care 2018, 31, 538–550. [Google Scholar] [CrossRef]
- Sugita, K.; Akdis, C.A. Recent developments and advances in atopic dermatitis and food allergy. Allergol. Int. 2020, 69, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Guttman-Yassky, E.; Diaz, A.; Pavel, A.B.; Fernandes, M.; Lefferdink, R.; Erickson, T.; Canter, T.; Rangel, S.; Peng, X.; Li, R.; et al. Use of Tape Strips to Detect Immune and Barrier Abnormalities in the Skin of Children With Early-Onset Atopic Dermatitis. JAMA Dermatol. 2019, 155, 1358–1370. [Google Scholar] [CrossRef]
- Brunner, P.M. Early immunologic changes during the onset of atopic dermatitis. Ann. Allergy Asthma Immunol. 2019, 123, 152–157. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Kim, H.S. Microbiome of the Skin and Gut in Atopic Dermatitis (AD): Understanding the Pathophysiology and Finding Novel Management Strategies. J. Clin. Med. 2019, 8, 444. [Google Scholar] [CrossRef] [Green Version]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Mancabelli, L.; Milani, C.; Lugli, G.A.; Turroni, F.; Cocconi, D.; van Sinderen, D.; Ventura, M. Identification of universal gut microbial biomarkers of common human intestinal diseases by meta-analysis. FEMS Microbiol. Ecol. 2017, 93, fix153. [Google Scholar] [CrossRef] [Green Version]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [Green Version]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef] [Green Version]
- Nylund, L.; Nermes, M.; Isolauri, E.; Salminen, S.; de Vos, W.M.; Satokari, R. Severity of atopic disease inversely correlates with intestinal microbiota diversity and butyrate-producing bacteria. Allergy 2015, 70, 241–244. [Google Scholar] [CrossRef]
- Song, H.; Yoo, Y.; Hwang, J.; Na, Y.C.; Kim, H.S. Faecalibacterium prausnitzii subspecies—Level dysbiosis in the human gut microbiome underlying atopic dermatitis. J. Allergy Clin. Immunol. 2016, 137, 852–860. [Google Scholar] [CrossRef] [Green Version]
- Ward, T.L.; Dominguez-Bello, M.G.; Heisel, T.; Al-Ghalith, G.; Knights, D.; Gale, C.A. Development of the Human Mycobiome over the First Month of Life and across Body Sites. mSystems 2018, 3, e00140-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, C.J.; Nelson, A.; Scribbins, D.; Marrs, E.C.L.; Lanyon, C.; Perry, J.D.; Embleton, N.D.; Cummings, S.P.; Berrington, J.E. Bacterial and fungal viability in the preterm gut: NEC and sepsis. Arch. Dis. Child. Fetal Neonatal Ed. 2013, 98, F298–F303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliev, I.D.; Funari, V.A.; Taylor, K.D.; Nguyen, Q.; Reyes, C.N.; Strom, S.P.; Brown, J.; Becker, C.A.; Fleshner, P.R.; Dubinsky, M.; et al. Interactions Between Commensal Fungi and the C-Type Lectin Receptor Dectin-1 Influence Colitis. Science 2012, 336, 1314–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laforest-Lapointe, I.; Arrieta, M.C. Patterns of Early-Life Gut Microbial Colonization during Human Immune Development: An Ecological Perspective. Front. Immunol. 2017, 8, 788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [Green Version]
- Nash, A.K.; Auchtung, T.A.; Wong, M.C.; Smith, D.P.; Gesell, J.R.; Ross, M.C.; Stewart, C.J.; Metcalf, G.A.; Muzny, D.M.; Gibbs, R.A.; et al. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 2017, 5, 153. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Lee, E.; Park, Y.M.; Hong, S.-J. Microbiome in the Gut-Skin Axis in Atopic Dermatitis. Allergy Asthma Immunol. Res. 2018, 10, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Lemoinne, S.; Kemgang, A.; Belkacem, K.B.; Straube, M.; Jegou, S.; Corpechot, C.; Network, S.A.I.; Chazouillères, O.; Housset, C.; Sokol, H. Fungi participate in the dysbiosis of gut microbiota in patients with primary sclerosing cholangitis. Gut 2020, 69, 92–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limon, J.J.; Tang, J.; Li, D.; Wolf, A.J.; Michelsen, K.S.; Funari, V.; Gargus, M.; Nguyen, C.; Sharma, P.; Maymi, V.I.; et al. Malassezia Is Associated with Crohn’s Disease and Exacerbates Colitis in Mouse Models. Cell Host Microbe 2019, 25, 377–388.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrieta, M.C.; Arévalo, A.; Stiemsma, L.; Dimitriu, P.; Chico, M.E.; Loor, S.; Vaca, M.; Boutin, R.C.; Morien, E.; Jin, M.; et al. Associations between infant fungal and bacterial dysbiosis and childhood atopic wheeze in a nonindustrialized setting. J. Allergy Clin. Immunol. 2018, 142, 424–434.e10. [Google Scholar] [CrossRef] [Green Version]
- Petriz, B.A.; Franco, O.L. Metaproteomics as a Complementary Approach to Gut Microbiota in Health and Disease. Front. Chem. 2017, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Raethong, N.; Nakphaichit, M.; Suratannon, N.; Sathitkowitchai, W.; Weerapakorn, W.; Keawsompong, S.; Vongsangnak, W. Analysis of Human Gut Microbiome: Taxonomy and Metabolic Functions in Thai Adults. Genes 2021, 12, 331. [Google Scholar] [CrossRef]
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Kingkaw, A.; Nakphaichit, M.; Suratannon, N.; Nitisinprasert, S.; Wongoutong, C.; Chatchatee, P.; Krobthong, S.; Charoenlappanit, S.; Roytrakul, S.; Vongsangnak, W. Analysis of the infant gut microbiome reveals metabolic functional roles associated with healthy infants and infants with atopic dermatitis using metaproteomics. PeerJ 2020, 8, e9988. [Google Scholar] [CrossRef] [PubMed]
- Kolmeder, C.A.; Salojärvi, J.; Ritari, J.; De Been, M.; Raes, J.; Falony, G.; Vieira-Silva, S.; Kekkonen, R.A.; Corthals, G.L.; Palva, A.; et al. Faecal Metaproteomic Analysis Reveals a Personalized and Stable Functional Microbiome and Limited Effects of a Probiotic Intervention in Adults. PLoS ONE 2016, 11, e0153294. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; Ren, H.; Lu, Y.; Fang, C.; Hou, G.; Yang, Z.; Chen, B.; Yang, F.; Zhao, Y.; Shi, Z.; et al. Distinct gut metagenomics and metaproteomics signatures in prediabetics and treatment-naïve type 2 diabetics. EBioMedicine 2019, 47, 373–383. [Google Scholar] [CrossRef]
- Eichenfield, L.F.; Ahluwalia, J.; Waldman, A.; Borok, J.; Udkoff, J.; Boguniewicz, M. Current guidelines for the evaluation and management of atopic dermatitis: A comparison of the Joint Task Force Practice Parameter and American Academy of Dermatology guidelines. J. Allergy Clin. Immunol. 2017, 139, S49–S57. [Google Scholar] [CrossRef] [Green Version]
- Costea, P.I.; Zeller, G.; Sunagawa, S.; Pelletier, E.; Alberti, A.; Levenez, F.; Tramontano, M.; Driessen, M.; Hercog, R.; Jung, F.E.; et al. Towards standards for human fecal sample processing in metagenomic studies. Nat. Biotechnol. 2017, 35, 1069–1076. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.J.W.T.; Taylor, J. Amplification And Direct Sequencing of Fungal Ribosomal Rna Genes for Phylogenetics. In PCR Protocols; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar]
- BBMap. Available online: http://sourceforge.net/projects/bbmap/ (accessed on 10 May 2020).
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Edgar, R.C. SINTAX: A simple non-Bayesian taxonomy classifier for 16S and ITS sequences. bioRxiv 2016, 074161. [Google Scholar] [CrossRef] [Green Version]
- Abarenkov, K.; Allan, Z.; Piirmann, T.; Pöhönen, R.; Ivanov, F.; Nilsson, R.H.; Kõljalg, U. UNITE USEARCH/UTAX release for Fungi. Version 04.02.2020. UNITE Community 2020. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [Green Version]
- Froger, A.; Hall, J.E. Transformation of plasmid DNA into E. coli using the heat shock method. J. Vis. Exp. 2007, 6, 253. [Google Scholar] [CrossRef] [PubMed]
- Losuwannarak, N.; Maiuthed, A.; Kitkumthorn, N.; Leelahavanichkul, A.; Roytrakul, S.; Chanvorachote, P. Gigantol Targets Cancer Stem Cells and Destabilizes Tumors via the Suppression of the PI3K/AKT and JAK/STAT Pathways in Ectopic Lung Cancer Xenografts. Cancers 2019, 11, 2032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Bardou, P.; Mariette, J.; Escudié, F.; Djemiel, C.; Klopp, C. Jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Krieger, A.; Yekutieli, D. Adaptive linear step-up false discovery rate controlling procedures. Biometrika 2006, 93, 491–507. [Google Scholar] [CrossRef]
- Novicki, T.J.; LaFe, K.; Bui, L.; Bui, U.; Geise, R.; Marr, K.; Cookson, B.T. Genetic Diversity among Clinical Isolates of Acremonium Strictum Determined during an Investigation of a Fatal Mycosis. J. Clin. Microbiol. 2003, 41, 2623–2628. [Google Scholar] [CrossRef] [Green Version]
- Ott, S.J.; Kühbacher, T.; Musfeldt, M.; Rosenstiel, P.; Hellmig, S.; Rehman, A.; Drews, O.; Weichert, W.; Timmis, K.N.; Schreiber, S. Fungi and inflammatory bowel diseases: Alterations of composition and diversity. Scand. J. Gastroenterol. 2008, 43, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Arunasri, K.; Mahesh, M.; Sai Prashanthi, G.; Jayasudha, R.; Kalyana Chakravarthy, S.; Tyagi, M.; Pappuru, R.R.; Shivaji, S. Mycobiome changes in the vitreous of post fever retinitis patients. PLoS ONE 2020, 15, e0242138. [Google Scholar] [CrossRef] [PubMed]
- Anandakumar, A.; Pellino, G.; Tekkis, P.; Kontovounisios, C. Fungal microbiome in colorectal cancer: A systematic review. Updates Surg. 2019, 71, 625–630. [Google Scholar] [CrossRef]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2017, 66, 1039. [Google Scholar] [CrossRef] [Green Version]
- García-Hernández, Y.; Rodríguez, Z.; Brandão, L.R.; Rosa, C.A.; Nicoli, J.R.; Iglesias, A.E.; Peréz-Sanchez, T.; Salabarría, R.B.; Halaihel, N. Identification and in vitro screening of avian yeasts for use as probiotic. Res. Vet. Sci. 2012, 93, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Hjortmo, S.B.; Hellström, A.M.; Andlid, T.A. Production of folates by yeasts in Tanzanian fermented togwa. FEMS Yeast Res. 2008, 8, 781–787. [Google Scholar] [CrossRef] [Green Version]
- Fincher, R.M.; Fisher, J.F.; Lovell, R.D.; Newman, C.L.; Espinel-Ingroff, A.; Shadomy, H.J. Infection due to the fungus Acremonium (cephalosporium). Medicine 1991, 70, 398–409. [Google Scholar] [CrossRef]
- Nucci, M.; Anaissie, E.J. Chapter 13—Hyalohyphomycosis. In Clinical Mycology, 2nd ed.; Anaissie, E.J., McGinnis, M.R., Pfaller, M.A., Eds.; Churchill Livingstone: Edinburgh, UK, 2009; pp. 309–327. [Google Scholar]
- Moreno-Sabater, A.; Autaa, G.; Sterlin, D.; Jerbi, A.; Villette, R.; Holm, J.B.; Parizot, C.; Selim, S.; Senghor, Y.; Ghillani-Dalbin, P.; et al. Systemic anti-commensal response to fungi analyzed by flow cytometry is related to gut mycobiome ecology. Microbiome 2020, 8, 159. [Google Scholar] [CrossRef]
- Tuon, F.F.; Costa, S.F. Rhodotorula infection. A systematic review of 128 cases from literature. Rev. Iberoam. Micol. 2008, 25, 135–140. [Google Scholar] [CrossRef]
- Goravey, W.; Ali, G.A.; Abid, F.; Ibrahim, E.B.; Al Maslamani, M.A.; Abdel Hadi, H. Central line-associated Rhodotorula mucilaginosa fungemia in an immunocompetent host: Case report and review of the literature. Clin. Case Rep. 2021, 9, 2158–2161. [Google Scholar] [CrossRef] [PubMed]
- Mims, T.S.; Al Abdallah, Q.; Stewart, J.D.; Watts, S.P.; White, C.T.; Rousselle, T.V.; Gosain, A.; Bajwa, A.; Han, J.C.; Willis, K.A.; et al. The gut mycobiome of healthy mice is shaped by the environment and correlates with metabolic outcomes in response to diet. Commun. Biol. 2021, 4, 281. [Google Scholar] [CrossRef] [PubMed]
- Swift, C.L.; Louie, K.B.; Bowen, B.P.; Olson, H.M.; Purvine, S.O.; Salamov, A.; Mondo, S.J.; Solomon, K.V.; Wright, A.T.; Northen, T.R.; et al. Anaerobic gut fungi are an untapped reservoir of natural products. Proc. Natl. Acad. Sci. USA 2021, 118, e2019855118. [Google Scholar] [CrossRef] [PubMed]
- Buldain, I.; Pellon, A.; Zaldibar, B.; Antoran, A.; Martin-Souto, L.; Aparicio-Fernandez, L.; Areitio, M.; Mayayo, E.; Rementeria, A.; Hernando, F.L.; et al. Study of Humoral Responses against Lomentospora/Scedosporium spp. and Aspergillus fumigatus to Identify L. prolificans Antigens of Interest for Diagnosis and Treatment. Vaccines 2019, 7, 212. [Google Scholar] [CrossRef] [Green Version]
- Buldain, I.; Martin-Souto, L.; Antoran, A.; Areitio, M.; Aparicio-Fernandez, L.; Rementeria, A.; Hernando, F.L.; Ramirez-Garcia, A. The Host Immune Response to Scedosporium/Lomentospora. J. Fungi 2021, 7, 75. [Google Scholar] [CrossRef]
- Ferguson, A.C.; Salinas, F.A. Elevated IgG immune complexes in children with atopic eczema. J. Allergy Clin. Immunol. 1984, 74, 678–682. [Google Scholar] [CrossRef]
- Totté, J.E.E.; Pardo, L.M.; Fieten, K.B.; de Wit, J.; de Boer, D.V.; van Wamel, W.J.; Pasmans, S.G.M.A. IgG response against Staphylococcus aureus is associated with severe atopic dermatitis in children. Br. J. Dermatol. 2018, 179, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Bublin, M.; Eiwegger, T.; Breiteneder, H. Do lipids influence the allergic sensitization process? J. Allergy Clin. Immunol. 2014, 134, 521–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiaro, T.R.; Soto, R.; Stephens, W.Z.; Kubinak, J.L.; Petersen, C.; Gogokhia, L.; Bell, R.; Delgado, J.C.; Cox, J.; Voth, W.; et al. A member of the gut mycobiota modulates host purine metabolism exacerbating colitis in mice. Sci. Transl. Med. 2017, 9, eaaf9044. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay | Primer Sequence (5′ to 3′) | Annealing Temp (°C) | Size (bp) | Reference |
|---|---|---|---|---|
| Total fungi | ITS3: GCATCGATGAAGAACGCAGC ITS4-NGS: TCCTCCGCTTATTGATATGC | 56 | Variable | [2] |
| Rhodotorula sp. | Rho-F: TCCGGGATGGATAATGGTAGAGAG Rho-R: CCTAAGTGTGAAGAGGTCGAAAC | 54 | 129 | This study |
| Wickerhamomyces sp. | Wic-F: TGGGTTTGGCTCTTGTCTATG Wic-R: CGACTCTTCGATAGCACCTTAC | 54 | 104 | This study |
| Rhizopus sp. | Rz-F: CTGTAGAAGTGTTTTCCAGGCAAGCCG Rz-R: CCCAAACAACCTGACTCTT | 56 | 133 | This study |
| Kodamaea sp. | Kod-F: CGACCGTGTCTATGTTCCTTGGAAC Kod-R: ACTCGTCGAGAGCGCCTTAT | 56 | 95 | This study |
| Acremonium sp. | Acre-F: GAGACCTTAACCTACTAACTAGCCCGTAT Acre-R: GTTATTGCCTCAAACTTCCCTCGGCTTG Probe: GCTTTGGCAGTACGCTGGCTTCTTAG | 54 | 98 | This study |
| Parameter | Healthy | Outgrown | Persisted | p-Value |
|---|---|---|---|---|
| Sampling point (month) | 10.59 ± 1.54 | 10.29 ± 1.6 | 10.2 ± 1.55 | 0.79 |
| Sex (female/male) | 6/11 | 3/4 | 3/7 | 0.7581 |
| Gestational age (month) | 38.45 ± 2.02 | 39.14 ± 1.86 | 38.7 ± 2 | 0.5167 |
| Birth delivery mode (v/cs) | 12/5 | 7/0 | 5/5 | 0.0957 |
| Birth weight (g) | 3207 ± 215.2 | 3057 ± 239.5 | 2967 ± 411.1 | 0.2113 |
| Breastfeed duration (month) | 8.96 ± 4.27 | 6.71 ± 5.02 | 7.85 ± 4.44 | 0.4723 |
| Age of solid food introduction (month) | 5.64 ± 0.92 | 5.43 ± 0.98 | 5.4 ± 0.84 | 0.7512 |
| Severity of atopic dermatitis (mild/moderate) | − | 2/7 | 2/10 | 0.4593 |
| Fungi | Healthy (n = 11) | Outgrown (n = 7) | Persisted (n = 5) | p-Value |
|---|---|---|---|---|
| Total protein | 1341 | 858 | 1485 | |
| Average protein per sample | 122 | 143 | 297 | |
| Acremonium sp. | 339.6 ± 281 b | 160.9 ± 261.5 b | 889.5 ± 133.2 a | 0.0023 |
| Kodamaea sp. | 20.21 ± 13.28 a | 10.64 ± 12.26 a | 30.42 ± 3.43 a | 0.0784 |
| Wickerhamomyces sp. | 355.5 ± 294.5 b | 168 ± 270.9 b | 956.4 ± 151.4 a | 0.0021 |
| Rhodotorula sp. | 368.9 ± 299.7 b | 178.5 ± 292.5 b | 971.5 ± 143.6 a | 0.0026 |
| Rhizopus sp. | 351.4 ± 287 b | 162.4 ± 268.4 b | 940.8 ± 138.8 a | 0.0022 |
| Accession Number | Organism | Functional Category | Sub-Functional Category | Protein Name |
|---|---|---|---|---|
| A0A086TGW8 | Acremonium sp. | Metabolism | Amino acid metabolism | 4-hydroxyphenylpyruvate dioxygenase |
| A0A086T8N3 | Acremonium sp. | Metabolism | Carbohydrate metabolism | 1,3-beta-glucan synthase |
| A0A086SWD1 | Acremonium sp. | Organismal systems | Endocrine system | exocyst complex component 7 |
| A0A086TCH4 | Acremonium sp. | Genetic information processing | Folding, sorting, and degradation | ubiquitin-conjugating enzyme E2 |
| A0A086TBI1 | Acremonium sp. | Metabolism | Lipid metabolism | glycerophosphodiester phosphodiesterase |
| A0A086T3W0 | Acremonium sp. | Metabolism | Metabolism of cofactors and vitamins | 5-formyltetrahydrofolate cyclo-ligase |
| A0A086TD10 | Acremonium sp. | Genetic information processing | Replication and repair | bloom syndrome protein |
| A0A367JPM9 | Rhizopus sp. | Metabolism | Amino acid metabolism | gamma-glutamyl hercynylcysteine S-oxide synthase |
| A0A2G4SN35 | Rhizopus sp. | Genetic information processing | Folding, sorting, and degradation | 20S proteasome subunit alpha 4 |
| I1CUB3 | Rhizopus sp. | Genetic information processing | Translation | small subunit ribosomal protein |
| A0A0K3CQU6 | Rhodotorula sp. | Metabolism | Lipid metabolism | glycerol kinase |
| A0A511KMS2 | Rhodotorula sp. | Genetic information processing | Translation | H/ACA ribonucleoprotein complex subunit 4 |
| A0A511K989 | Rhodotorula sp. | Human disease | Cancer | RAN-binding protein 1 |
| K0KHA2 | Wickerhamomyces sp. | Genetic information processing | Folding, sorting, and degradation | YidC/Oxa1 family membrane protein insertase |
| A0A1E3NZ77 | Wickerhamomyces sp. | Genetic information processing | Replication and repair | DNA excision repair protein ERCC-6 |
| K0KHS6 | Wickerhamomyces sp. | Genetic information processing | Transcription | DNA-directed RNA polymerase I subunit RPA2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mok, K.; Suratanon, N.; Roytrakul, S.; Charoenlappanit, S.; Patumcharoenpol, P.; Chatchatee, P.; Vongsangnak, W.; Nakphaichit, M. ITS2 Sequencing and Targeted Meta-Proteomics of Infant Gut Mycobiome Reveal the Functional Role of Rhodotorula sp. during Atopic Dermatitis Manifestation. J. Fungi 2021, 7, 748. https://doi.org/10.3390/jof7090748
Mok K, Suratanon N, Roytrakul S, Charoenlappanit S, Patumcharoenpol P, Chatchatee P, Vongsangnak W, Nakphaichit M. ITS2 Sequencing and Targeted Meta-Proteomics of Infant Gut Mycobiome Reveal the Functional Role of Rhodotorula sp. during Atopic Dermatitis Manifestation. Journal of Fungi. 2021; 7(9):748. https://doi.org/10.3390/jof7090748
Chicago/Turabian StyleMok, Kevin, Narissara Suratanon, Sittiruk Roytrakul, Sawanya Charoenlappanit, Preecha Patumcharoenpol, Pantipa Chatchatee, Wanwipa Vongsangnak, and Massalin Nakphaichit. 2021. "ITS2 Sequencing and Targeted Meta-Proteomics of Infant Gut Mycobiome Reveal the Functional Role of Rhodotorula sp. during Atopic Dermatitis Manifestation" Journal of Fungi 7, no. 9: 748. https://doi.org/10.3390/jof7090748
APA StyleMok, K., Suratanon, N., Roytrakul, S., Charoenlappanit, S., Patumcharoenpol, P., Chatchatee, P., Vongsangnak, W., & Nakphaichit, M. (2021). ITS2 Sequencing and Targeted Meta-Proteomics of Infant Gut Mycobiome Reveal the Functional Role of Rhodotorula sp. during Atopic Dermatitis Manifestation. Journal of Fungi, 7(9), 748. https://doi.org/10.3390/jof7090748